Novel Insights into the Role of HDL-Associated Sphingosine-1-Phosphate in Cardiometabolic Diseases

 , , and
, , and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. DCM: A Risk Factor for HF
3. S1P Determines Cardiomyocyte Fate
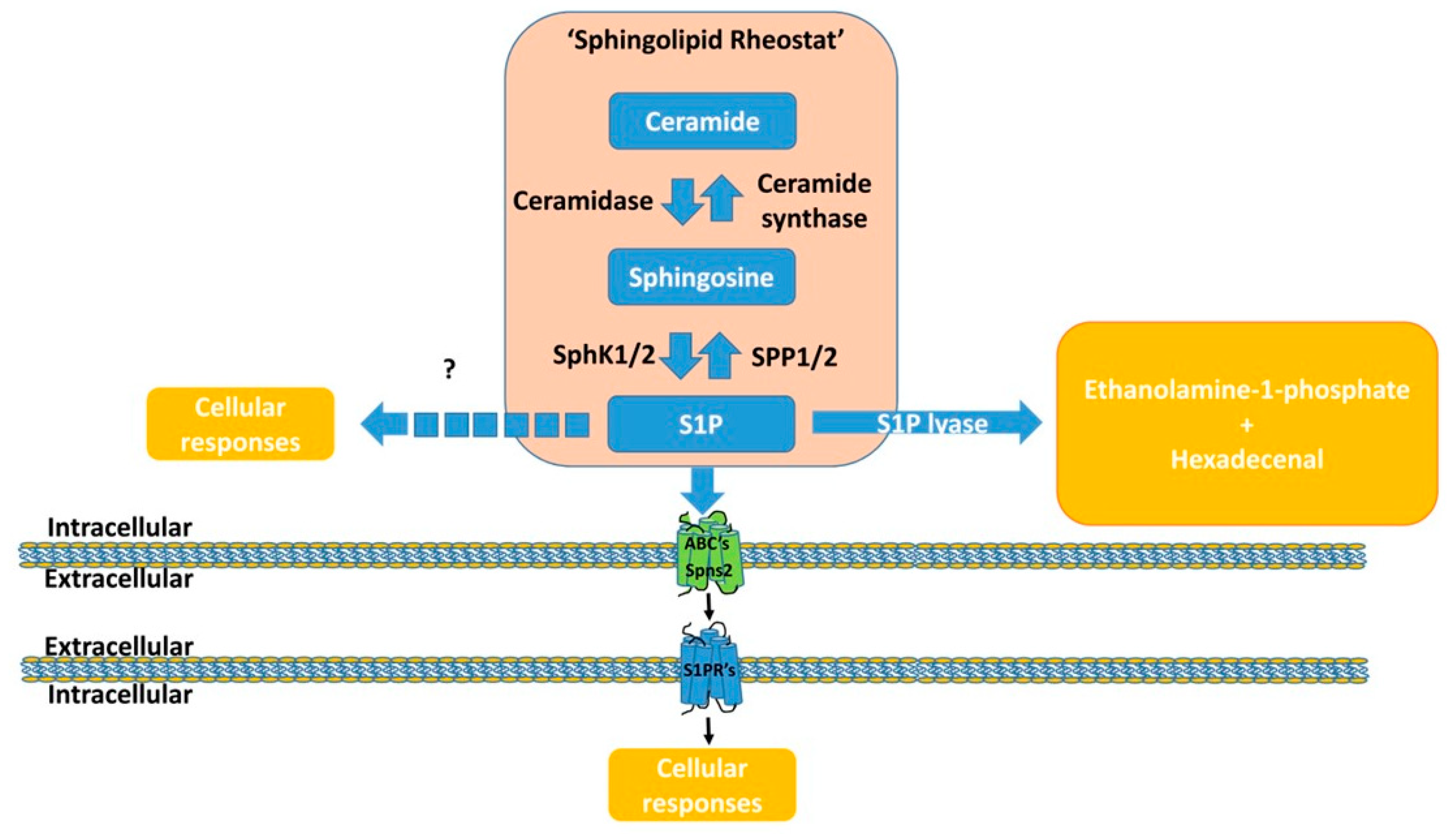
3.1. S1P Is an Intracellular Component of the “Sphingolipid Rheostat”
3.2. S1P Modulates Cellular Physiology
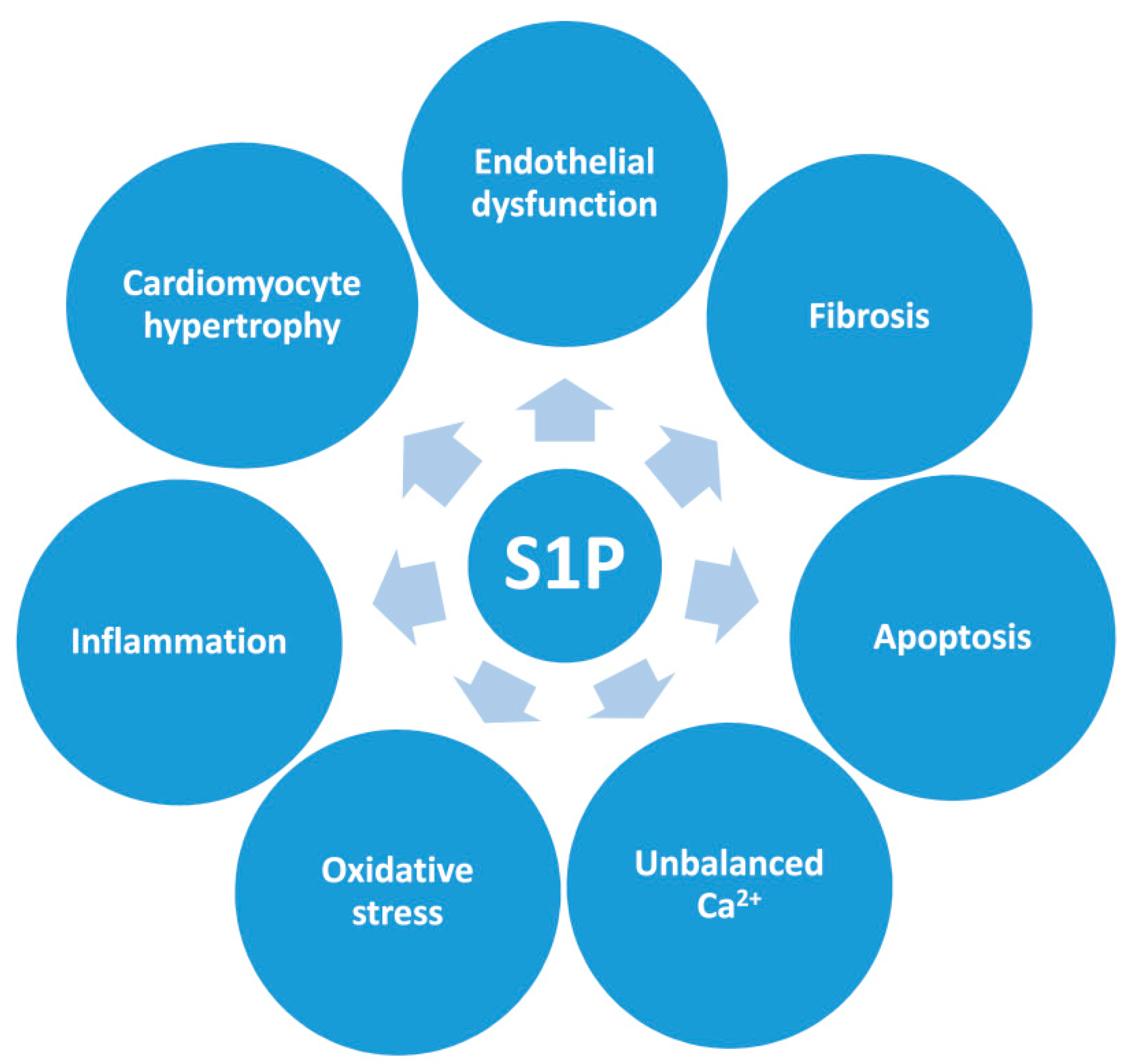
4. The Effect of S1P on the Cellular Mechanisms Involved in Cardiac Remodeling and Dysfunction
4.1. Oxidative Stress
4.2. Inflammation
4.3. Mitochondrial Dysfunction
4.4. Calcium Handling
4.5. Cardiomyocyte Hypertrophy
4.6. Autophagy
4.7. Apoptosis
4.8. Fibrosis
4.9. Endothelial Dysfunction
5. S1P in Circulation: HDL, More than a Cargo of S1P
5.1. HDL-Bound S1P and Apolipoprotein (Apo)M
5.2. Transporters Involved in Extracellular S1P Efflux and Signaling
5.2.1. Spinster2
5.2.2. ATP-Binding Cassette (ABC) Transporters
5.2.3. Scavenger Receptor Class B Type I (SR-BI)
5.3. Proteins Involved in Plasma HDL Remodeling
6. S1P in Cardiovascular Disease: A Role for HDL?
7. Assessment of Circulating S1P in Cardiometabolic Diseases
7.1. Circulating S1P/ApoM in Cardiac Diseases
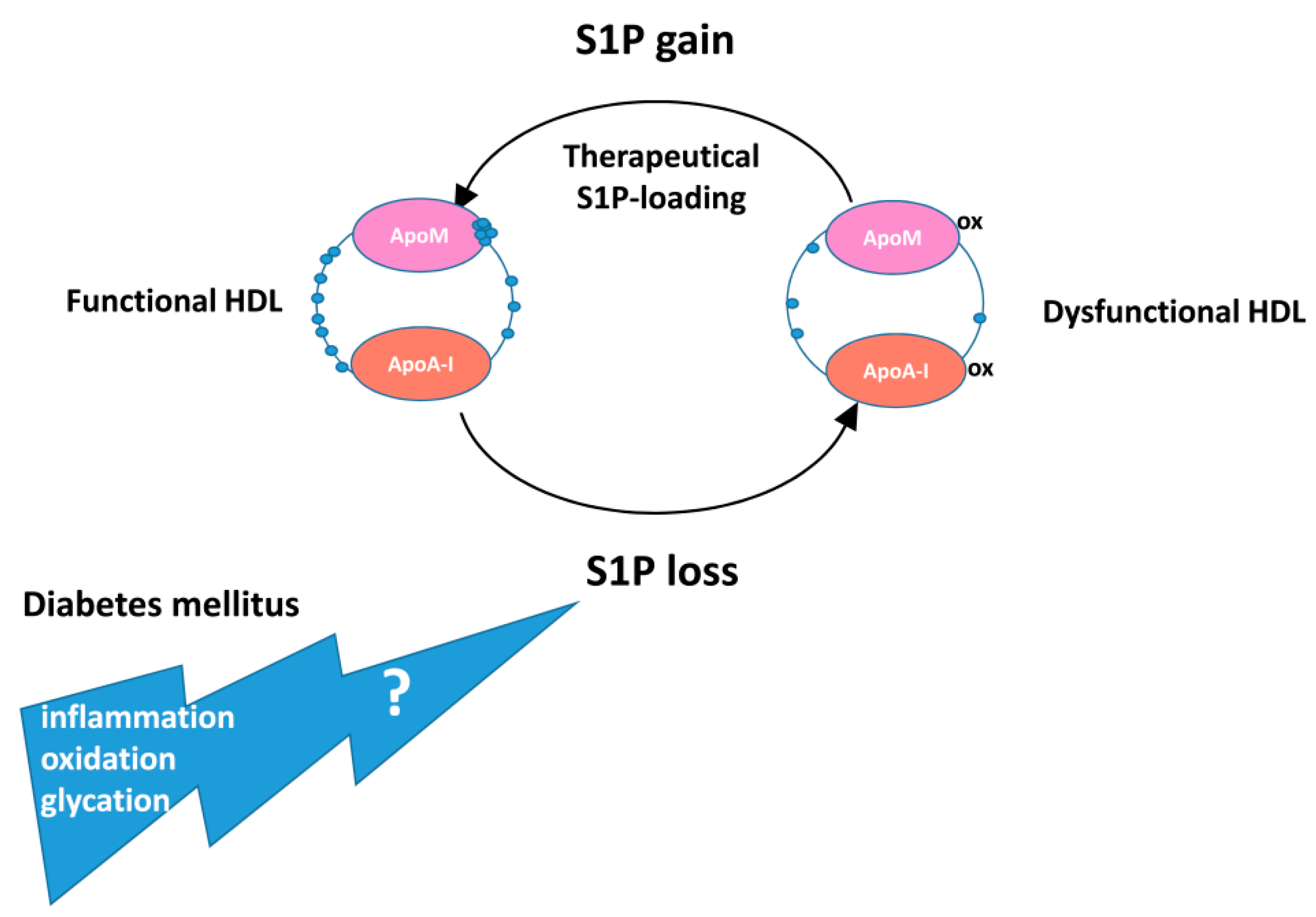
7.2. Circulating S1P/ApoM in Patients with Diabetes Mellitus and Relationship with Cardiac Disease
8. Assessment of S1P–S1PR-Based Experimental Pharmacological Strategies in Ischemic Heart Disease
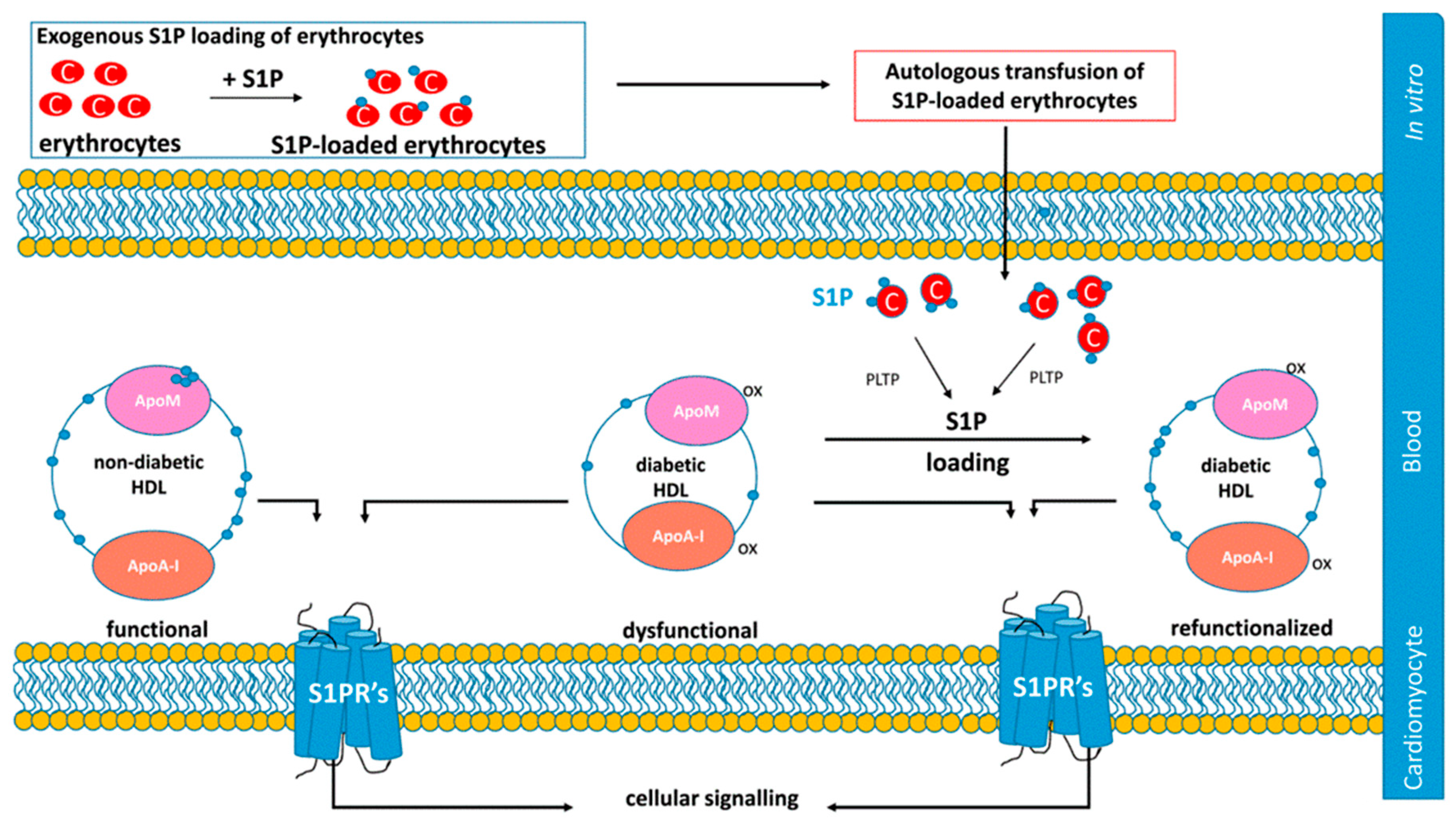
8.1. Strategies Targeting S1P Signaling to Improve Cardiac Dysfunction: A Role for HDL?
8.2. Pharmacological Approaches Targeting S1P Signaling
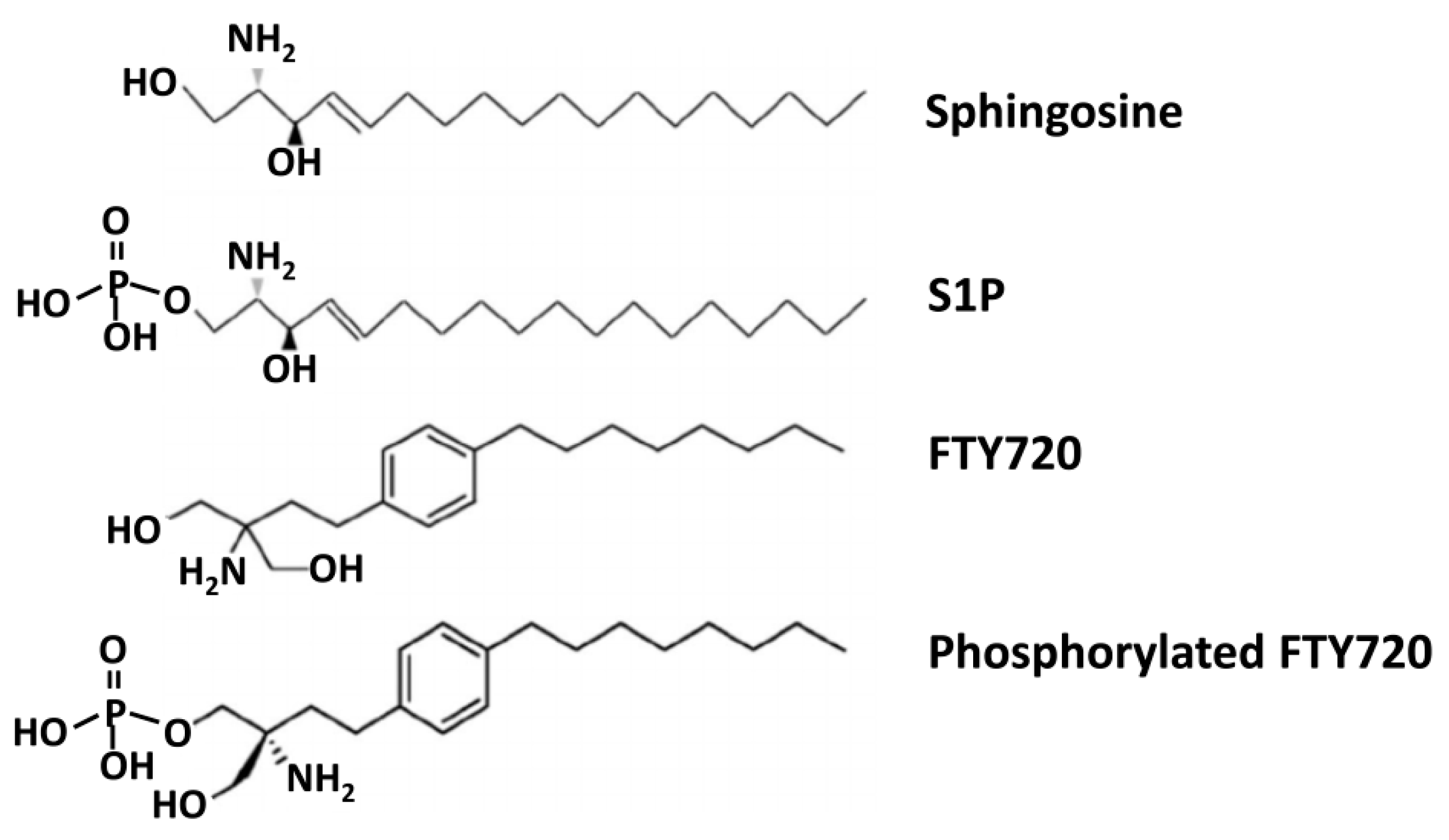
8.2.1. FTY720
8.2.2. SEW2871
8.3. Other Potential Pharmacological Strategies to Raise S1P Levels: Lipid-Modifying-Based Therapies
8.3.1. Statins
8.3.2. PPARγ
9. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| Apo | Apolipoprotein |
| eNOS | nitric oxide synthase |
| HDL | Plasma high-density lipoprotein fraction |
| HF | Heart failure |
| Non-HDL | Plasma non-high-density lipoprotein fraction |
| S1PR’s | S1P receptors |
| SphK | Sphingosine kinase |
| Spns | Spinster2 |
| SR-BI | Scavenger receptor class B, type I |
References
- Oudejans, I.; Mosterd, A.; Bloemen, J.A.; Valk, M.J.; van Velzen, E.; Wielders, J.P.; Zuithoff, N.P.; Rutten, F.H.; Hoes, A.W. Clinical evaluation of geriatric outpatients with suspected heart failure: Value of symptoms, signs, and additional tests. Eur. J. Heart Fail. 2011, 13, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; McGee, D.L. Diabetes and cardiovascular disease. The Framingham study. JAMA 1979, 241, 2035–2038. [Google Scholar] [CrossRef] [PubMed]
- Nichols, G.A.; Hillier, T.A.; Erbey, J.R.; Brown, J.B. Congestive heart failure in type 2 diabetes: Prevalence, incidence, and risk factors. Diabetes Care 2001, 24, 1614–1619. [Google Scholar] [CrossRef] [PubMed]
- Alonso, N.; Moliner, P.; Mauricio, D. Heart Failure: From Research to Clinical Practice; Islam, S., Ed.; Springer: Uppsala, Sweden, 2018; Volume 3, pp. 197–217. [Google Scholar]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef]
- Regan, T.J.; Lyons, M.M.; Ahmed, S.S.; Levinson, G.E.; Oldewurtel, H.A.; Ahmad, M.R.; Haider, B. Evidence for cardiomyopathy in familial diabetes mellitus. J. Clin. Investig. 1977, 60, 884–899. [Google Scholar] [CrossRef]
- Park, T.S.; Goldberg, I.J. Sphingolipids, lipotoxic cardiomyopathy, and cardiac failure. Heart Fail. Clin. 2012, 8, 633–641. [Google Scholar] [CrossRef]
- Russo, S.B.; Ross, J.S.; Cowart, L.A. Sphingolipids in obesity, type 2 diabetes, and metabolic disease. Handb. Exp. Pharmacol. 2013, 373–401. [Google Scholar] [CrossRef]
- Iqbal, J.; Walsh, M.T.; Hammad, S.M.; Hussain, M.M. Sphingolipids and Lipoproteins in Health and Metabolic Disorders. Trends Endocrinol. Metab. 2017, 28, 506–518. [Google Scholar] [CrossRef]
- Levkau, B. HDL-S1P: Cardiovascular functions, disease-associated alterations, and therapeutic applications. Front. Pharmacol. 2015, 6, 243. [Google Scholar] [CrossRef]
- Poti, F.; Simoni, M.; Nofer, J.R. Atheroprotective role of high-density lipoprotein (HDL)-associated sphingosine-1-phosphate (S1P). Cardiovasc. Res. 2014, 103, 395–404. [Google Scholar] [CrossRef]
- Srivastava, R.A.K. Dysfunctional HDL in diabetes mellitus and its role in the pathogenesis of cardiovascular disease. Mol. Cell. Biochem. 2018, 440, 167–187. [Google Scholar] [CrossRef] [PubMed]
- From, A.M.; Scott, C.G.; Chen, H.H. Changes in diastolic dysfunction in diabetes mellitus over time. Am. J. Cardiol. 2009, 103, 1463–1466. [Google Scholar] [CrossRef] [PubMed]
- Teupe, C.; Rosak, C. Diabetic cardiomyopathy and diastolic heart failure—Difficulties with relaxation. Diabetes Res. Clin. Pract. 2012, 97, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Velez, M.; Kohli, S.; Sabbah, H.N. Animal models of insulin resistance and heart failure. Heart Fail. Rev. 2014, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy revisited. Circulation 2007, 115, 3213–3223. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.S.; Jaferi, G.A.; Narang, R.M.; Regan, T.J. Preclinical abnormality of left ventricular function in diabetes mellitus. Am. Heart J. 1975, 89, 153–158. [Google Scholar] [CrossRef]
- Alonso, N.; Lupon, J.; Barallat, J.; de Antonio, M.; Domingo, M.; Zamora, E.; Moliner, P.; Galan, A.; Santesmases, J.; Pastor, C.; et al. Impact of diabetes on the predictive value of heart failure biomarkers. Cardiovasc. Diabetol. 2016, 15, 151. [Google Scholar] [CrossRef]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef]
- Karliner, J.S. Sphingosine kinase and sphingosine 1-phosphate in cardioprotection. J. Cardiovasc. Pharmacol. 2009, 53, 189–197. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Luberto, C.; Argraves, K.M. Enzymes of sphingolipid metabolism: From modular to integrative signaling. Biochemistry 2001, 40, 4893–4903. [Google Scholar] [CrossRef]
- D’Angelo, G.; Uemura, T.; Chuang, C.C.; Polishchuk, E.; Santoro, M.; Ohvo-Rekila, H.; Sato, T.; Di Tullio, G.; Varriale, A.; D’Auria, S.; et al. Vesicular and non-vesicular transport feed distinct glycosylation pathways in the Golgi. Nature 2013, 501, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Adada, M.; Luberto, C.; Canals, D. Inhibitors of the sphingomyelin cycle: Sphingomyelin synthases and sphingomyelinases. Chem. Phys. Lipids 2016, 197, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Mandala, S.M.; Thornton, R.; Galve-Roperh, I.; Poulton, S.; Peterson, C.; Olivera, A.; Bergstrom, J.; Kurtz, M.B.; Spiegel, S. Molecular cloning and characterization of a lipid phosphohydrolase that degrades sphingosine-1- phosphate and induces cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 7859–7864. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, C.; Kihara, A.; Gokoh, M.; Igarashi, Y. Identification and characterization of a novel human sphingosine-1-phosphate phosphohydrolase, hSPP2. J. Biol. Chem. 2003, 278, 1268–1272. [Google Scholar] [CrossRef] [PubMed]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef] [PubMed]
- Karliner, J.S.; Honbo, N.; Summers, K.; Gray, M.O.; Goetzl, E.J. The lysophospholipids sphingosine-1-phosphate and lysophosphatidic acid enhance survival during hypoxia in neonatal rat cardiac myocytes. J. Mol. Cell. Cardiol. 2001, 33, 1713–1717. [Google Scholar] [CrossRef] [PubMed]
- Delpy, E.; Hatem, S.N.; Andrieu, N.; de Vaumas, C.; Henaff, M.; Rucker-Martin, C.; Jaffrezou, J.P.; Laurent, G.; Levade, T.; Mercadier, J.J. Doxorubicin induces slow ceramide accumulation and late apoptosis in cultured adult rat ventricular myocytes. Cardiovasc. Res. 1999, 43, 398–407. [Google Scholar] [CrossRef]
- Zhang, D.X.; Fryer, R.M.; Hsu, A.K.; Zou, A.P.; Gross, G.J.; Campbell, W.B.; Li, P.L. Production and metabolism of ceramide in normal and ischemic-reperfused myocardium of rats. Basic Res. Cardiol. 2001, 96, 267–274. [Google Scholar] [CrossRef]
- Murata, N.; Sato, K.; Kon, J.; Tomura, H.; Yanagita, M.; Kuwabara, A.; Ui, M.; Okajima, F. Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem. J. 2000, 352, 809–815. [Google Scholar] [CrossRef]
