The Extraordinary Role of Extracellular RNA in Arteriogenesis, the Growth of Collateral Arteries





Abstract
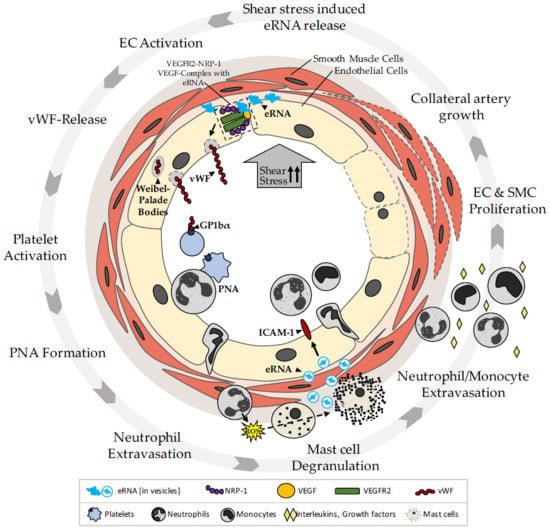
{kind=link}
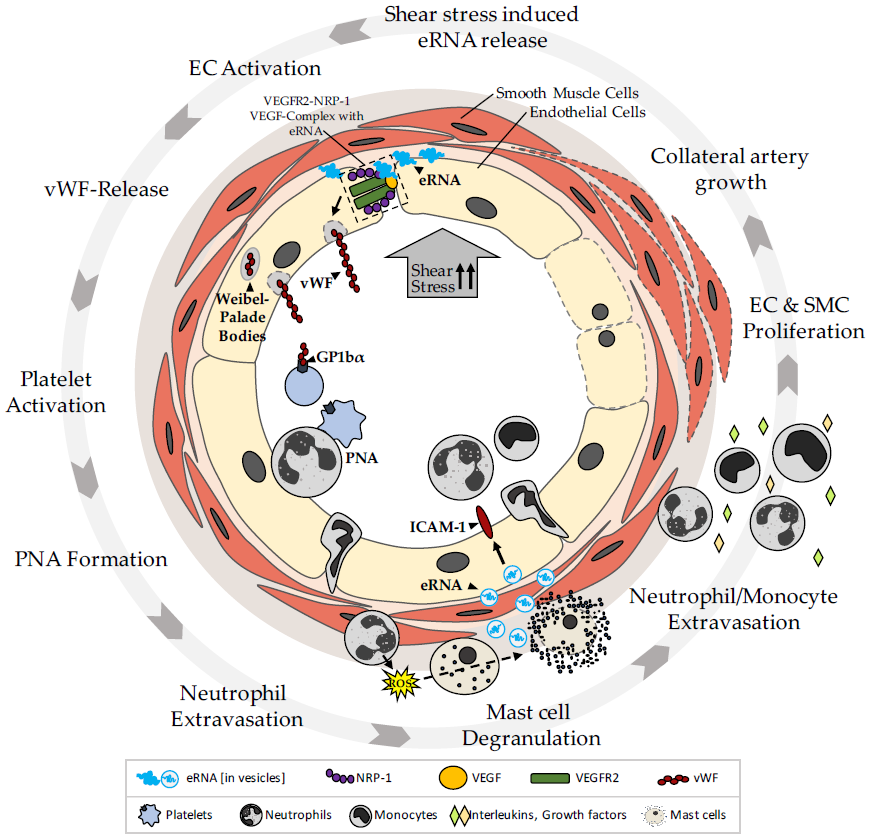
{kind=link}
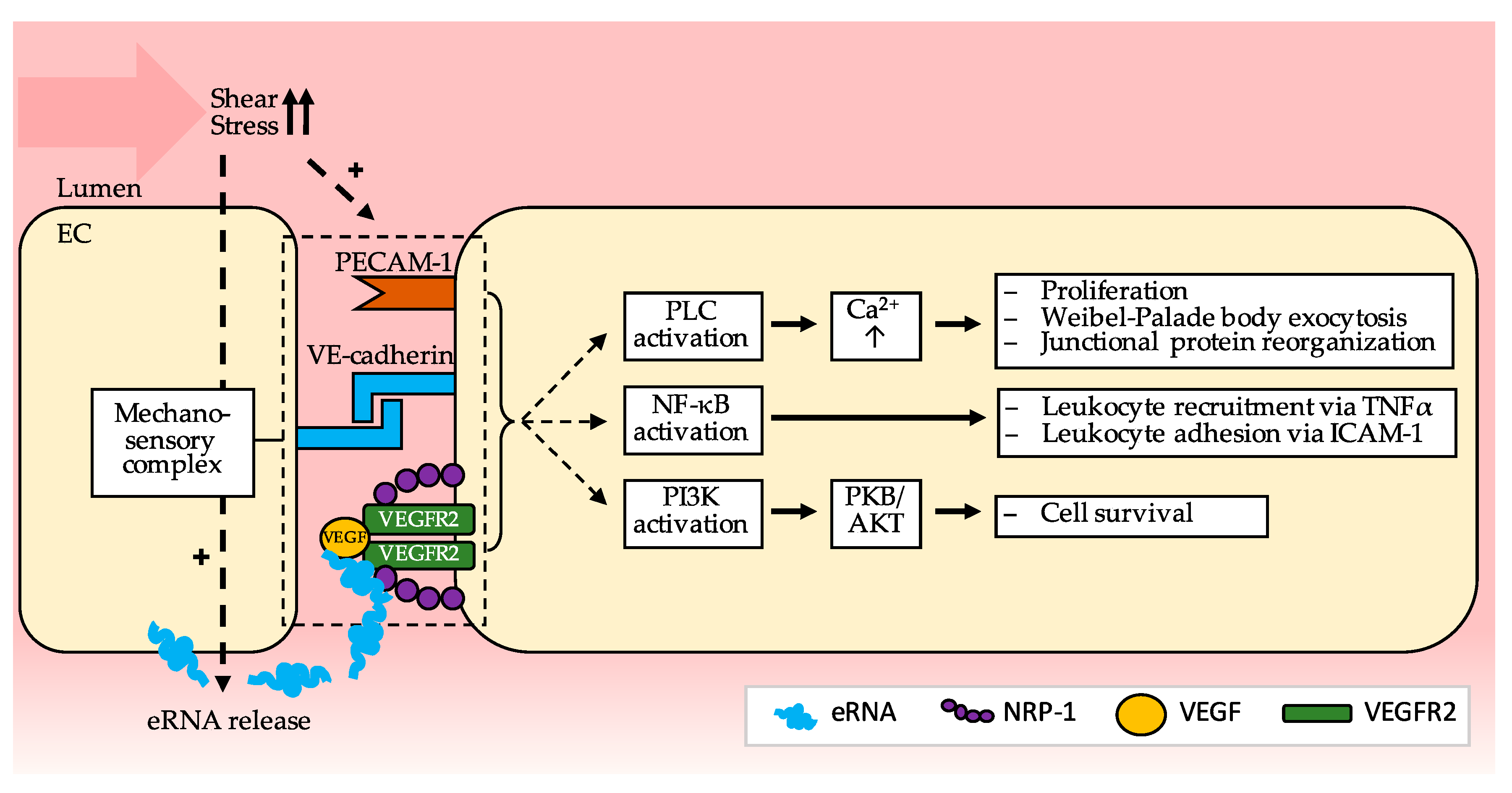
{kind=link}
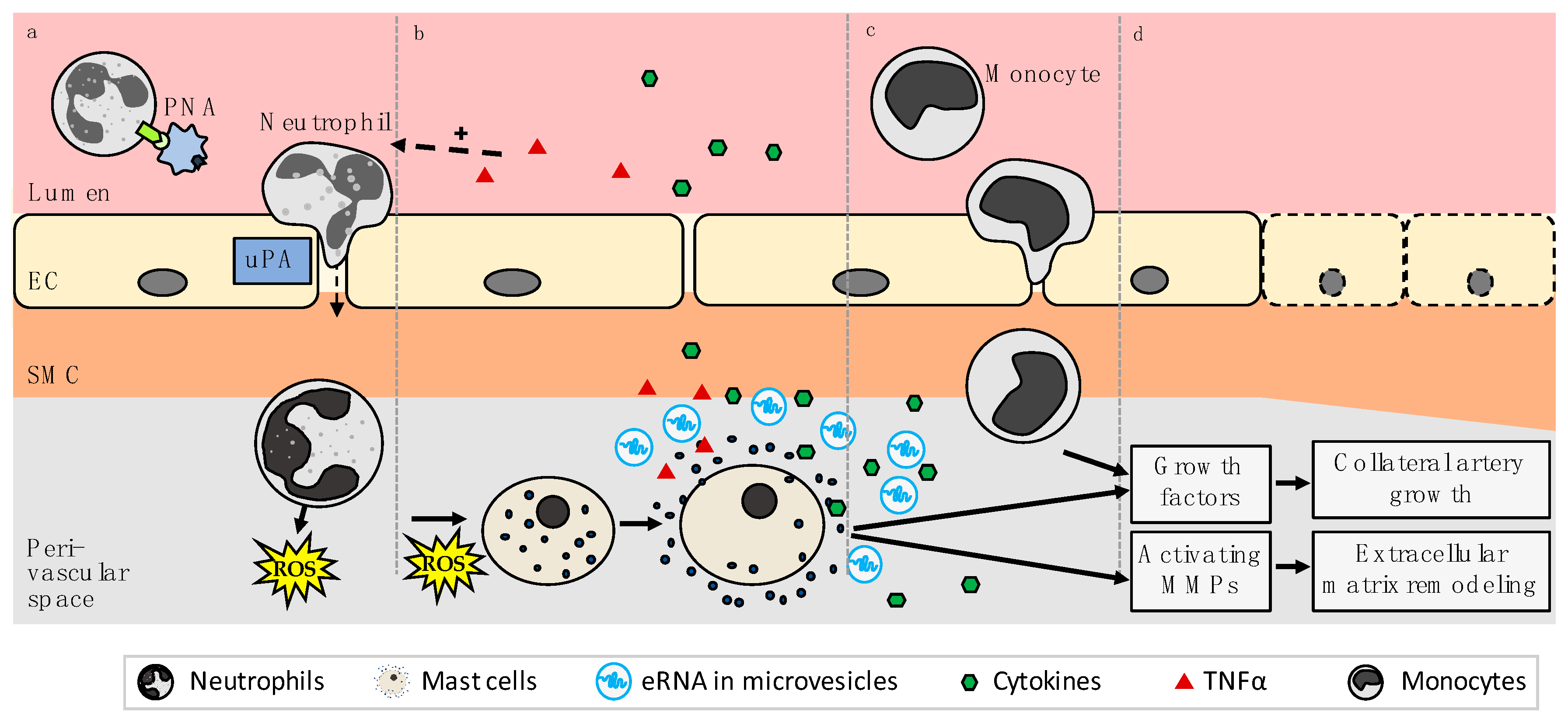{kind=link}
1. Introduction
2. The Role of eRNA in Arteriogenesis
2.1. eRNA Acts as a Translator of Shear Stress during Arteriogenesis through an Endothelial Mechanosensory Complex
2.1.1. eRNA Initiates Arteriogenesis by Locally Enhancing VEGF/VEGFR2 Signaling
2.1.2. Contribution of eRNA in the Promotion of Collateral Remodeling through Stimulation of Endothelial NFκB Signaling
2.2. eRNA Is Relevant for Mast Cell Degranulation during Arteriogenesis by Stimulating vWF Release and PNA Formation
2.2.1. eRNA Mediates vWF Release from Endothelial Weibel–Palade Bodies by Promoting VEGF/ VEGFR2 Signaling
2.2.2. eRNA-Mediated vWF Release Is Pivotal for PNA Formation
2.2.3. The Possible Role of ADAMTS13 in the Suppression of Thrombus Formation in Arteriogenesis
2.2.4. eRNA is Relevant for Mast Cell Degranulation Following PNA Formation
2.3. eRNA Released from Mast Cells Provides a Second Stimulatory Boost for Collateral Remodeling
3. eRNA in Other Forms of Vessel Growth
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADAMTS13 | A disintegrin and metalloprotease with a thrombospondin type 1 motif, member 13 |
| eRNA | Extracellular RNA |
| ICAM-1 | Intercellular adhesion molecule 1 |
| Mac-1 | Macrophage-1 antigen |
| MCP-1 | Monocyte chemoattractant protein 1 |
| MMP | Matrix metalloprotease |
| MV | Microvesicle |
| NFkB | Nuclear factor kB |
| Nox2 | NADPH oxidase 2 |
| NRP-1 | Neuropilin-1 |
| PECAM-1 | Platelet endothelial cell adhesion molecule 1 |
| PLC | Phospholipase C |
| PNA | Platelet-neutrophil aggregate |
| ROS | Reactive oxygen species |
| TACE | TNFa converting enzyme |
| TNFa | Tumor necrosis factor a |
| uPA | Urokinase-type plasminogen activator |
| VE-cadherin | Vascular endothelial cell cadherin |
| VEGF | Vascular endothelial growth factor |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
| vWF | Von Willebrand factor |
References
- GBD 2017 Causes of Death Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [CrossRef]
- Deindl, E.; Schaper, W. The art of arteriogenesis. Cell Biochem. Biophys. 2005, 43, 1–15. [Google Scholar] [CrossRef]
- Lasch, M.; Kleinert, E.C.; Meister, S.; Kumaraswami, K.; Buchheim, J.I.; Grantzow, T.; Lautz, T.; Salpisti, S.; Fischer, S.; Troidl, K.; et al. Extracellular RNA released due to shear stress controls natural bypass growth by mediating mechanotransduction in mice. Blood 2019, 134, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Fuentes, H.A.; Ruiz-Meana, M.; Simsekyilmaz, S.; Kostin, S.; Inserte, J.; Saffarzadeh, M.; Galuska, S.P.; Vijayan, V.; Barba, I.; Barreto, G.; et al. RNase1 prevents the damaging interplay between extracellular RNA and tumour necrosis factor-alpha in cardiac ischaemia/reperfusion injury. Thromb. Haemost. 2014, 112, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.P.; Zhou, J.P.; Zhang, J.X.; Wang, J.Y.; Wang, Z.Y.; Pan, M.; Li, L.F.; Li, C.C.; Wang, K.K.; Bai, Y.P.; et al. MiR-15b-5p Regulates Collateral Artery Formation by Targeting AKT3 (Protein Kinase B-3). Arterioscler. Thromb. Vasc. Biol. 2017, 37, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Cai, B.; Wu, X.; Peng, S.; Gan, L.; Huang, D.; Liu, G.; Dong, L.; Xiao, L.; Liu, J.; et al. microRNA-352 regulates collateral vessel growth induced by elevated fluid shear stress in the rat hind limb. Sci. Rep. 2017, 7, 6643. [Google Scholar] [CrossRef] [PubMed]
