The Phosphatase SHP-2 Activates HIF-1α in Wounds In Vivo by Inhibition of 26S Proteasome Activity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
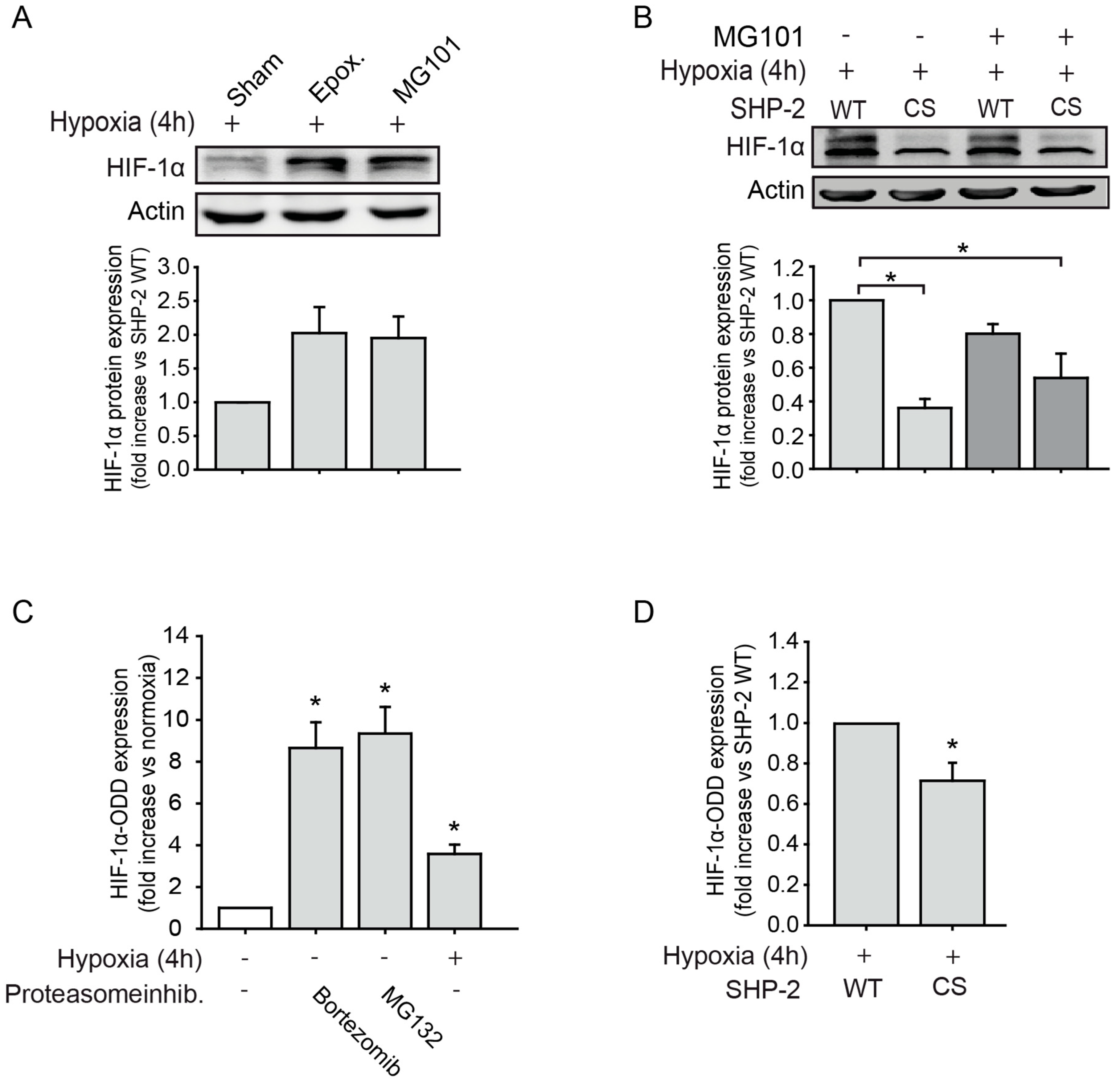
2.1. SHP-2 Inactivation Leads to Increased Proteasome Dependent HIF-1α Degradation during Hypoxia
2.2. SHP-2 Regulates Proteasomal Degradation of HIF-1α in Hypoxic Wounds In Vivo
2.3. The Proteasomal Degradation of HIF-1α is Dependent on Src Kinase and p38 MAPK Activation
2.4. SHP-2 Activity Inhibits the Chymotrypsin-Like Activity of the 26S Proteasome upon Hypoxia
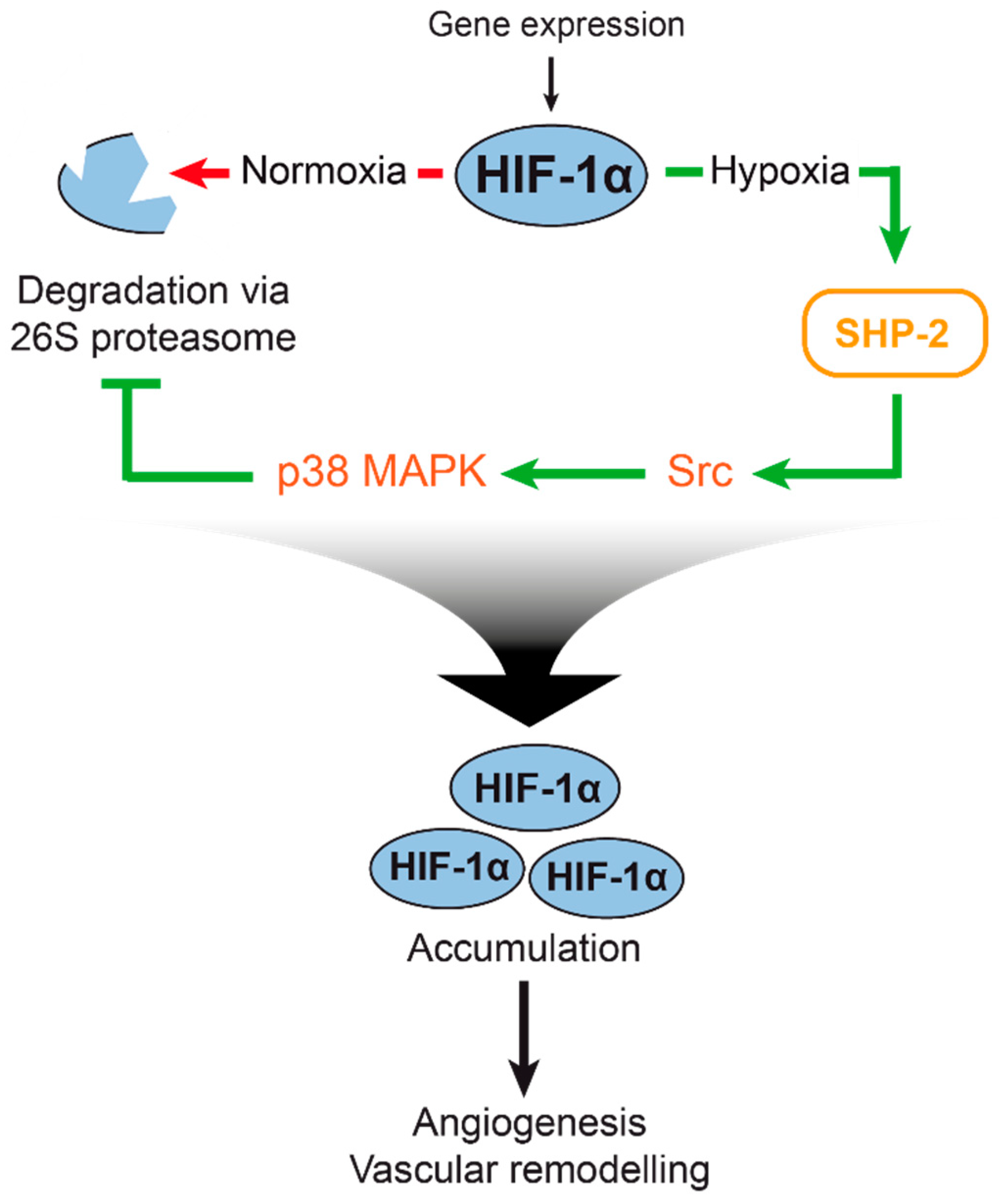
3. Discussion
4. Materials and Methods
4.1. Antibodies and Chemicals
4.2. Human Microvascular Endothelial Cell (HMEC) Culture
4.3. Lentiviral Constructs and Transductions
4.4. Hypoxia Treatment
4.5. In Vitro 26S Proteasome Activity
4.6. In Vitro Luciferase Assay
4.7. In Vivo Transduction and Luciferase Imaging
4.8. Immunoblotting
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ctrl-Luc | Lentiviral control luciferase reporter construct lacking the HIF-1α ODD |
| HIF1-ODD-Luc | Lentiviral construct containing the oxygen-dependent degradation (ODD) domain of HIF-1α fused to luciferase |
| HMEC | Human microvascular endothelial cells |
| SHP-2 | Src homology domain containing tyrosine phosphatase 2 |
| SHP-2 WT | SHP-2 wildtype construct |
| SHP-2 CS | Dominant negative SHP-2 construct where Cys459 was exchanged to Ser459 |
| SHP-2 E76A | Constitutively active SHP-2 construct where Glu76 was exchanged to Ala76 |
References
- Semenza, G.L. Targeting hypoxia-inducible factor 1 to stimulate tissue vascularization. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2016, 64, 361–363. [Google Scholar] [CrossRef] [PubMed]
- Rey, S.; Semenza, G.L. Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc. Res. 2010, 86, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, K.; Fox-Talbot, K.; Steenbergen, C.; Bosch-Marce, M.; Semenza, G.L. Adenoviral transfer of HIF-1alpha enhances vascular responses to critical limb ischemia in diabetic mice. Proc. Natl. Acad. Sci. USA 2009, 106, 18769–18774. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.H.; Kimura, H.; Weiss, C.R.; Semenza, G.L.; Hofmann, L.V. Constitutively active HIF-1alpha improves perfusion and arterial remodeling in an endovascular model of limb ischemia. Cardiovasc Res. 2005, 68, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Botusan, I.R.; Sunkari, V.G.; Savu, O.; Catrina, A.I.; Grunler, J.; Lindberg, S.; Pereira, T.; Yla-Herttuala, S.; Poellinger, L.; Brismar, K.; et al. Stabilization of HIF-1alpha is critical to improve wound healing in diabetic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 19426–19431. [Google Scholar] [CrossRef] [PubMed]
- Mace, K.A.; Yu, D.H.; Paydar, K.Z.; Boudreau, N.; Young, D.M. Sustained expression of Hif-1α in the diabetic environment promotes angiogenesis and cutaneous wound repair. Wound Repair Regen. 2007, 15, 636–645. [Google Scholar] [CrossRef]
- Walshe, T.E.; D’Amore, P.A. The role of hypoxia in vascular injury and repair. Annu Rev. Pathol. 2008, 3, 615–643. [Google Scholar] [CrossRef]
- Bilton, R.L.; Booker, G.W. The subtle side to hypoxia inducible factor (HIFalpha) regulation. Eur. J. Biochem. 2003, 270, 791–798. [Google Scholar] [CrossRef]
- Zhou, J.; Kohl, R.; Herr, B.; Frank, R.; Brune, B. Calpain mediates a von Hippel-Lindau protein-independent destruction of hypoxia-inducible factor-1alpha. Mol. Biol. Cell 2006, 17, 1549–1558. [Google Scholar] [CrossRef]
- Mannell, H.; Hellwig, N.; Gloe, T.; Plank, C.; Sohn, H.Y.; Groesser, L.; Walzog, B.; Pohl, U.; Krötz, F. Inhibition of the tyrosine phosphatase SHP-2 suppresses angiogenesis in vitro and in vivo. J. Vasc. Res. 2008, 45, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Heun, Y.; Pogoda, K.; Anton, M.; Pircher, J.; Pfeifer, A.; Woernle, M.; Ribeiro, A.; Kameritsch, P.; Mykhaylyk, O.; Plank, C.; et al. HIF-1alpha Dependent Wound Healing Angiogenesis In Vivo Can Be Controlled by Site-Specific Lentiviral Magnetic Targeting of SHP-2. Mol. Ther. 2017, 25, 1616–1627. [Google Scholar] [CrossRef] [PubMed]
- Rieck, S.; Heun, Y.; Heidsieck, A.; Mykhaylyk, O.; Pfeifer, A.; Gleich, B.; Mannell, H.; Wenzel, D. Local anti-angiogenic therapy by magnet-assisted downregulation of SHP2 phosphatase. J. Control. Release 2019, 305, 155–164. [Google Scholar] [CrossRef]
- Jang, H.H. Regulation of Protein Degradation by Proteasomes in Cancer. J. Cancer Prev. 2018, 23, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Huang, X.; Chen, M.J. Reversible phosphorylation of the 26S proteasome. Protein Cell 2017, 8, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Kimbrel, E.A.; Davis, T.N.; Bradner, J.E.; Kung, A.L. In vivo pharmacodynamic imaging of proteasome inhibition. Mol. Imaging 2009, 8, 140–147. [Google Scholar] [CrossRef]