- Takabe, K.; Paugh, S.W.; Milstien, S.; Spiegel, S. “Inside-out” signaling of sphingosine-1-phosphate: Therapeutic targets. Pharmacol. Rev. 2008, 60, 181–195. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Merrill, A.H., Jr. Sphingolipid metabolism and cell growth regulation. FASEB J. 1996, 10, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Van Brocklyn, J.R.; Thangada, S.; Liu, C.H.; Hand, A.R.; Menzeleev, R.; Spiegel, S.; Hla, T. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science 1998, 279, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Mao, J.; Redfield, S.; Mo, Y.; Lage, J.M.; Zhou, X. Systemic distribution, subcellular localization and differential expression of sphingosine-1-phosphate receptors in benign and malignant human tissues. Exp. Mol. Pathol. 2014, 97, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Yi, S.; Zhuang, H.; Wu, L.; Wang, D.W.; Jiang, J. Sphingosine-1-phosphate ameliorates the cardiac hypertrophic response through inhibiting the activity of histone deacetylase-2. Int. J. Mol. Med. 2018, 41, 1704–1714. [Google Scholar] [CrossRef]
- Nakajima, N.; Cavalli, A.L.; Biral, D.; Glembotski, C.C.; McDonough, P.M.; Ho, P.D.; Betto, R.; Sandona, D.; Palade, P.T.; Dettbarn, C.A.; et al. Expression and characterization of Edg-1 receptors in rat cardiomyocytes: Calcium deregulation in response to sphingosine 1-phosphate. Eur. J. Biochem. 2000, 267, 5679–5686. [Google Scholar] [CrossRef]
- Robert, P.; Tsui, P.; Laville, M.P.; Livi, G.P.; Sarau, H.M.; Bril, A.; Berrebi-Bertrand, I. EDG1 receptor stimulation leads to cardiac hypertrophy in rat neonatal myocytes. J. Mol. Cell. Cardiol. 2001, 33, 1589–1606. [Google Scholar] [CrossRef]
- Mazurais, D.; Robert, P.; Gout, B.; Berrebi-Bertrand, I.; Laville, M.P.; Calmels, T. Cell type-specific localization of human cardiac S1P receptors. J. Histochem. Cytochem. 2002, 50, 661–670. [Google Scholar] [CrossRef]
- Rivera, R.; Chun, J. Biological effects of lysophospholipids. Rev. Physiol. Biochem. Pharmacol. 2008, 160, 25–46. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: Signaling inside and out. FEBS Lett. 2000, 476, 55–57. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Sphingosine 1-phosphate, a key cell signaling molecule. J. Biol. Chem. 2002, 277, 25851–25854. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S. Sphingosine 1-phosphate: A ligand for the EDG-1 family of G-protein-coupled receptors. Ann. N. Y. Acad. Sci. 2000, 905, 54–60. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Functions of a new family of sphingosine-1-phosphate receptors. Biochim. Biophys. Acta 2000, 1484, 107–116. [Google Scholar] [CrossRef]
- Pyne, S.; Pyne, N. Sphingosine 1-phosphate signalling via the endothelial differentiation gene family of G-protein-coupled receptors. Pharmacol. Ther. 2000, 88, 115–131. [Google Scholar] [CrossRef]
- Hla, T.; Lee, M.J.; Ancellin, N.; Thangada, S.; Liu, C.H.; Kluk, M.; Chae, S.S.; Wu, M.T. Sphingosine-1-phosphate signaling via the EDG-1 family of G-protein-coupled receptors. Ann. N. Y. Acad. Sci. 2000, 905, 16–24. [Google Scholar] [CrossRef]
- Harnett, M.M. Sphingolipid Signaling. In Cell Signaling in Vascular Inflammation; Bhattacharya, J., Ed.; Humana Press: Totowa, NJ, USA, 2005; Volume 10, pp. 91–101. [Google Scholar] [CrossRef]
- Okamoto, Y.; Wang, F.; Yoshioka, K.; Takuwa, N.; Takuwa, Y. Sphingosine-1-phosphate-specific G protein -coupled receptors as novel therapeutic targets for atherosclerosis. Pharmaceuticals 2011, 4, 117–137. [Google Scholar] [CrossRef]
- Alewijnse, A.E.; Peters, S.L.; Michel, M.C. Cardiovascular effects of sphingosine-1-phosphate and other sphingomyelin metabolites. Br. J. Pharmacol. 2004, 143, 666–684. [Google Scholar] [CrossRef]
- Sattler, K.J.; Elbasan, S.; Keul, P.; Elter-Schulz, M.; Bode, C.; Graler, M.H.; Brocker-Preuss, M.; Budde, T.; Erbel, R.; Heusch, G.; et al. Sphingosine 1-phosphate levels in plasma and HDL are altered in coronary artery disease. Basic Res. Cardiol. 2010, 105, 821–832. [Google Scholar] [CrossRef]
- Awojoodu, A.O.; Ogle, M.E.; Sefcik, L.S.; Bowers, D.T.; Martin, K.; Brayman, K.L.; Lynch, K.R.; Peirce-Cottler, S.M.; Botchwey, E. Sphingosine 1-phosphate receptor 3 regulates recruitment of anti-inflammatory monocytes to microvessels during implant arteriogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 13785–13790. [Google Scholar] [CrossRef] [PubMed]
- Bot, M.; Van Veldhoven, P.P.; de Jager, S.C.; Johnson, J.; Nijstad, N.; Van Santbrink, P.J.; Westra, M.M.; Van Der Hoeven, G.; Gijbels, M.J.; Muller-Tidow, C.; et al. Hematopoietic sphingosine 1-phosphate lyase deficiency decreases atherosclerotic lesion development in LDL-receptor deficient mice. PLoS ONE 2013, 8, e63360. [Google Scholar] [CrossRef] [PubMed]
- Waeber, C.; Walther, T. Sphingosine-1-phosphate as a potential target for the treatment of myocardial infarction. Circ. J. 2014, 78, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Sattler, K.; Graler, M.; Keul, P.; Weske, S.; Reimann, C.M.; Jindrova, H.; Kleinbongard, P.; Sabbadini, R.; Brocker-Preuss, M.; Erbel, R.; et al. Defects of High-Density Lipoproteins in Coronary Artery Disease Caused by Low Sphingosine-1-Phosphate Content: Correction by Sphingosine-1-Phosphate-Loading. J. Am. Coll. Cardiol. 2015, 66, 1470–1485. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Linardi, D.; Muhammad, N.; Chiamulera, C.; Fumagalli, G.; Biagio, L.S.; Gebrie, M.A.; Aslam, M.; Luciani, G.B.; Faggian, G.; et al. Sphingosine 1-Phosphate Receptor Modulator Fingolimod (FTY720) Attenuates Myocardial Fibrosis in Post-heterotopic Heart Transplantation. Front. Pharmacol. 2017, 8, 645. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.; Qian, A.S.; Tahir, U.; Yu, P.; Trigatti, B.L. Sphingosine-1-Phosphate Receptor 1, Expressed in Myeloid Cells, Slows Diet-Induced Atherosclerosis and Protects against Macrophage Apoptosis in Ldlr KO Mice. Int. J. Mol. Sci. 2017, 18, 2721. [Google Scholar] [CrossRef]
- Polzin, A.; Piayda, K.; Keul, P.; Dannenberg, L.; Mohring, A.; Graler, M.; Zeus, T.; Kelm, M.; Levkau, B. Plasma sphingosine-1-phosphate concentrations are associated with systolic heart failure in patients with ischemic heart disease. J. Mol. Cell. Cardiol. 2017, 110, 35–37. [Google Scholar] [CrossRef]
- Chen, R.; Cai, X.; Liu, J.; Bai, B.; Li, X. Sphingosine 1-phosphate promotes mesenchymal stem cell-mediated cardioprotection against myocardial infarction via ERK1/2-MMP-9 and Akt signaling axis. Life Sci. 2018, 215, 31–42. [Google Scholar] [CrossRef]
- Deshpande, G.P.; Imamdin, A.; Lecour, S.; Opie, L.H. Sphingosine-1-phosphate (S1P) activates STAT3 to protect against de novo acute heart failure (AHF). Life Sci. 2018, 196, 127–132. [Google Scholar] [CrossRef]
- Feuerborn, R.; Besser, M.; Poti, F.; Burkhardt, R.; Weissen-Plenz, G.; Ceglarek, U.; Simoni, M.; Proia, R.L.; Freise, H.; Nofer, J.R. Elevating Endogenous Sphingosine-1-Phosphate (S1P) Levels Improves Endothelial Function and Ameliorates Atherosclerosis in Low Density Lipoprotein Receptor-Deficient (LDL-R-/-) Mice. Thromb. Haemost. 2018, 118, 1470–1480. [Google Scholar] [CrossRef]
- Nofer, J.R. High-density lipoprotein, sphingosine 1-phosphate, and atherosclerosis. J. Clin. Lipidol. 2008, 2, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Keul, P.; Sattler, K.; Levkau, B. HDL and its sphingosine-1-phosphate content in cardioprotection. Heart Fail. Rev. 2007, 12, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Karliner, J.S. Sphingosine kinase regulation and cardioprotection. Cardiovasc. Res. 2009, 82, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Karliner, J.S. Sphingosine kinase and sphingosine 1-phosphate in the heart: A decade of progress. Biochim. Biophys. Acta 2013, 1831, 203–212. [Google Scholar] [CrossRef]