- Heuslein, J.L.; McDonnell, S.P.; Song, J.; Annex, B.H.; Price, R.J. MicroRNA-146a Regulates Perfusion Recovery in Response to Arterial Occlusion via Arteriogenesis. Front. Bioeng. Biotechnol. 2018, 6, 1. [Google Scholar] [CrossRef]
- Lei, Z.; van Mil, A.; Brandt, M.M.; Grundmann, S.; Hoefer, I.; Smits, M.; El Azzouzi, H.; Fukao, T.; Cheng, C.; Doevendans, P.A.; et al. MicroRNA-132/212 family enhances arteriogenesis after hindlimb ischaemia through modulation of the Ras-MAPK pathway. J. Cell. Mol. Med. 2015, 19, 1994–2005. [Google Scholar] [CrossRef]
- Stieger, P.; Daniel, J.M.; Tholen, C.; Dutzmann, J.; Knopp, K.; Gunduz, D.; Aslam, M.; Kampschulte, M.; Langheinrich, A.; Fischer, S.; et al. Targeting of Extracellular RNA Reduces Edema Formation and Infarct Size and Improves Survival After Myocardial Infarction in Mice. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Kleinert, E.; Langenmayer, M.C.; Reichart, B.; Kindermann, J.; Griemert, B.; Blutke, A.; Troidl, K.; Mayr, T.; Grantzow, T.; Noyan, F.; et al. Ribonuclease (RNase) Prolongs Survival of Grafts in Experimental Heart Transplantation. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Simsekyilmaz, S.; Cabrera-Fuentes, H.A.; Meiler, S.; Kostin, S.; Baumer, Y.; Liehn, E.A.; Weber, C.; Boisvert, W.A.; Preissner, K.T.; Zernecke, A. Role of extracellular RNA in atherosclerotic plaque formation in mice. Circulation 2014, 129, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Gerriets, T.; Wessels, C.; Walberer, M.; Kostin, S.; Stolz, E.; Zheleva, K.; Hocke, A.; Hippenstiel, S.; Preissner, K.T. Extracellular RNA mediates endothelial-cell permeability via vascular endothelial growth factor. Blood 2007, 110, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Kannemeier, C.; Shibamiya, A.; Nakazawa, F.; Trusheim, H.; Ruppert, C.; Markart, P.; Song, Y.; Tzima, E.; Kennerknecht, E.; Niepmann, M.; et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc. Natl. Acad. Sci. USA 2007, 104, 6388–6393. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Grantzow, T.; Pagel, J.I.; Tschernatsch, M.; Sperandio, M.; Preissner, K.T.; Deindl, E. Extracellular RNA promotes leukocyte recruitment in the vascular system by mobilising proinflammatory cytokines. Thromb. Haemost. 2012, 108, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Pipp, F.; Boehm, S.; Cai, W.J.; Adili, F.; Ziegler, B.; Karanovic, G.; Ritter, R.; Balzer, J.; Scheler, C.; Schaper, W.; et al. Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Deindl, E.; Buschmann, I.; Hoefer, I.E.; Podzuweit, T.; Boengler, K.; Vogel, S.; van Royen, N.; Fernandez, B.; Schaper, W. Role of ischemia and of hypoxia-inducible genes in arteriogenesis after femoral artery occlusion in the rabbit. Circ. Res. 2001, 89, 779–786. [Google Scholar] [CrossRef]
- Busse, R.; Fleming, I. Regulation of endothelium-derived vasoactive autacoid production by hemodynamic forces. Trends Pharmacol. Sci. 2003, 24, 24–29. [Google Scholar] [CrossRef]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef]
- Shay-Salit, A.; Shushy, M.; Wolfovitz, E.; Yahav, H.; Breviario, F.; Dejana, E.; Resnick, N. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2002, 99, 9462–9467. [Google Scholar] [CrossRef]
- Carmeliet, P.; Lampugnani, M.G.; Moons, L.; Breviario, F.; Compernolle, V.; Bono, F.; Balconi, G.; Spagnuolo, R.; Oosthuyse, B.; Dewerchin, M.; et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 1999, 98, 147–157. [Google Scholar] [CrossRef]