- Thobe, B.M.; Frink, M.; Choudhry, M.A.; Schwacha, M.G.; Bland, K.I.; Chaudry, I.H. Src family kinases regulate p38 MAPK-mediated IL-6 production in Kupffer cells following hypoxia. Am. J. Physiol. 2006, 291, C476–C482. [Google Scholar] [CrossRef]
- Wojcik, C.; Di Napoli, M. Ubiquitin-proteasome system and proteasome inhibition: New strategies in stroke therapy. Stroke 2004, 35, 1506–1518. [Google Scholar] [CrossRef]
- Grundler, K.; Rotter, R.; Tilley, S.; Pircher, J.; Czermak, T.; Yakac, M.; Gaitzsch, E.; Massberg, S.; Krotz, F.; Sohn, H.Y.; et al. The proteasome regulates collagen-induced platelet aggregation via nuclear-factor-kappa-B (NFkB) activation. Thromb. Res. 2016, 148, 15–22. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, Y.; Yoon, S.K.; Yoon, J.B. Osmotic stress inhibits proteasome by p38 MAPK-dependent phosphorylation. J. Biol. Chem. 2010, 285, 41280–41289. [Google Scholar] [CrossRef]
- Zong, C.; Gomes, A.V.; Drews, O.; Li, X.; Young, G.W.; Berhane, B.; Qiao, X.; French, S.W.; Bardag-Gorce, F.; Ping, P. Regulation of murine cardiac 20S proteasomes: Role of associating partners. Circ. Res. 2006, 99, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Ades, E.W.; Candal, F.J.; Swerlick, R.A.; George, V.G.; Summers, S.; Bosse, D.C.; Lawley, T.J. HMEC-1: Establishment of an immortalized human microvascular endothelial cell line. J. Investig. Derm. 1992, 99, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Mannell, H.K.; Pircher, J.; Chaudhry, D.I.; Alig, S.K.; Koch, E.G.; Mettler, R.; Pohl, U.; Krötz, F. ARNO regulates VEGF-dependent tissue responses by stabilizing endothelial VEGFR-2 surface expression. Cardiovasc. Res. 2012, 93, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Kontaridis, M.I.; Liu, X.; Zhang, L.; Bennett, A.M. Role of SHP-2 in fibroblast growth factor receptor-mediated suppression of myogenesis in C2C12 myoblasts. Mol. Cell. Biol. 2002, 22, 3875–3891. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.; Wenzel, D.; Becher, U.M.; Freitag, D.F.; Klein, A.M.; Eberbeck, D.; Schulte, M.; Zimmermann, K.; Bergemann, C.; Gleich, B.; et al. Combined targeting of lentiviral vectors and positioning of transduced cells by magnetic nanoparticles. Proc. Natl. Acad. Sci. USA 2009, 106, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Alig, S.K.; Stampnik, Y.; Pircher, J.; Rotter, R.; Gaitzsch, E.; Ribeiro, A.; Wornle, M.; Krötz, F.; Mannell, H. The Tyrosine Phosphatase SHP-1 Regulates Hypoxia Inducible Factor-1alpha (HIF-1alpha) Protein Levels in Endothelial Cells under Hypoxia. PLoS ONE 2015, 10, e0121113. [Google Scholar] [CrossRef] [PubMed]
- Mannell, H.; Pircher, J.; Fochler, F.; Stampnik, Y.; Räthel, T.; Gleich, B.; Plank, C.; Mykhaylyk, O.; Dahmani, C.; Wornle, M.; et al. Site directed vascular gene delivery in vivo by ultrasonic destruction of magnetic nanoparticle coated microbubbles. Nanomedicine 2012, 8, 1309–1318. [Google Scholar] [CrossRef]
- Krötz, F.; Engelbrecht, B.; Buerkle, M.A.; Bassermann, F.; Bridell, H.; Gloe, T.; Duyster, J.; Pohl, U.; Sohn, H.Y. The tyrosine phosphatase, SHP-1, is a negative regulator of endothelial superoxide formation. J. Am. Coll. Cardiol. 2005, 45, 1700–1706. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heun, Y.; Grundler Groterhorst, K.; Pogoda, K.; Kraemer, B.F.; Pfeifer, A.; Pohl, U.; Mannell, H. The Phosphatase SHP-2 Activates HIF-1α in Wounds In Vivo by Inhibition of 26S Proteasome Activity. Int. J. Mol. Sci. 2019, 20, 4404. https://doi.org/10.3390/ijms20184404
Heun Y, Grundler Groterhorst K, Pogoda K, Kraemer BF, Pfeifer A, Pohl U, Mannell H. The Phosphatase SHP-2 Activates HIF-1α in Wounds In Vivo by Inhibition of 26S Proteasome Activity. International Journal of Molecular Sciences. 2019; 20(18):4404. https://doi.org/10.3390/ijms20184404
Chicago/Turabian StyleHeun, Yvonn, Katharina Grundler Groterhorst, Kristin Pogoda, Bjoern F Kraemer, Alexander Pfeifer, Ulrich Pohl, and Hanna Mannell. 2019. "The Phosphatase SHP-2 Activates HIF-1α in Wounds In Vivo by Inhibition of 26S Proteasome Activity" International Journal of Molecular Sciences 20, no. 18: 4404. https://doi.org/10.3390/ijms20184404
APA StyleHeun, Y., Grundler Groterhorst, K., Pogoda, K., Kraemer, B. F., Pfeifer, A., Pohl, U., & Mannell, H. (2019). The Phosphatase SHP-2 Activates HIF-1α in Wounds In Vivo by Inhibition of 26S Proteasome Activity. International Journal of Molecular Sciences, 20(18), 4404. https://doi.org/10.3390/ijms20184404