- Cannavo, A.; Rengo, G.; Liccardo, D.; Pagano, G.; Zincarelli, C.; De Angelis, M.C.; Puglia, R.; Di Pietro, E.; Rabinowitz, J.E.; Barone, M.V.; et al. beta1-adrenergic receptor and sphingosine-1-phosphate receptor 1 (S1PR1) reciprocal downregulation influences cardiac hypertrophic response and progression to heart failure: Protective role of S1PR1 cardiac gene therapy. Circulation 2013, 128, 1612–1622. [Google Scholar] [CrossRef]
- Means, C.K.; Xiao, C.Y.; Li, Z.; Zhang, T.; Omens, J.H.; Ishii, I.; Chun, J.; Brown, J.H. Sphingosine 1-phosphate S1P2 and S1P3 receptor-mediated Akt activation protects against in vivo myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2944–H2951. [Google Scholar] [CrossRef]
- Cannavo, A.; Liccardo, D.; Komici, K.; Corbi, G.; de Lucia, C.; Femminella, G.D.; Elia, A.; Bencivenga, L.; Ferrara, N.; Koch, W.J.; et al. Sphingosine Kinases and Sphingosine 1-Phosphate Receptors: Signaling and Actions in the Cardiovascular System. Front. Pharmacol. 2017, 8, 556. [Google Scholar] [CrossRef]
- Seferovic, P.M.; Paulus, W.J. Clinical diabetic cardiomyopathy: A two-faced disease with restrictive and dilated phenotypes. Eur. Heart J. 2015, 36, 1718–1727. [Google Scholar] [CrossRef]
- Zhang, J.; Honbo, N.; Goetzl, E.J.; Chatterjee, K.; Karliner, J.S.; Gray, M.O. Signals from type 1 sphingosine 1-phosphate receptors enhance adult mouse cardiac myocyte survival during hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3150–H3158. [Google Scholar] [CrossRef]
- Spillmann, F.; Van Linthout, S.; Tschope, C. Cardiac effects of HDL and its components on diabetic cardiomyopathy. Endocr. Metab. Immune Disord. Drug Targets 2012, 12, 132–147. [Google Scholar] [CrossRef]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Ravassa, S.; Beaumont, J.; Lopez, B.; Diez, J. New targets to treat the structural remodeling of the myocardium. J. Am. Coll. Cardiol. 2011, 58, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Bielawska, A.E.; Shapiro, J.P.; Jiang, L.; Melkonyan, H.S.; Piot, C.; Wolfe, C.L.; Tomei, L.D.; Hannun, Y.A.; Umansky, S.R. Ceramide is involved in triggering of cardiomyocyte apoptosis induced by ischemia and reperfusion. Am. J. Pathol. 1997, 151, 1257–1263. [Google Scholar]
- Pchejetski, D.; Kunduzova, O.; Dayon, A.; Calise, D.; Seguelas, M.H.; Leducq, N.; Seif, I.; Parini, A.; Cuvillier, O. Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circ. Res. 2007, 100, 41–49. [Google Scholar] [CrossRef]
- Pyszko, J.; Strosznajder, J.B. Sphingosine kinase 1 and sphingosine-1-phosphate in oxidative stress evoked by 1-methyl-4-phenylpyridinium (MPP+) in human dopaminergic neuronal cells. Mol. Neurobiol. 2014, 50, 38–48. [Google Scholar] [CrossRef]
- Taha, T.A.; Osta, W.; Kozhaya, L.; Bielawski, J.; Johnson, K.R.; Gillanders, W.E.; Dbaibo, G.S.; Hannun, Y.A.; Obeid, L.M. Down-regulation of sphingosine kinase-1 by DNA damage: Dependence on proteases and p53. J. Biol. Chem. 2004, 279, 20546–20554. [Google Scholar] [CrossRef]
- Pchejetski, D.; Golzio, M.; Bonhoure, E.; Calvet, C.; Doumerc, N.; Garcia, V.; Mazerolles, C.; Rischmann, P.; Teissie, J.; Malavaud, B.; et al. Sphingosine kinase-1 as a chemotherapy sensor in prostate adenocarcinoma cell and mouse models. Cancer Res. 2005, 65, 11667–11675. [Google Scholar] [CrossRef]
- Bouras, G.; Giannopoulos, G.; Hatzis, G.; Alexopoulos, D.; Leventopoulos, G.; Deftereos, S. Inflammation and chronic heart failure: From biomarkers to novel anti-inflammatory therapeutic strategies. Med. Chem. 2014, 10, 682–699. [Google Scholar] [CrossRef]
- Weigert, A.; Weis, N.; Brune, B. Regulation of macrophage function by sphingosine-1-phosphate. Immunobiology 2009, 214, 748–760. [Google Scholar] [CrossRef]
- Duenas, A.I.; Aceves, M.; Fernandez-Pisonero, I.; Gomez, C.; Orduna, A.; Crespo, M.S.; Garcia-Rodriguez, C. Selective attenuation of Toll-like receptor 2 signalling may explain the atheroprotective effect of sphingosine 1-phosphate. Cardiovasc. Res. 2008, 79, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Theilmeier, G.; Schmidt, C.; Herrmann, J.; Keul, P.; Schafers, M.; Herrgott, I.; Mersmann, J.; Larmann, J.; Hermann, S.; Stypmann, J.; et al. High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation 2006, 114, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Keul, P.; Lucke, S.; von Wnuck Lipinski, K.; Bode, C.; Graler, M.; Heusch, G.; Levkau, B. Sphingosine-1-phosphate receptor 3 promotes recruitment of monocyte/macrophages in inflammation and atherosclerosis. Circ. Res. 2011, 108, 314–323. [Google Scholar] [CrossRef]
- Skoura, A.; Michaud, J.; Im, D.S.; Thangada, S.; Xiong, Y.; Smith, J.D.; Hla, T. Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Nakazawa, T.; Cho, A.; Dastvan, F.; Shilling, D.; Daum, G.; Reidy, M.A. Sphingosine 1-phosphate receptor 2 negatively regulates neointimal formation in mouse arteries. Circ. Res. 2007, 101, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Grabski, A.D.; Shimizu, T.; Deou, J.; Mahoney, W.M., Jr.; Reidy, M.A.; Daum, G. Sphingosine-1-phosphate receptor-2 regulates expression of smooth muscle alpha-actin after arterial injury. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1644–1650. [Google Scholar] [CrossRef] [PubMed]
- Jezek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef]
- Moris, D.; Spartalis, M.; Spartalis, E.; Karachaliou, G.S.; Karaolanis, G.I.; Tsourouflis, G.; Tsilimigras, D.I.; Tzatzaki, E.; Theocharis, S. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann. Transl Med. 2017, 5, 326. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, D.; Zhang, L.; Ye, F.; Li, M.; Wen, K. Role of JAK-STAT pathway in reducing cardiomyocytes hypoxia/reoxygenation injury induced by S1P postconditioning. Eur. J. Pharmacol. 2016, 784, 129–136. [Google Scholar] [CrossRef]
- Ke, M.; Tang, Q.; Pan, Z.; Yin, Y.; Zhang, L.; Wen, K. Sphingosine-1-phosphate attenuates hypoxia/reoxygenation-induced cardiomyocyte injury via a mitochondrial pathway. Biochem. Biophys. Res. Commun. 2019, 510, 142–148. [Google Scholar] [CrossRef]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011, 25, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Maier, L.S.; Hasenfuss, G. Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart. Heart Fail. Rev. 2009, 14, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Egom, E.E.; Bae, J.S.; Capel, R.; Richards, M.; Ke, Y.; Pharithi, R.B.; Maher, V.; Kruzliak, P.; Lei, M. Effect of sphingosine-1-phosphate on L-type calcium current and Ca(2+) transient in rat ventricular myocytes. Mol. Cell. Biochem. 2016, 419, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, A.; Yatomi, Y.; Ozaki, Y.; Hashimoto, K. Sphingosine 1-phosphate induces sinus tachycardia and coronary vasoconstriction in the canine heart. Cardiovasc. Res. 2000, 46, 119–125. [Google Scholar] [CrossRef][Green Version]
- Valverde, C.A.; Kornyeyev, D.; Ferreiro, M.; Petrosky, A.D.; Mattiazzi, A.; Escobar, A.L. Transient Ca2+ depletion of the sarcoplasmic reticulum at the onset of reperfusion. Cardiovasc. Res. 2010, 85, 671–680. [Google Scholar] [CrossRef]
- Ai, X.; Curran, J.W.; Shannon, T.R.; Bers, D.M.; Pogwizd, S.M. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 2005, 97, 1314–1322. [Google Scholar] [CrossRef]
- Yan, W.; Zhang, F.; Zhang, R.; Zhang, X.; Wang, Y.; Zhou, F.; Xia, Y.; Liu, P.; Gao, C.; Wang, H.; et al. Adiponectin regulates SR Ca(2+) cycling following ischemia/reperfusion via sphingosine 1-phosphate-CaMKII signaling in mice. J. Mol. Cell. Cardiol. 2014, 74, 183–192. [Google Scholar] [CrossRef]
- Keul, P.; van Borren, M.M.; Ghanem, A.; Muller, F.U.; Baartscheer, A.; Verkerk, A.O.; Stumpel, F.; Schulte, J.S.; Hamdani, N.; Linke, W.A.; et al. Sphingosine-1-Phosphate Receptor 1 Regulates Cardiac Function by Modulating Ca2+ Sensitivity and Na+/H+ Exchange and Mediates Protection by Ischemic Preconditioning. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Yokoyama, T.; Arai, M.; Sekiguchi, K.; Tanaka, T.; Kanda, T.; Suzuki, T.; Nagai, R. Tumor necrosis factor-alpha decreases the phosphorylation levels of phospholamban and troponin I in spontaneously beating rat neonatal cardiac myocytes. J. Mol. Cell. Cardiol. 1999, 31, 261–273. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Wang, F.; Wang, H.J.; Liu, H.B. Sphingosine 1 phosphate receptor-1 (S1PR1) signaling protects cardiac function by inhibiting cardiomyocyte autophagy. J. Geriatr. Cardiol. 2018, 15, 334–345. [Google Scholar] [CrossRef]