- Chen, Z.; Rubin, J.; Tzima, E. Role of PECAM-1 in arteriogenesis and specification of preexisting collaterals. Circ. Res. 2010, 107, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Koh, D.R. Neutrophils promote inflammatory angiogenesis via release of preformed VEGF in an in vivo corneal model. Cell Tissue Res. 2010, 339, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, M.; Giladi, A.; Leibovich, S.J. Regulation of vascular endothelial growth factor gene expression in murine macrophages by nitric oxide and hypoxia. Exp. Biol. Med. 2003, 228, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.R.; Yarovinsky, T.O.; Young, B.D.; Moraes, F.; Ross, T.D.; Ceneri, N.; Zhang, J.; Zhuang, Z.W.; Sinusas, A.J.; Pardi, R.; et al. Chemokine-coupled beta2 integrin-induced macrophage Rac2-Myosin IIA interaction regulates VEGF-A mRNA stability and arteriogenesis. J. Exp. Med. 2014, 211, 1957–1968. [Google Scholar] [CrossRef] [PubMed]
- Lautz, T.; Lasch, M.; Borgolte, J.; Troidl, K.; Pagel, J.I.; Caballero-Martinez, A.; Kleinert, E.C.; Walzog, B.; Deindl, E. Midkine Controls Arteriogenesis by Regulating the Bioavailability of Vascular Endothelial Growth Factor A and the Expression of Nitric Oxide Synthase 1 and 3. EBioMedicine 2018, 27, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Connolly, D.T.; Heuvelman, D.M.; Nelson, R.; Olander, J.V.; Eppley, B.L.; Delfino, J.J.; Siegel, N.R.; Leimgruber, R.M.; Feder, J. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J. Clin. Investig. 1989, 84, 1470–1478. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Chen, Y.; Dellian, M.; Safabakhsh, N.; Ferrara, N.; Jain, R.K. Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proc. Natl. Acad. Sci. USA 1996, 93, 14765–14770. [Google Scholar] [CrossRef]
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef]
- Pipp, F.; Heil, M.; Issbrucker, K.; Ziegelhoeffer, T.; Martin, S.; van den Heuvel, J.; Weich, H.; Fernandez, B.; Golomb, G.; Carmeliet, P.; et al. VEGFR-1-selective VEGF homologue PlGF is arteriogenic: Evidence for a monocyte-mediated mechanism. Circ. Res. 2003, 92, 378–385. [Google Scholar] [CrossRef]
- Lloyd, P.G.; Prior, B.M.; Li, H.; Yang, H.T.; Terjung, R.L. VEGF receptor antagonism blocks arteriogenesis, but only partially inhibits angiogenesis, in skeletal muscle of exercise-trained rats. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H759–H768. [Google Scholar] [CrossRef]
- Toyota, E.; Warltier, D.C.; Brock, T.; Ritman, E.; Kolz, C.; O’Malley, P.; Rocic, P.; Focardi, M.; Chilian, W.M. Vascular endothelial growth factor is required for coronary collateral growth in the rat. Circulation 2005, 112, 2108–2113. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect. Med. 2012, 2, a006502. [Google Scholar] [CrossRef] [PubMed]
- Lanahan, A.; Zhang, X.; Fantin, A.; Zhuang, Z.; Rivera-Molina, F.; Speichinger, K.; Prahst, C.; Zhang, J.; Wang, Y.; Davis, G.; et al. The neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent arteriogenesis. Dev. Cell 2013, 25, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Lanahan, A.A.; Hermans, K.; Claes, F.; Kerley-Hamilton, J.S.; Zhuang, Z.W.; Giordano, F.J.; Carmeliet, P.; Simons, M. VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev. Cell 2010, 18, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Nishio, M.; Peters, S.C.; Tschernatsch, M.; Walberer, M.; Weidemann, S.; Heidenreich, R.; Couraud, P.O.; Weksler, B.B.; Romero, I.A.; et al. Signaling mechanism of extracellular RNA in endothelial cells. FASEB J. 2009, 23, 2100–2109. [Google Scholar] [CrossRef]