- Zhang, F.; Xia, Y.; Yan, W.; Zhang, H.; Zhou, F.; Zhao, S.; Wang, W.; Zhu, D.; Xin, C.; Lee, Y.; et al. Sphingosine 1-phosphate signaling contributes to cardiac inflammation, dysfunction, and remodeling following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H250–H261. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, S.I.; Usui, S.; Takashima, S.I.; Takuwa, N.; Yoshioka, K.; Okamoto, Y.; Inagaki, Y.; Sugimoto, N.; Kitano, T.; Takamura, M.; et al. Augmented sphingosine 1 phosphate receptor-1 signaling in cardiac fibroblasts induces cardiac hypertrophy and fibrosis through angiotensin II and interleukin-6. PLoS ONE 2017, 12, e0182329. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, A.B.; Gottlieb, R.A. Recycle or die: The role of autophagy in cardioprotection. J. Mol. Cell. Cardiol. 2008, 44, 654–661. [Google Scholar] [CrossRef]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef]
- Jimenez, R.E.; Kubli, D.A.; Gustafsson, A.B. Autophagy and mitophagy in the myocardium: Therapeutic potential and concerns. Br. J. Pharmacol. 2014, 171, 1907–1916. [Google Scholar] [CrossRef]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Investig. 2007, 117, 1782–1793. [Google Scholar] [CrossRef]
- Wang, X.; Cui, T. Autophagy modulation: A potential therapeutic approach in cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H304–H319. [Google Scholar] [CrossRef]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef]
- Lavieu, G.; Scarlatti, F.; Sala, G.; Carpentier, S.; Levade, T.; Ghidoni, R.; Botti, J.; Codogno, P. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J. Biol. Chem. 2006, 281, 8518–8527. [Google Scholar] [CrossRef]
- Lavieu, G.; Scarlatti, F.; Sala, G.; Levade, T.; Ghidoni, R.; Botti, J.; Codogno, P. Is autophagy the key mechanism by which the sphingolipid rheostat controls the cell fate decision? Autophagy 2007, 3, 45–47. [Google Scholar] [CrossRef]
- Xie, Z.; He, C.; Zou, M.H. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy 2011, 7, 1254–1255. [Google Scholar] [CrossRef]
- Xie, Z.; Lau, K.; Eby, B.; Lozano, P.; He, C.; Pennington, B.; Li, H.; Rathi, S.; Dong, Y.; Tian, R.; et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 2011, 60, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Liang, Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim. Biophys. Acta 2015, 1852, 252–261. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhu, H.; Li, H.; Zou, M.H.; Xie, Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes 2013, 62, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- Lecour, S.; Smith, R.M.; Woodward, B.; Opie, L.H.; Rochette, L.; Sack, M.N. Identification of a novel role for sphingolipid signaling in TNF alpha and ischemic preconditioning mediated cardioprotection. J. Mol. Cell. Cardiol. 2002, 34, 509–518. [Google Scholar] [CrossRef]
- Tao, R.; Zhang, J.; Vessey, D.A.; Honbo, N.; Karliner, J.S. Deletion of the sphingosine kinase-1 gene influences cell fate during hypoxia and glucose deprivation in adult mouse cardiomyocytes. Cardiovasc. Res. 2007, 74, 56–63. [Google Scholar] [CrossRef]
- Wang, S.; Lin, X.; Wang, L.Y.; Ruan, K.F.; Feng, Y.; Li, X.Y. A polysaccharides MDG-1 augments survival in the ischemic heart by inducing S1P release and S1P1 expression. Int. J. Biol. Macromol. 2012, 50, 734–740. [Google Scholar] [CrossRef]
- Sun, J.; Yan, G.; Ren, A.; You, B.; Liao, J.K. FHL2/SLIM3 decreases cardiomyocyte survival by inhibitory interaction with sphingosine kinase-1. Circ. Res. 2006, 99, 468–476. [Google Scholar] [CrossRef]
- Ho, C.Y.; Lopez, B.; Coelho-Filho, O.R.; Lakdawala, N.K.; Cirino, A.L.; Jarolim, P.; Kwong, R.; Gonzalez, A.; Colan, S.D.; Seidman, J.G.; et al. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N. Engl. J. Med. 2010, 363, 552–563. [Google Scholar] [CrossRef]
- Gulati, A.; Japp, A.G.; Raza, S.; Halliday, B.P.; Jones, D.A.; Newsome, S.; Ismail, N.A.; Morarji, K.; Khwaja, J.; Spath, N.; et al. Absence of Myocardial Fibrosis Predicts Favorable Long-Term Survival in New-Onset Heart Failure. Circ. Cardiovasc. Imaging 2018, 11, e007722. [Google Scholar] [CrossRef]
- Wu, C.K.; Su, M.M.; Wu, Y.F.; Hwang, J.J.; Lin, L.Y. Combination of Plasma Biomarkers and Clinical Data for the Detection of Myocardial Fibrosis or Aggravation of Heart Failure Symptoms in Heart Failure with Preserved Ejection Fraction Patients. J. Clin. Med. 2018, 7, 427. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; He, X.; Zeng, M. The Role of S1P and the Related Signaling Pathway in the Development of Tissue Fibrosis. Front. Pharmacol. 2018, 9, 1504. [Google Scholar] [CrossRef] [PubMed]
- Gellings Lowe, N.; Swaney, J.S.; Moreno, K.M.; Sabbadini, R.A. Sphingosine-1-phosphate and sphingosine kinase are critical for transforming growth factor-beta-stimulated collagen production by cardiac fibroblasts. Cardiovasc. Res. 2009, 82, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, N.; Ohkura, S.; Takashima, S.; Ohtani, K.; Okamoto, Y.; Tanaka, T.; Hirano, K.; Usui, S.; Wang, F.; Du, W.; et al. S1P3-mediated cardiac fibrosis in sphingosine kinase 1 transgenic mice involves reactive oxygen species. Cardiovasc. Res. 2010, 85, 484–493. [Google Scholar] [CrossRef]
- Krenning, G.; Zeisberg, E.M.; Kalluri, R. The origin of fibroblasts and mechanism of cardiac fibrosis. J. Cell. Physiol. 2010, 225, 631–637. [Google Scholar] [CrossRef]
- Levick, J.R.; Michel, C.C. Microvascular fluid exchange and the revised Starling principle. Cardiovasc. Res. 2010, 87, 198–210. [Google Scholar] [CrossRef]
- Leucker, T.M.; Jones, S.P. Endothelial dysfunction as a nexus for endothelial cell-cardiomyocyte miscommunication. Front. Physiol. 2014, 5, 328. [Google Scholar] [CrossRef]
- Wilkerson, B.A.; Argraves, K.M. The role of sphingosine-1-phosphate in endothelial barrier function. Biochim. Biophys. Acta 2014, 1841, 1403–1412. [Google Scholar] [CrossRef]
- Zhang, G.; Xu, S.; Qian, Y.; He, P. Sphingosine-1-phosphate prevents permeability increases via activation of endothelial sphingosine-1-phosphate receptor 1 in rat venules. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1494–H1504. [Google Scholar] [CrossRef]
- Kurano, M.; Yatomi, Y. Sphingosine 1-Phosphate and Atherosclerosis. J. Atheroscler. Thromb. 2018, 25, 16–26. [Google Scholar] [CrossRef]
- Igarashi, J.; Bernier, S.G.; Michel, T. Sphingosine 1-phosphate and activation of endothelial nitric-oxide synthase. differential regulation of Akt and MAP kinase pathways by EDG and bradykinin receptors in vascular endothelial cells. J. Biol. Chem. 2001, 276, 12420–12426. [Google Scholar] [CrossRef] [PubMed]
- Nofer, J.R.; van der Giet, M.; Tolle, M.; Wolinska, I.; von Wnuck Lipinski, K.; Baba, H.A.; Tietge, U.J.; Godecke, A.; Ishii, I.; Kleuser, B.; et al. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J. Clin. Investig. 2004, 113, 569–581. [Google Scholar] [CrossRef] [PubMed]
- De Palma, C.; Meacci, E.; Perrotta, C.; Bruni, P.; Clementi, E. Endothelial nitric oxide synthase activation by tumor necrosis factor alpha through neutral sphingomyelinase 2, sphingosine kinase 1, and sphingosine 1 phosphate receptors: A novel pathway relevant to the pathophysiology of endothelium. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Adamson, R.H.; Curry, F.R.; Tarbell, J.M. Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H363–H372. [Google Scholar] [CrossRef]
- Hammad, S.M.; Pierce, J.S.; Soodavar, F.; Smith, K.J.; Al Gadban, M.M.; Rembiesa, B.; Klein, R.L.; Hannun, Y.A.; Bielawski, J.; Bielawska, A. Blood sphingolipidomics in healthy humans: Impact of sample collection methodology. J. Lipid Res. 2010, 51, 3074–3087. [Google Scholar] [CrossRef]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L.; et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef]