- Landre, J.B.; Hewett, P.W.; Olivot, J.M.; Friedl, P.; Ko, Y.; Sachinidis, A.; Moenner, M. Human endothelial cells selectively express large amounts of pancreatic-type ribonuclease (RNase 1). J. Cell. Biochem. 2002, 86, 540–552. [Google Scholar] [CrossRef]
- Tsui, N.B.; Ng, E.K.; Lo, Y.M. Stability of endogenous and added RNA in blood specimens, serum, and plasma. Clin. Chem. 2002, 48, 1647–1653. [Google Scholar]
- Arras, M.; Ito, W.D.; Scholz, D.; Winkler, B.; Schaper, J.; Schaper, W. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J. Clin. Investig. 1998, 101, 40–50. [Google Scholar] [CrossRef]
- Hoefer, I.E.; Grundmann, S.; van Royen, N.; Voskuil, M.; Schirmer, S.H.; Ulusans, S.; Bode, C.; Buschmann, I.R.; Piek, J.J. Leukocyte subpopulations and arteriogenesis: Specific role of monocytes, lymphocytes and granulocytes. Atherosclerosis 2005, 181, 285–293. [Google Scholar] [CrossRef]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- Hoefer, I.E.; van Royen, N.; Rectenwald, J.E.; Deindl, E.; Hua, J.; Jost, M.; Grundmann, S.; Voskuil, M.; Ozaki, C.K.; Piek, J.J.; et al. Arteriogenesis proceeds via ICAM-1/Mac-1- mediated mechanisms. Circ. Res. 2004, 94, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Scholz, D.; Ito, W.; Fleming, I.; Deindl, E.; Sauer, A.; Wiesnet, M.; Busse, R.; Schaper, J.; Schaper, W. Ultrastructure and molecular histology of rabbit hind-limb collateral artery growth (arteriogenesis). Virchows Arch. 2000, 436, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Zoja, C.; Figliuzzi, M.; Foppolo, M.; Micheletti, G.; Bontempelli, M.; Saronni, M.; Remuzzi, G.; Remuzzi, A. Fluid shear stress modulates surface expression of adhesion molecules by endothelial cells. Blood 1995, 85, 1696–1703. [Google Scholar] [CrossRef] [PubMed]
- Elsemuller, A.K.; Tomalla, V.; Gartner, U.; Troidl, K.; Jeratsch, S.; Graumann, J.; Baal, N.; Hackstein, H.; Lasch, M.; Deindl, E.; et al. Characterization of mast cell-derived rRNA-containing microvesicles and their inflammatory impact on endothelial cells. FASEB J. 2019, 33, 5457–5467. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.P.; Li, B.; Hylton, D.; Detmar, M.; Yancopoulos, G.D.; Rudge, J.S. Transgenic delivery of VEGF to mouse skin leads to an inflammatory condition resembling human psoriasis. Blood 2003, 102, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, S.; Li, M.; Wu, D.; Liu, F.; Yang, R.; Ji, S.; Ji, A.; Li, Y. The Neuropilin-1 Inhibitor, ATWLPPR Peptide, Prevents Experimental Diabetes-Induced Retinal Injury by Preserving Vascular Integrity and Decreasing Oxidative Stress. PLoS ONE 2015, 10, e0142571. [Google Scholar] [CrossRef]
- Numata, T.; Ito, T.; Maeda, T.; Egusa, C.; Tsuboi, R. IL-33 promotes ICAM-1 expression via NF-kB in murine mast cells. Allergol. Int. 2016, 65, 158–165. [Google Scholar] [CrossRef]
- Unthank, J.L.; McClintick, J.N.; Labarrere, C.A.; Li, L.; Distasi, M.R.; Miller, S.J. Molecular basis for impaired collateral artery growth in the spontaneously hypertensive rat: Insight from microarray analysis. Physiol. Rep. 2013, 1, e0005. [Google Scholar] [CrossRef]
- Huang, S.; Pettaway, C.A.; Uehara, H.; Bucana, C.D.; Fidler, I.J. Blockade of NF-kappaB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene 2001, 20, 4188–4197. [Google Scholar] [CrossRef]
- Hunt, B.J.; Jurd, K.M. Endothelial cell activation. A central pathophysiological process. Bmj (Clin. Res. Ed.) 1998, 316, 1328–1329. [Google Scholar] [CrossRef]
- Wagner, D.D.; Olmsted, J.B.; Marder, V.J. Immunolocalization of von Willebrand protein in Weibel-Palade bodies of human endothelial cells. J. Cell Biol 1982, 95, 355–360. [Google Scholar] [CrossRef] [PubMed]