- Hammad, S.M.; Al Gadban, M.M.; Semler, A.J.; Klein, R.L. Sphingosine 1-phosphate distribution in human plasma: Associations with lipid profiles. J. Lipids 2012, 2012, 180705. [Google Scholar] [CrossRef]
- Kimura, T.; Sato, K.; Kuwabara, A.; Tomura, H.; Ishiwara, M.; Kobayashi, I.; Ui, M.; Okajima, F. Sphingosine 1-phosphate may be a major component of plasma lipoproteins responsible for the cytoprotective actions in human umbilical vein endothelial cells. J. Biol. Chem. 2001, 276, 31780–31785. [Google Scholar] [CrossRef]
- Sato, K.; Okajima, F. Role of sphingosine 1-phosphate in anti-atherogenic actions of high-density lipoprotein. World J. Biol. Chem. 2010, 1, 327–337. [Google Scholar] [CrossRef]
- Iqbal, J.; Walsh, M.T.; Hammad, S.M.; Cuchel, M.; Tarugi, P.; Hegele, R.A.; Davidson, N.O.; Rader, D.J.; Klein, R.L.; Hussain, M.M. Microsomal Triglyceride Transfer Protein Transfers and Determines Plasma Concentrations of Ceramide and Sphingomyelin but Not Glycosylceramide. J. Biol. Chem. 2015, 290, 25863–25875. [Google Scholar] [CrossRef]
- Yu, Y.; Guo, S.; Feng, Y.; Feng, L.; Cui, Y.; Song, G.; Luo, T.; Zhang, K.; Wang, Y.; Jiang, X.C.; et al. Phospholipid transfer protein deficiency decreases the content of S1P in HDL via the loss of its transfer capability. Lipids 2014, 49, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Kurano, M.; Hara, M.; Ikeda, H.; Tsukamoto, K.; Yatomi, Y. Involvement of CETP (Cholesteryl Ester Transfer Protein) in the Shift of Sphingosine-1-Phosphate Among Lipoproteins and in the Modulation of its Functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Tamama, K.; Okajima, F. Sphingosine 1-phosphate signaling in atherosclerosis and vascular biology. Curr. Opin. Lipidol. 2002, 13, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Schwab, S.R.; Pereira, J.P.; Matloubian, M.; Xu, Y.; Huang, Y.; Cyster, J.G. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science 2005, 309, 1735–1739. [Google Scholar] [CrossRef]
- Zhang, B.; Tomura, H.; Kuwabara, A.; Kimura, T.; Miura, S.; Noda, K.; Okajima, F.; Saku, K. Correlation of high density lipoprotein (HDL)-associated sphingosine 1-phosphate with serum levels of HDL-cholesterol and apolipoproteins. Atherosclerosis 2005, 178, 199–205. [Google Scholar] [CrossRef]
- Schwab, S.R.; Cyster, J.G. Finding a way out: Lymphocyte egress from lymphoid organs. Nat. Immunol. 2007, 8, 1295–1301. [Google Scholar] [CrossRef]
- Sachinidis, A.; Kettenhofen, R.; Seewald, S.; Gouni-Berthold, I.; Schmitz, U.; Seul, C.; Ko, Y.; Vetter, H. Evidence that lipoproteins are carriers of bioactive factors. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2412–2421. [Google Scholar] [CrossRef]
- Kontush, A.; Therond, P.; Zerrad, A.; Couturier, M.; Negre-Salvayre, A.; de Souza, J.A.; Chantepie, S.; Chapman, M.J. Preferential sphingosine-1-phosphate enrichment and sphingomyelin depletion are key features of small dense HDL3 particles: Relevance to antiapoptotic and antioxidative activities. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1843–1849. [Google Scholar] [CrossRef]
- Ksiazek, M.; Chacinska, M.; Chabowski, A.; Baranowski, M. Sources, metabolism, and regulation of circulating sphingosine-1-phosphate. J. Lipid Res. 2015, 56, 1271–1281. [Google Scholar] [CrossRef]
- Proia, R.L.; Hla, T. Emerging biology of sphingosine-1-phosphate: Its role in pathogenesis and therapy. J. Clin. Investig. 2015, 125, 1379–1387. [Google Scholar] [CrossRef]
- Vaidya, M.; Jentsch, J.A.; Peters, S.; Keul, P.; Weske, S.; Graler, M.H.; Mladenov, E.; Iliakis, G.; Heusch, G.; Levkau, B. Regulation of ABCA1-mediated cholesterol efflux by sphingosine-1-phosphate signaling in macrophages. J. Lipid Res. 2019, 60, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Christensen, P.M.; Bosteen, M.H.; Hajny, S.; Nielsen, L.B.; Christoffersen, C. Apolipoprotein M mediates sphingosine-1-phosphate efflux from erythrocytes. Sci. Rep. 2017, 7, 14983. [Google Scholar] [CrossRef] [PubMed]
- Sevvana, M.; Ahnstrom, J.; Egerer-Sieber, C.; Lange, H.A.; Dahlback, B.; Muller, Y.A. Serendipitous fatty acid binding reveals the structural determinants for ligand recognition in apolipoprotein M. J. Mol. Biol. 2009, 393, 920–936. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnstrom, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B.; et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 2011, 108, 9613–9618. [Google Scholar] [CrossRef] [PubMed]
- Okajima, F. Distribution of sphingosine 1-phosphate in plasma lipoproteins and its role in the regulation of the vascular cell functions. Tanpakushitsu Kakusan Koso 2002, 47, 480–487. [Google Scholar] [PubMed]
- Christoffersen, C.; Nielsen, L.B.; Axler, O.; Andersson, A.; Johnsen, A.H.; Dahlback, B. Isolation and characterization of human apolipoprotein M-containing lipoproteins. J. Lipid Res. 2006, 47, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Silva, R.A.; Chantepie, S.; Lagor, W.R.; Chapman, M.J.; Kontush, A. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: Relevance to antioxidative function. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 870–876. [Google Scholar] [CrossRef]
- De Souza, J.A.; Vindis, C.; Negre-Salvayre, A.; Rye, K.A.; Couturier, M.; Therond, P.; Chantepie, S.; Salvayre, R.; Chapman, M.J.; Kontush, A. Small, dense HDL 3 particles attenuate apoptosis in endothelial cells: Pivotal role of apolipoprotein A-I. J. Cell. Mol. Med. 2010, 14, 608–620. [Google Scholar] [CrossRef]
- Kontush, A.; Lhomme, M.; Chapman, M.J. Unraveling the complexities of the HDL lipidome. J. Lipid Res. 2013, 54, 2950–2963. [Google Scholar] [CrossRef]
- Persegol, L.; Darabi, M.; Dauteuille, C.; Lhomme, M.; Chantepie, S.; Rye, K.A.; Therond, P.; Chapman, M.J.; Salvayre, R.; Negre-Salvayre, A.; et al. Small dense HDLs display potent vasorelaxing activity, reflecting their elevated content of sphingosine-1-phosphate. J. Lipid Res. 2018, 59, 25–34. [Google Scholar] [CrossRef]
- Kurano, M.; Tsukamoto, K.; Ohkawa, R.; Hara, M.; Iino, J.; Kageyama, Y.; Ikeda, H.; Yatomi, Y. Liver involvement in sphingosine 1-phosphate dynamism revealed by adenoviral hepatic overexpression of apolipoprotein M. Atherosclerosis 2013, 229, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Seo, J.; Allegood, J.; Bi, X.; Zhu, X.; Boudyguina, E.; Gebre, A.K.; Avni, D.; Shah, D.; Sorci-Thomas, M.G.; et al. Hepatic apolipoprotein M (apoM) overexpression stimulates formation of larger apoM/sphingosine 1-phosphate-enriched plasma high density lipoprotein. J. Biol. Chem. 2014, 289, 2801–2814. [Google Scholar] [CrossRef] [PubMed]
- Karuna, R.; Park, R.; Othman, A.; Holleboom, A.G.; Motazacker, M.M.; Sutter, I.; Kuivenhoven, J.A.; Rohrer, L.; Matile, H.; Hornemann, T.; et al. Plasma levels of sphingosine-1-phosphate and apolipoprotein M in patients with monogenic disorders of HDL metabolism. Atherosclerosis 2011, 219, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, C.; Nielsen, L.B. Apolipoprotein M: Bridging HDL and endothelial function. Curr. Opin. Lipidol. 2013, 24, 295–300. [Google Scholar] [CrossRef]
- Arkensteijn, B.W.; Berbee, J.F.; Rensen, P.C.; Nielsen, L.B.; Christoffersen, C. The apolipoprotein m-sphingosine-1-phosphate axis: Biological relevance in lipoprotein metabolism, lipid disorders and atherosclerosis. Int. J. Mol. Sci. 2013, 14, 4419–4431. [Google Scholar] [CrossRef]
- Ahnstrom, J.; Axler, O.; Jauhiainen, M.; Salomaa, V.; Havulinna, A.S.; Ehnholm, C.; Frikke-Schmidt, R.; Tybjaerg-Hansen, A.; Dahlback, B. Levels of apolipoprotein M are not associated with the risk of coronary heart disease in two independent case-control studies. J. Lipid Res. 2008, 49, 1912–1917. [Google Scholar] [CrossRef]
- Saier, M.H., Jr.; Beatty, J.T.; Goffeau, A.; Harley, K.T.; Heijne, W.H.; Huang, S.C.; Jack, D.L.; Jahn, P.S.; Lew, K.; Liu, J.; et al. The major facilitator superfamily. J. Mol. Microbiol. Biotechnol. 1999, 1, 257–279. [Google Scholar]
- Nagahashi, M.; Kim, E.Y.; Yamada, A.; Ramachandran, S.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Milstien, S.; Takabe, K.; Spiegel, S. Spns2, a transporter of phosphorylated sphingoid bases, regulates their blood and lymph levels, and the lymphatic network. FASEB J. 2013, 27, 1001–1011. [Google Scholar] [CrossRef]