- McEver, R.P.; Beckstead, J.H.; Moore, K.L.; Marshall-Carlson, L.; Bainton, D.F. GMP-140, a platelet alpha-granule membrane protein, is also synthesized by vascular endothelial cells and is localized in Weibel-Palade bodies. J. Clin. Investig. 1989, 84, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Utgaard, J.O.; Jahnsen, F.L.; Bakka, A.; Brandtzaeg, P.; Haraldsen, G. Rapid secretion of prestored interleukin 8 from Weibel-Palade bodies of microvascular endothelial cells. J. Exp. Med. 1998, 188, 1751–1756. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Yamakuchi, M.; Morrell, C.N.; Ozaki, M.; O’Rourke, B.; Irani, K.; Lowenstein, C.J. Vascular endothelial growth factor regulation of Weibel-Palade-body exocytosis. Blood 2005, 105, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Vischer, U.M.; Jornot, L.; Wollheim, C.B.; Theler, J.M. Reactive oxygen intermediates induce regulated secretion of von Willebrand factor from cultured human vascular endothelial cells. Blood 1995, 85, 3164–3172. [Google Scholar] [CrossRef]
- Van den Biggelaar, M.; Hernandez-Fernaud, J.R.; van den Eshof, B.L.; Neilson, L.J.; Meijer, A.B.; Mertens, K.; Zanivan, S. Quantitative phosphoproteomics unveils temporal dynamics of thrombin signaling in human endothelial cells. Blood 2014, 123, e22–e36. [Google Scholar] [CrossRef]
- Crawley, J.T.; de Groot, R.; Xiang, Y.; Luken, B.M.; Lane, D.A. Unraveling the scissile bond: How ADAMTS13 recognizes and cleaves von Willebrand factor. Blood 2011, 118, 3212–3221. [Google Scholar] [CrossRef]
- Shim, K.; Anderson, P.J.; Tuley, E.A.; Wiswall, E.; Sadler, J.E. Platelet-VWF complexes are preferred substrates of ADAMTS13 under fluid shear stress. Blood 2008, 111, 651–657. [Google Scholar] [CrossRef]
- Chandraratne, S.; von Bruehl, M.L.; Pagel, J.I.; Stark, K.; Kleinert, E.; Konrad, I.; Farschtschi, S.; Coletti, R.; Gartner, F.; Chillo, O.; et al. Critical role of platelet glycoprotein ibalpha in arterial remodeling. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 589–597. [Google Scholar] [CrossRef]
- Chillo, O.; Kleinert, E.C.; Lautz, T.; Lasch, M.; Pagel, J.I.; Heun, Y.; Troidl, K.; Fischer, S.; Caballero-Martinez, A.; Mauer, A.; et al. Perivascular Mast Cells Govern Shear Stress-Induced Arteriogenesis by Orchestrating Leukocyte Function. Cell Rep. 2016, 16, 2197–2207. [Google Scholar] [CrossRef]
- Page, C.; Pitchford, S. Neutrophil and platelet complexes and their relevance to neutrophil recruitment and activation. Int. Immunopharmacol. 2013, 17, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Deindl, E.; Ziegelhoffer, T.; Kanse, S.M.; Fernandez, B.; Neubauer, E.; Carmeliet, P.; Preissner, K.T.; Schaper, W. Receptor-independent role of the urokinase-type plasminogen activator during arteriogenesis. FASEB J. 2003, 17, 1174–1176. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Sadler, J.E. Contact—How platelets touch von Willebrand factor. Science 2002, 297, 1128–1129. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.K.; Gardiner, E.E.; Shen, Y.; Whisstock, J.C.; Berndt, M.C. Glycoprotein Ib-IX-V. Int. J. Biochem. Cell Biol. 2003, 35, 1170–1174. [Google Scholar] [CrossRef]
- Savage, B.; Saldivar, E.; Ruggeri, Z.M. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 1996, 84, 289–297. [Google Scholar] [CrossRef]
- Goto, S.; Ichikawa, N.; Lee, M.; Goto, M.; Sakai, H.; Kim, J.J.; Yoshida, M.; Handa, M.; Ikeda, Y.; Handa, S. Platelet surface P-selectin molecules increased after exposing platelet to a high shear flow. Int. Angiol. 2000, 19, 147–151. [Google Scholar]