- Hisano, Y.; Kobayashi, N.; Yamaguchi, A.; Nishi, T. Mouse SPNS2 functions as a sphingosine-1-phosphate transporter in vascular endothelial cells. PLoS ONE 2012, 7, e38941. [Google Scholar] [CrossRef]
- Thuy, A.V.; Reimann, C.M.; Hemdan, N.Y.; Graler, M.H. Sphingosine 1-phosphate in blood: Function, metabolism, and fate. Cell. Physiol. Biochem. 2014, 34, 158–171. [Google Scholar] [CrossRef]
- Bode, C.; Sensken, S.C.; Peest, U.; Beutel, G.; Thol, F.; Levkau, B.; Li, Z.; Bittman, R.; Huang, T.; Tolle, M.; et al. Erythrocytes serve as a reservoir for cellular and extracellular sphingosine 1-phosphate. J. Cell. Biochem. 2010, 109, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Nishi, T.; Hirata, T.; Kihara, A.; Sano, T.; Igarashi, Y.; Yamaguchi, A. Sphingosine 1-phosphate is released from the cytosol of rat platelets in a carrier-mediated manner. J. Lipid Res. 2006, 47, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Yamaguchi, A.; Nishi, T. Characterization of the ATP-dependent sphingosine 1-phosphate transporter in rat erythrocytes. J. Biol. Chem. 2009, 284, 21192–21200. [Google Scholar] [CrossRef] [PubMed]
- Mitra, P.; Oskeritzian, C.A.; Payne, S.G.; Beaven, M.A.; Milstien, S.; Spiegel, S. Role of ABCC1 in export of sphingosine-1-phosphate from mast cells. Proc. Natl. Acad. Sci. USA 2006, 103, 16394–16399. [Google Scholar] [CrossRef] [PubMed]
- Takabe, K.; Kim, R.H.; Allegood, J.C.; Mitra, P.; Ramachandran, S.; Nagahashi, M.; Harikumar, K.B.; Hait, N.C.; Milstien, S.; Spiegel, S. Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J. Biol. Chem. 2010, 285, 10477–10486. [Google Scholar] [CrossRef]
- Japtok, L.; Schaper, K.; Baumer, W.; Radeke, H.H.; Jeong, S.K.; Kleuser, B. Sphingosine 1-phosphate modulates antigen capture by murine Langerhans cells via the S1P2 receptor subtype. PLoS ONE 2012, 7, e49427. [Google Scholar] [CrossRef]
- Cartwright, T.A.; Campos, C.R.; Cannon, R.E.; Miller, D.S. Mrp1 is essential for sphingolipid signaling to p-glycoprotein in mouse blood-brain and blood-spinal cord barriers. J. Cereb. Blood Flow Metab. 2013, 33, 381–388. [Google Scholar] [CrossRef]
- Ito, S.; Iwaki, S.; Koike, K.; Yuda, Y.; Nagasaki, A.; Ohkawa, R.; Yatomi, Y.; Furumoto, T.; Tsutsui, H.; Sobel, B.E.; et al. Increased plasma sphingosine-1-phosphate in obese individuals and its capacity to increase the expression of plasminogen activator inhibitor-1 in adipocytes. Coron. Artery Dis. 2013, 24, 642–650. [Google Scholar] [CrossRef]
- Camont, L.; Chapman, M.J.; Kontush, A. Biological activities of HDL subpopulations and their relevance to cardiovascular disease. Trends Mol. Med. 2011, 17, 594–603. [Google Scholar] [CrossRef]
- Sato, K.; Malchinkhuu, E.; Horiuchi, Y.; Mogi, C.; Tomura, H.; Tosaka, M.; Yoshimoto, Y.; Kuwabara, A.; Okajima, F. Critical role of ABCA1 transporter in sphingosine 1-phosphate release from astrocytes. J. Neurochem. 2007, 103, 2610–2619. [Google Scholar] [CrossRef]
- Sato, K.; Malchinkhuu, E.; Horiuchi, Y.; Mogi, C.; Tomura, H.; Tosaka, M.; Yoshimoto, Y.; Kuwabara, A.; Okajima, F. HDL-like lipoproteins in cerebrospinal fluid affect neural cell activity through lipoprotein-associated sphingosine 1-phosphate. Biochem. Biophys. Res. Commun. 2007, 359, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Y.; Ren, J.H.; Tao, N.N.; Ran, L.K.; Chen, X.; Zhou, H.Z.; Liu, B.; Li, X.S.; Huang, A.L.; Chen, J. The SIRT1 inhibitor, nicotinamide, inhibits hepatitis B virus replication in vitro and in vivo. Arch. Virol. 2016, 161, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Venkataraman, K.; Hwang, S.I.; Han, D.K.; Hla, T. A novel method to quantify sphingosine 1-phosphate by immobilized metal affinity chromatography (IMAC). Prostaglandins Other Lipid Mediat. 2007, 84, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Walsh, M.T.; Hammad, S.M.; Cuchel, M.; Rader, D.J.; Hussain, M.M. ATP binding cassette family A protein 1 determines hexosylceramide and sphingomyelin levels in human and mouse plasma. J. Lipid Res. 2018, 59, 2084–2097. [Google Scholar] [CrossRef]
- Mineo, C.; Shaul, P.W. Novel biological functions of high-density lipoprotein cholesterol. Circ. Res. 2012, 111, 1079–1090. [Google Scholar] [CrossRef]
- Tolle, M.; Pawlak, A.; Schuchardt, M.; Kawamura, A.; Tietge, U.J.; Lorkowski, S.; Keul, P.; Assmann, G.; Chun, J.; Levkau, B.; et al. HDL-associated lysosphingolipids inhibit NAD(P)H oxidase-dependent monocyte chemoattractant protein-1 production. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1542–1548. [Google Scholar] [CrossRef]
- Lee, M.H.; Appleton, K.M.; El-Shewy, H.M.; Sorci-Thomas, M.G.; Thomas, M.J.; Lopes-Virella, M.F.; Luttrell, L.M.; Hammad, S.M.; Klein, R.L. S1P in HDL promotes interaction between SR-BI and S1PR1 and activates S1PR1-mediated biological functions: Calcium flux and S1PR1 internalization. J. Lipid Res. 2017, 58, 325–338. [Google Scholar] [CrossRef]
- Ren, K.; Lu, Y.J.; Mo, Z.C.; Liu, X.; Tang, Z.L.; Jiang, Y.; Peng, X.S.; Li, L.; Zhang, Q.H.; Yi, G.H. ApoA-I/SR-BI modulates S1P/S1PR2-mediated inflammation through the PI3K/Akt signaling pathway in HUVECs. J. Physiol. Biochem. 2017, 73, 287–296. [Google Scholar] [CrossRef]
- Sattler, K.; Levkau, B. Sphingosine-1-phosphate as a mediator of high-density lipoprotein effects in cardiovascular protection. Cardiovasc. Res. 2009, 82, 201–211. [Google Scholar] [CrossRef]
- Argraves, K.M.; Sethi, A.A.; Gazzolo, P.J.; Wilkerson, B.A.; Remaley, A.T.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Yeatts, S.D.; Nicholas, K.S.; Barth, J.L.; et al. S1P, dihydro-S1P and C24:1-ceramide levels in the HDL-containing fraction of serum inversely correlate with occurrence of ischemic heart disease. Lipids Health Dis. 2011, 10, 70. [Google Scholar] [CrossRef]
- Wolfrum, C.; Poy, M.N.; Stoffel, M. Apolipoprotein M is required for prebeta-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nat. Med. 2005, 11, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Brulhart-Meynet, M.C.; Braunersreuther, V.; Brinck, J.; Montecucco, F.; Prost, J.C.; Thomas, A.; Galan, K.; Pelli, G.; Pedretti, S.; Vuilleumier, N.; et al. Improving reconstituted HDL composition for efficient post-ischemic reduction of ischemia reperfusion injury. PLoS ONE 2015, 10, e0119664. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Peng, H.; Liu, D.; Ji, L.; Niu, C.; Ren, J.; Pan, B.; Hu, J.; Zheng, L.; Huang, Y. High-density lipoprotein of patients with type 2 diabetes mellitus upregulates cyclooxgenase-2 expression and prostacyclin I-2 release in endothelial cells: Relationship with HDL-associated sphingosine-1-phosphate. Cardiovasc. Diabetol. 2013, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Frias, M.A.; Lang, U.; Gerber-Wicht, C.; James, R.W. Native and reconstituted HDL protect cardiomyocytes from doxorubicin-induced apoptosis. Cardiovasc. Res. 2010, 85, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.; Okada, H.; Dahlback, B. HDL-associated ApoM is anti-apoptotic by delivering sphingosine 1-phosphate to S1P1 & S1P3 receptors on vascular endothelium. Lipids Health Dis. 2017, 16, 36. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, C.; Jauhiainen, M.; Moser, M.; Porse, B.; Ehnholm, C.; Boesl, M.; Dahlback, B.; Nielsen, L.B. Effect of apolipoprotein M on high density lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knock-out mice. J. Biol. Chem. 2008, 283, 1839–1847. [Google Scholar] [CrossRef]
- Christoffersen, C.; Pedersen, T.X.; Gordts, P.L.; Roebroek, A.J.; Dahlback, B.; Nielsen, L.B. Opposing effects of apolipoprotein m on catabolism of apolipoprotein B-containing lipoproteins and atherosclerosis. Circ. Res. 2010, 106, 1624–1634. [Google Scholar] [CrossRef]
- Richter, S.; Shih, D.Q.; Pearson, E.R.; Wolfrum, C.; Fajans, S.S.; Hattersley, A.T.; Stoffel, M. Regulation of apolipoprotein M gene expression by MODY3 gene hepatocyte nuclear factor-1alpha: Haploinsufficiency is associated with reduced serum apolipoprotein M levels. Diabetes 2003, 52, 2989–2995. [Google Scholar] [CrossRef]
- Elsoe, S.; Christoffersen, C.; Luchoomun, J.; Turner, S.; Nielsen, L.B. Apolipoprotein M promotes mobilization of cellular cholesterol in vivo. Biochim. Biophys. Acta 2013, 1831, 1287–1292. [Google Scholar] [CrossRef]
- Elsoe, S.; Ahnstrom, J.; Christoffersen, C.; Hoofnagle, A.N.; Plomgaard, P.; Heinecke, J.W.; Binder, C.J.; Bjorkbacka, H.; Dahlback, B.; Nielsen, L.B. Apolipoprotein M binds oxidized phospholipids and increases the antioxidant effect of HDL. Atherosclerosis 2012, 221, 91–97. [Google Scholar] [CrossRef]
- Brinck, J.W.; Thomas, A.; Lauer, E.; Jornayvaz, F.R.; Brulhart-Meynet, M.C.; Prost, J.C.; Pataky, Z.; Lofgren, P.; Hoffstedt, J.; Eriksson, M.; et al. Diabetes Mellitus Is Associated With Reduced High-Density Lipoprotein Sphingosine-1-Phosphate Content and Impaired High-Density Lipoprotein Cardiac Cell Protection. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Frej, C.; Mendez, A.J.; Ruiz, M.; Castillo, M.; Hughes, T.A.; Dahlback, B.; Goldberg, R.B. A Shift in ApoM/S1P Between HDL-Particles in Women With Type 1 Diabetes Mellitus Is Associated With Impaired Anti-Inflammatory Effects of the ApoM/S1P Complex. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Knapp, M.; Baranowski, M.; Czarnowski, D.; Lisowska, A.; Zabielski, P.; Gorski, J.; Musial, W. Plasma sphingosine-1-phosphate concentration is reduced in patients with myocardial infarction. Med. Sci. Monit. 2009, 15, CR490–CR493. [Google Scholar] [PubMed]
- Deutschman, D.H.; Carstens, J.S.; Klepper, R.L.; Smith, W.S.; Page, M.T.; Young, T.R.; Gleason, L.A.; Nakajima, N.; Sabbadini, R.A. Predicting obstructive coronary artery disease with serum sphingosine-1-phosphate. Am. Heart J. 2003, 146, 62–68. [Google Scholar] [CrossRef]
- Argraves, K.M.; Wilkerson, B.A.; Argraves, W.S.; Fleming, P.A.; Obeid, L.M.; Drake, C.J. Sphingosine-1-phosphate signaling promotes critical migratory events in vasculogenesis. J. Biol. Chem. 2004, 279, 50580–50590. [Google Scholar] [CrossRef]
- Sattler, K.; Lehmann, I.; Graler, M.; Brocker-Preuss, M.; Erbel, R.; Heusch, G.; Levkau, B. HDL-bound sphingosine 1-phosphate (S1P) predicts the severity of coronary artery atherosclerosis. Cell. Physiol. Biochem. 2014, 34, 172–184. [Google Scholar] [CrossRef]
- Jing, X.D.; Wei, X.M.; Deng, S.B.; Du, J.L.; Liu, Y.J.; She, Q. The relationship between the high-density lipoprotein (HDL)-associated sphingosine-1-phosphate (S1P) and coronary in-stent restenosis. Clin. Chim. Acta 2015, 446, 248–252. [Google Scholar] [CrossRef]
- Nielsen, L.B.; Christoffersen, C.; Ahnstrom, J.; Dahlback, B. ApoM: Gene regulation and effects on HDL metabolism. Trends Endocrinol. Metab. 2009, 20, 66–71. [Google Scholar] [CrossRef]
- Dullaart, R.P.; Plomgaard, P.; de Vries, R.; Dahlback, B.; Nielsen, L.B. Plasma apolipoprotein M is reduced in metabolic syndrome but does not predict intima media thickness. Clin. Chim. Acta 2009, 406, 129–133. [Google Scholar] [CrossRef]
- Randriamboavonjy, V.; Badenhoop, K.; Schmidt, H.; Geisslinger, G.; Fisslthaler, B.; Fleming, I. The S1P(2) receptor expressed in human platelets is linked to the RhoA-Rho kinase pathway and is down regulated in type 2 diabetes. Basic Res. Cardiol. 2009, 104, 333–340. [Google Scholar] [CrossRef]
- Fox, T.E.; Bewley, M.C.; Unrath, K.A.; Pedersen, M.M.; Anderson, R.E.; Jung, D.Y.; Jefferson, L.S.; Kim, J.K.; Bronson, S.K.; Flanagan, J.M.; et al. Circulating sphingolipid biomarkers in models of type 1 diabetes. J. Lipid Res. 2011, 52, 509–517. [Google Scholar] [CrossRef]
- Vaisar, T.; Couzens, E.; Hwang, A.; Russell, M.; Barlow, C.E.; DeFina, L.F.; Hoofnagle, A.N.; Kim, F. Type 2 diabetes is associated with loss of HDL endothelium protective functions. PLoS ONE 2018, 13, e0192616. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.S.; Lu, F.; Zhang, Y.; Du, J.L.; Deng, S.B.; She, Q. The effects of sphingosine-1-phosphate (S1P) and CD69 on the pathogenesis of type 2 diabetes complicated by coronary heart disease. Int. J. Clin. Exp. Med. 2018, 11, 9621–9629. [Google Scholar]
- Kowalski, G.M.; Carey, A.L.; Selathurai, A.; Kingwell, B.A.; Bruce, C.R. Plasma sphingosine-1-phosphate is elevated in obesity. PLoS ONE 2013, 8, e72449. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Kanazawa, I.; Sugimoto, T. Visceral fat accumulation is associated with increased plasma sphingosine-1-phosphate levels in type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2018, 143, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Gao, J.; Pu, C.; Feng, G.; Wang, L.; Huang, L.; Tao, Q.; Zhang, Y. Effects of hyperlipidaemia on plasma apolipoprotein M levels in patients with type 2 diabetes mellitus: An independent case-control study. Lipids Health Dis. 2016, 15, 158. [Google Scholar] [CrossRef]
- Keul, P.; Polzin, A.; Kaiser, K.; Graler, M.; Dannenberg, L.; Daum, G.; Heusch, G.; Levkau, B. Potent anti-inflammatory properties of HDL in vascular smooth muscle cells mediated by HDL-S1P and their impairment in coronary artery disease due to lower HDL-S1P: A new aspect of HDL dysfunction and its therapy. FASEB J. 2019, 33, 1482–1495. [Google Scholar] [CrossRef]
- Kontush, A.; Chapman, M.J. Why is HDL functionally deficient in type 2 diabetes? Curr. Diabetes Rep. 2008, 8, 51–59. [Google Scholar] [CrossRef]
- Denimal, D.; Monier, S.; Brindisi, M.C.; Petit, J.M.; Bouillet, B.; Nguyen, A.; Demizieux, L.; Simoneau, I.; Pais de Barros, J.P.; Verges, B.; et al. Impairment of the Ability of HDL From Patients With Metabolic Syndrome but Without Diabetes Mellitus to Activate eNOS: Correction by S1P Enrichment. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 804–811. [Google Scholar] [CrossRef]
- Tong, X.; Lv, P.; Mathew, A.V.; Liu, D.; Niu, C.; Wang, Y.; Ji, L.; Li, J.; Fu, Z.; Pan, B.; et al. The compensatory enrichment of sphingosine -1- phosphate harbored on glycated high-density lipoprotein restores endothelial protective function in type 2 diabetes mellitus. Cardiovasc. Diabetol. 2014, 13, 82. [Google Scholar] [CrossRef]
- Mishima, Y.; Kurano, M.; Kobayashi, T.; Nishikawa, M.; Ohkawa, R.; Tozuka, M.; Yatomi, Y. Dihydro-sphingosine 1-phosphate interacts with carrier proteins in a manner distinct from that of sphingosine 1-phosphate. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.; Frej, C.; Holmer, A.; Guo, L.J.; Tran, S.; Dahlback, B. High-Density Lipoprotein-Associated Apolipoprotein M Limits Endothelial Inflammation by Delivering Sphingosine-1-Phosphate to the Sphingosine-1-Phosphate Receptor 1. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Selim, S.; Sunkara, M.; Salous, A.K.; Leung, S.W.; Berdyshev, E.V.; Bailey, A.; Campbell, C.L.; Charnigo, R.; Morris, A.J.; Smyth, S.S. Plasma levels of sphingosine 1-phosphate are strongly correlated with haematocrit, but variably restored by red blood cell transfusions. Clin. Sci. 2011, 121, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Chew, W.S.; Wang, W.; Herr, D.R. To fingolimod and beyond: The rich pipeline of drug candidates that target S1P signaling. Pharmacol. Res. 2016, 113, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Cusack, K.P.; Stoffel, R.H. S1P(1) receptor agonists: Assessment of selectivity and current clinical activity. Curr. Opin. Drug Discov. Dev. 2010, 13, 481–488. [Google Scholar]
- Vestri, A.; Pierucci, F.; Frati, A.; Monaco, L.; Meacci, E. Sphingosine 1-Phosphate Receptors: Do They Have a Therapeutic Potential in Cardiac Fibrosis? Front. Pharmacol. 2017, 8, 296. [Google Scholar] [CrossRef]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef]
- Vessey, D.A.; Kelley, M.; Zhang, J.; Li, L.; Tao, R.; Karliner, J.S. Dimethylsphingosine and FTY720 inhibit the SK1 form but activate the SK2 form of sphingosine kinase from rat heart. J. Biochem. Mol. Toxicol. 2007, 21, 273–279. [Google Scholar] [CrossRef]
- Kawahara, A.; Nishi, T.; Hisano, Y.; Fukui, H.; Yamaguchi, A.; Mochizuki, N. The sphingolipid transporter spns2 functions in migration of zebrafish myocardial precursors. Science 2009, 323, 524–527. [Google Scholar] [CrossRef]