- Moore, K.L.; Patel, K.D.; Bruehl, R.E.; Li, F.; Johnson, D.A.; Lichenstein, H.S.; Cummings, R.D.; Bainton, D.F.; McEver, R.P. P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. J. Cell Biol. 1995, 128, 661–671. [Google Scholar] [CrossRef]
- Xiao, Z.; Goldsmith, H.L.; McIntosh, F.A.; Shankaran, H.; Neelamegham, S. Biomechanics of P-selectin PSGL-1 bonds: Shear threshold and integrin-independent cell adhesion. Biophys. J. 2006, 90, 2221–2234. [Google Scholar] [CrossRef]
- Dole, V.S.; Bergmeier, W.; Mitchell, H.A.; Eichenberger, S.C.; Wagner, D.D. Activated platelets induce Weibel-Palade-body secretion and leukocyte rolling in vivo: Role of P-selectin. Blood 2005, 106, 2334–2339. [Google Scholar] [CrossRef]
- De Wit, T.R.; van Mourik, J.A. Biosynthesis, processing and secretion of von Willebrand factor: Biological implications. Best Pract. Res. Clin. Haematol. 2001, 14, 241–255. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Levy, G.G.; Nichols, W.C.; Lian, E.C.; Foroud, T.; McClintick, J.N.; McGee, B.M.; Yang, A.Y.; Siemieniak, D.R.; Stark, K.R.; Gruppo, R.; et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001, 413, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Feys, H.B.; Anderson, P.J.; Sadler, J.E. Binding of ADAMTS13 to Von Willebrand Factor Under Static Conditions and Under Fluid Shear Stress. Blood 2008, 112, 258. [Google Scholar] [CrossRef]
- Chauhan, A.K.; Motto, D.G.; Lamb, C.B.; Bergmeier, W.; Dockal, M.; Plaimauer, B.; Scheiflinger, F.; Ginsburg, D.; Wagner, D.D. Systemic antithrombotic effects of ADAMTS13. J. Exp. Med. 2006, 203, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cao, Y.; Yang, X.; Cai, P.; Kang, L.; Zhu, X.; Luo, H.; Lu, L.; Wei, L.; Bai, X.; et al. ADAMTS13 controls vascular remodeling by modifying VWF reactivity during stroke recovery. Blood 2017, 130, 11–22. [Google Scholar] [CrossRef]
- Lee, M.; Keener, J.; Xiao, J.; Long Zheng, X.; Rodgers, G.M. ADAMTS13 and its variants promote angiogenesis via upregulation of VEGF and VEGFR2. Cell. Mol. Life Sci. 2015, 72, 349–356. [Google Scholar] [CrossRef]
- Tang, H.; Lee, M.; Kim, E.H.; Bishop, D.; Rodgers, G.M. siRNA-knockdown of ADAMTS-13 modulates endothelial cell angiogenesis. Microvasc. Res. 2017, 113, 65–70. [Google Scholar] [CrossRef]
- Wheway, J.; Latham, S.L.; Combes, V.; Grau, G.E. Endothelial microparticles interact with and support the proliferation of T cells. J.Immunol. 2014, 193, 3378–3387. [Google Scholar] [CrossRef]
- Mesri, M.; Altieri, D.C. Endothelial cell activation by leukocyte microparticles. J. Immunol. 1998, 161, 4382–4387. [Google Scholar]
- Baj-Krzyworzeka, M.; Majka, M.; Pratico, D.; Ratajczak, J.; Vilaire, G.; Kijowski, J.; Reca, R.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Platelet-derived microparticles stimulate proliferation, survival, adhesion, and chemotaxis of hematopoietic cells. Exp. Hematol. 2002, 30, 450–459. [Google Scholar] [CrossRef]
- Kunder, C.A.; St John, A.L.; Li, G.; Leong, K.W.; Berwin, B.; Staats, H.F.; Abraham, S.N. Mast cell-derived particles deliver peripheral signals to remote lymph nodes. J. Exp. Med. 2009, 206, 2455–2467. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, E.; Boing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef] [PubMed]
- Shai, E.; Varon, D. Development, cell differentiation, angiogenesis--microparticles and their roles in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Bowers, E.; Slaughter, A.; Frenette, P.S.; Kuick, R.; Pello, O.M.; Lucas, D. Granulocyte-derived TNFalpha promotes vascular and hematopoietic regeneration in the bone marrow. Nat. Med. 2018, 24, 95–102. [Google Scholar] [CrossRef]