- Hisano, Y.; Kobayashi, N.; Kawahara, A.; Yamaguchi, A.; Nishi, T. The sphingosine 1-phosphate transporter, SPNS2, functions as a transporter of the phosphorylated form of the immunomodulating agent FTY720. J. Biol. Chem. 2011, 286, 1758–1766. [Google Scholar] [CrossRef]
- Keul, P.; Tolle, M.; Lucke, S.; von Wnuck Lipinski, K.; Heusch, G.; Schuchardt, M.; van der Giet, M.; Levkau, B. The sphingosine-1-phosphate analogue FTY720 reduces atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Nofer, J.R.; Bot, M.; Brodde, M.; Taylor, P.J.; Salm, P.; Brinkmann, V.; van Berkel, T.; Assmann, G.; Biessen, E.A. FTY720, a synthetic sphingosine 1 phosphate analogue, inhibits development of atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2007, 115, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Camm, J.; Hla, T.; Bakshi, R.; Brinkmann, V. Cardiac and vascular effects of fingolimod: Mechanistic basis and clinical implications. Am. Heart J. 2014, 168, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Fan, L.; Wei, L.; Gao, H.; Zhang, R.; Tao, L.; Cao, F.; Wang, H. FTY720 protects cardiac microvessels of diabetes: A critical role of S1P1/3 in diabetic heart disease. PLoS ONE 2012, 7, e42900. [Google Scholar] [CrossRef]
- Aytan, N.; Choi, J.K.; Carreras, I.; Brinkmann, V.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. Fingolimod modulates multiple neuroinflammatory markers in a mouse model of Alzheimer’s disease. Sci. Rep. 2016, 6, 24939. [Google Scholar] [CrossRef]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef]
- DiMarco, J.P.; O’Connor, P.; Cohen, J.A.; Reder, A.T.; Zhang-Auberson, L.; Tang, D.; Collins, W.; Kappos, L. First-dose effects of fingolimod: Pooled safety data from three phase 3 studies. Mult. Scler. Relat. Disord. 2014, 3, 629–638. [Google Scholar] [CrossRef]
- Cohen, J.A.; Barkhof, F.; Comi, G.; Hartung, H.P.; Khatri, B.O.; Montalban, X.; Pelletier, J.; Capra, R.; Gallo, P.; Izquierdo, G.; et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 402–415. [Google Scholar] [CrossRef]
- Kappos, L.; Radue, E.W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef]
- Sanna, M.G.; Liao, J.; Jo, E.; Alfonso, C.; Ahn, M.Y.; Peterson, M.S.; Webb, B.; Lefebvre, S.; Chun, J.; Gray, N.; et al. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J. Biol. Chem. 2004, 279, 13839–13848. [Google Scholar] [CrossRef]
- Jo, E.; Sanna, M.G.; Gonzalez-Cabrera, P.J.; Thangada, S.; Tigyi, G.; Osborne, D.A.; Hla, T.; Parrill, A.L.; Rosen, H. S1P1-selective in vivo-active agonists from high-throughput screening: Off-the-shelf chemical probes of receptor interactions, signaling, and fate. Chem. Biol. 2005, 12, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Cyster, J.G.; Hla, T. FTY720: Sphingosine 1-phosphate receptor-1 in the control of lymphocyte egress and endothelial barrier function. Am. J. Transplant. 2004, 4, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, Y.T.; Sanna, M.G.; Rosen, H.; Gottlieb, R.A. S1P1-selective agonist SEW2871 exacerbates reperfusion arrhythmias. J. Cardiovasc. Pharmacol. 2007, 50, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.J. Pitavastatin: Novel effects on lipid parameters. Atheroscler. Suppl. 2011, 12, 277–284. [Google Scholar] [CrossRef]
- Kishida, K.; Funahashi, T.; Shimomura, I. Importance of assessing the effect of statins on the function of high- density lipoproteins on coronary plaque. Cardiovasc. Hematol. Disord. Drug Targets 2012, 12, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, J.; Miyoshi, M.; Hashimoto, T.; Kubota, Y.; Kosaka, H. Statins induce S1P1 receptors and enhance endothelial nitric oxide production in response to high-density lipoproteins. Br. J. Pharmacol. 2007, 150, 470–479. [Google Scholar] [CrossRef]
- Kimura, T.; Mogi, C.; Tomura, H.; Kuwabara, A.; Im, D.S.; Sato, K.; Kurose, H.; Murakami, M.; Okajima, F. Induction of scavenger receptor class B type I is critical for simvastatin enhancement of high-density lipoprotein-induced anti-inflammatory actions in endothelial cells. J. Immunol. 2008, 181, 7332–7340. [Google Scholar] [CrossRef]
- Koch, A.; Volzke, A.; Wunsche, C.; Meyer zu Heringdorf, D.; Huwiler, A.; Pfeilschifter, J. Thiazolidinedione-dependent activation of sphingosine kinase 1 causes an anti-fibrotic effect in renal mesangial cells. Br. J. Pharmacol. 2012, 166, 1018–1032. [Google Scholar] [CrossRef][Green Version]
- Luo, G.; Feng, Y.; Zhang, J.; Mu, Q.; Shi, Y.; Qin, L.; Zheng, L.; Berggren-Soderlund, M.; Nilsson-Ehle, P.; Zhang, X.; et al. Rosiglitazone enhances apolipoprotein M (Apom) expression in rat’s liver. Int. J. Med. Sci. 2014, 11, 1015–1021. [Google Scholar] [CrossRef]
- Parham, K.A.; Zebol, J.R.; Tooley, K.L.; Sun, W.Y.; Moldenhauer, L.M.; Cockshell, M.P.; Gliddon, B.L.; Moretti, P.A.; Tigyi, G.; Pitson, S.M.; et al. Sphingosine 1-phosphate is a ligand for peroxisome proliferator-activated receptor-gamma that regulates neoangiogenesis. FASEB J. 2015, 29, 3638–3653. [Google Scholar] [CrossRef]
- Chen, J.; Wang, W.; Qi, Y.; Kaczorowski, D.; McCaughan, G.W.; Gamble, J.R.; Don, A.S.; Gao, X.; Vadas, M.A.; Xia, P. Deletion of sphingosine kinase 1 ameliorates hepatic steatosis in diet-induced obese mice: Role of PPARgamma. Biochim. Biophys. Acta 2016, 1861, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Kurano, M.; Ikeda, H.; Iso, O.N.; Hara, M.; Tsukamoto, K.; Yatomi, Y. Regulation of the metabolism of apolipoprotein M and sphingosine 1-phosphate by hepatic PPARgamma activity. Biochem. J. 2018, 475, 2009–2024. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Summers, S.A. A ceramide-centric view of insulin resistance. Cell Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, R.N.; Yu, C.; Hoofnagle, A.; Hari, N.; Jensen, P.N.; Fretts, A.M.; Umans, J.G.; Howard, B.V.; Sitlani, C.M.; Siscovick, D.S.; et al. Circulating Sphingolipids, Insulin, HOMA-IR, and HOMA-B: The Strong Heart Family Study. Diabetes 2018, 67, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.N.; Fretts, A.M.; Yu, C.; Hoofnagle, A.N.; Umans, J.G.; Howard, B.V.; Sitlani, C.M.; Siscovick, D.S.; King, I.B.; Sotoodehnia, N.; et al. Circulating sphingolipids, fasting glucose, and impaired fasting glucose: The Strong Heart Family Study. EBioMedicine 2019, 41, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Turpin-Nolan, S.M.; Hammerschmidt, P.; Chen, W.; Jais, A.; Timper, K.; Awazawa, M.; Brodesser, S.; Bruning, J.C. CerS1-Derived C18:0 Ceramide in Skeletal Muscle Promotes Obesity-Induced Insulin Resistance. Cell Rep. 2019, 26, 1–10. [Google Scholar] [CrossRef]
- Laaksonen, R.; Ekroos, K.; Sysi-Aho, M.; Hilvo, M.; Vihervaara, T.; Kauhanen, D.; Suoniemi, M.; Hurme, R.; Marz, W.; Scharnagl, H.; et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur. Heart J. 2016, 37, 1967–1976. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diarte-Añazco, E.M.G.; Méndez-Lara, K.A.; Pérez, A.; Alonso, N.; Blanco-Vaca, F.; Julve, J. Novel Insights into the Role of HDL-Associated Sphingosine-1-Phosphate in Cardiometabolic Diseases. Int. J. Mol. Sci. 2019, 20, 6273. https://doi.org/10.3390/ijms20246273
Diarte-Añazco EMG, Méndez-Lara KA, Pérez A, Alonso N, Blanco-Vaca F, Julve J. Novel Insights into the Role of HDL-Associated Sphingosine-1-Phosphate in Cardiometabolic Diseases. International Journal of Molecular Sciences. 2019; 20(24):6273. https://doi.org/10.3390/ijms20246273
Chicago/Turabian StyleDiarte-Añazco, Elena M. G., Karen Alejandra Méndez-Lara, Antonio Pérez, Núria Alonso, Francisco Blanco-Vaca, and Josep Julve. 2019. "Novel Insights into the Role of HDL-Associated Sphingosine-1-Phosphate in Cardiometabolic Diseases" International Journal of Molecular Sciences 20, no. 24: 6273. https://doi.org/10.3390/ijms20246273
APA StyleDiarte-Añazco, E. M. G., Méndez-Lara, K. A., Pérez, A., Alonso, N., Blanco-Vaca, F., & Julve, J. (2019). Novel Insights into the Role of HDL-Associated Sphingosine-1-Phosphate in Cardiometabolic Diseases. International Journal of Molecular Sciences, 20(24), 6273. https://doi.org/10.3390/ijms20246273