- Tanaka, H.; Sukhova, G.; Schwartz, D.; Libby, P. Proliferating arterial smooth muscle cells after balloon injury express TNF-alpha but not interleukin-1 or basic fibroblast growth factor. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 12–18. [Google Scholar] [CrossRef]
- Buschmann, I.R.; Hoefer, I.E.; van Royen, N.; Katzer, E.; Braun-Dulleaus, R.; Heil, M.; Kostin, S.; Bode, C.; Schaper, W. GM-CSF: A strong arteriogenic factor acting by amplification of monocyte function. Atherosclerosis 2001, 159, 343–356. [Google Scholar] [CrossRef]
- Griffin, G.K.; Newton, G.; Tarrio, M.L.; Bu, D.X.; Maganto-Garcia, E.; Azcutia, V.; Alcaide, P.; Grabie, N.; Luscinskas, F.W.; Croce, K.J.; et al. IL-17 and TNF-alpha sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J. Immunol. 2012, 188, 6287–6299. [Google Scholar] [CrossRef]
- Stabile, E.; Burnett, M.S.; Watkins, C.; Kinnaird, T.; Bachis, A.; la Sala, A.; Miller, J.M.; Shou, M.; Epstein, S.E.; Fuchs, S. Impaired arteriogenic response to acute hindlimb ischemia in CD4-knockout mice. Circulation 2003, 108, 205–210. [Google Scholar] [CrossRef]
- Stabile, E.; Kinnaird, T.; la Sala, A.; Hanson, S.K.; Watkins, C.; Campia, U.; Shou, M.; Zbinden, S.; Fuchs, S.; Kornfeld, H.; et al. CD8+ T lymphocytes regulate the arteriogenic response to ischemia by infiltrating the site of collateral vessel development and recruiting CD4+ mononuclear cells through the expression of interleukin-16. Circulation 2006, 113, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Hoefer, I.E.; van Royen, N.; Rectenwald, J.E.; Bray, E.J.; Abouhamze, Z.; Moldawer, L.L.; Voskuil, M.; Piek, J.J.; Buschmann, I.R.; Ozaki, C.K. Direct evidence for tumor necrosis factor-alpha signaling in arteriogenesis. Circulation 2002, 105, 1639–1641. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, S.; Hoefer, I.; Ulusans, S.; van Royen, N.; Schirmer, S.H.; Ozaki, C.K.; Bode, C.; Piek, J.J.; Buschmann, I. Anti-tumor necrosis factor-{alpha} therapies attenuate adaptive arteriogenesis in the rabbit. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1497–H1505. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, S.H.; Buschmann, I.R.; Jost, M.M.; Hoefer, I.E.; Grundmann, S.; Andert, J.P.; Ulusans, S.; Bode, C.; Piek, J.J.; van Royen, N. Differential effects of MCP-1 and leptin on collateral flow and arteriogenesis. Cardiovasc. Res. 2004, 64, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Ito, W.D.; Arras, M.; Winkler, B.; Scholz, D.; Schaper, J.; Schaper, W. Monocyte chemotactic protein-1 increases collateral and peripheral conductance after femoral artery occlusion. Circ. Res. 1997, 80, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Hoefer, I.E.; van Royen, N.; Buschmann, I.R.; Piek, J.J.; Schaper, W. Time course of arteriogenesis following femoral artery occlusion in the rabbit. Cardiovasc. Res. 2001, 49, 609–617. [Google Scholar] [CrossRef]
- Gunn, M.D.; Nelken, N.A.; Liao, X.; Williams, L.T. Monocyte chemoattractant protein-1 is sufficient for the chemotaxis of monocytes and lymphocytes in transgenic mice but requires an additional stimulus for inflammatory activation. J. Immunol. 1997, 158, 376–383. [Google Scholar]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Kinoshita, M.; Okada, M.; Hara, M.; Furukawa, Y.; Matsumori, A. Mast cell tryptase in mast cell granules enhances MCP-1 and interleukin-8 production in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1858–1863. [Google Scholar] [CrossRef]
- Cushing, S.D.; Berliner, J.A.; Valente, A.J.; Territo, M.C.; Navab, M.; Parhami, F.; Gerrity, R.; Schwartz, C.J.; Fogelman, A.M. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc. Natl. Acad. Sci. USA 1990, 87, 5134–5138. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Gesierich, S.; Griemert, B.; Schanzer, A.; Acker, T.; Augustin, H.G.; Olsson, A.K.; Preissner, K.T. Extracellular RNA liberates tumor necrosis factor-alpha to promote tumor cell trafficking and progression. Cancer Res. 2013, 73, 5080–5089. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E. NF-kappaB activation. Crit. Care Med. 2000, 28, N100–N104. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Fuentes, H.A.; Lopez, M.L.; McCurdy, S.; Fischer, S.; Meiler, S.; Baumer, Y.; Galuska, S.P.; Preissner, K.T.; Boisvert, W.A. Regulation of monocyte/macrophage polarisation by extracellular RNA. Thromb. Haemost. 2015, 113, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Troidl, C.; Jung, G.; Troidl, K.; Hoffmann, J.; Mollmann, H.; Nef, H.; Schaper, W.; Hamm, C.W.; Schmitz-Rixen, T. The temporal and spatial distribution of macrophage subpopulations during arteriogenesis. Curr. Vasc. Pharmacol. 2013, 11, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Sharifpanah, F.; De Silva, S.; Bekhite, M.M.; Hurtado-Oliveros, J.; Preissner, K.T.; Wartenberg, M.; Sauer, H. Stimulation of vasculogenesis and leukopoiesis of embryonic stem cells by extracellular transfer RNA and ribosomal RNA. Free Radic. Biol. Med. 2015, 89, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Bekhite, M.M.; Finkensieper, A.; Binas, S.; Muller, J.; Wetzker, R.; Figulla, H.R.; Sauer, H.; Wartenberg, M. VEGF-mediated PI3K class IA and PKC signaling in cardiomyogenesis and vasculogenesis of mouse embryonic stem cells. J. Cell Sci. 2011, 124, 1819–1830. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Urao, N. Novel role of NADPH oxidase in angiogenesis and stem/progenitor cell function. Antioxid. Redox Signal. 2009, 11, 2517–2533. [Google Scholar] [CrossRef]
- Lu, L.; Li, J.; Moussaoui, M.; Boix, E. Immune Modulation by Human Secreted RNases at the Extracellular Space. Front. Immunol. 2018, 9, 1012. [Google Scholar] [CrossRef]
- Fett, J.W.; Strydom, D.J.; Lobb, R.R.; Alderman, E.M.; Bethune, J.L.; Riordan, J.F.; Vallee, B.L. Isolation and characterization of angiogenin, an angiogenic protein from human carcinoma cells. Biochemistry 1985, 24, 5480–5486. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kluever, A.-K.; Braumandl, A.; Fischer, S.; Preissner, K.T.; Deindl, E. The Extraordinary Role of Extracellular RNA in Arteriogenesis, the Growth of Collateral Arteries. Int. J. Mol. Sci. 2019, 20, 6177. https://doi.org/10.3390/ijms20246177
Kluever A-K, Braumandl A, Fischer S, Preissner KT, Deindl E. The Extraordinary Role of Extracellular RNA in Arteriogenesis, the Growth of Collateral Arteries. International Journal of Molecular Sciences. 2019; 20(24):6177. https://doi.org/10.3390/ijms20246177
Chicago/Turabian StyleKluever, Anna-Kristina, Anna Braumandl, Silvia Fischer, Klaus T. Preissner, and Elisabeth Deindl. 2019. "The Extraordinary Role of Extracellular RNA in Arteriogenesis, the Growth of Collateral Arteries" International Journal of Molecular Sciences 20, no. 24: 6177. https://doi.org/10.3390/ijms20246177
APA StyleKluever, A.-K., Braumandl, A., Fischer, S., Preissner, K. T., & Deindl, E. (2019). The Extraordinary Role of Extracellular RNA in Arteriogenesis, the Growth of Collateral Arteries. International Journal of Molecular Sciences, 20(24), 6177. https://doi.org/10.3390/ijms20246177