Comparative Transcriptome Analysis between a Novel Allohexaploid Cotton Progeny CMS Line LD6A and Its Maintainer Line LD6B

 , ,
, ,
Abstract
1. Introduction
2. Results
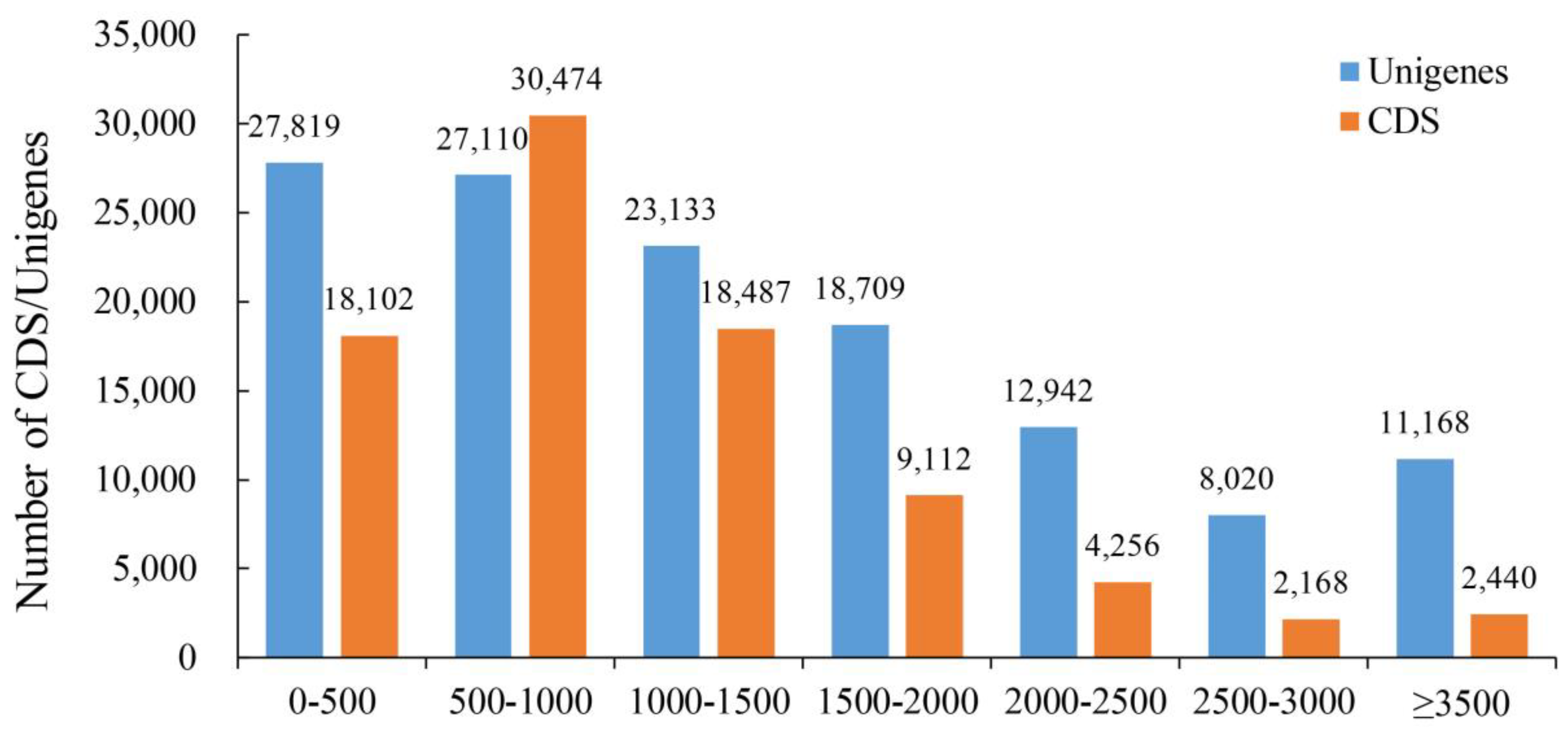
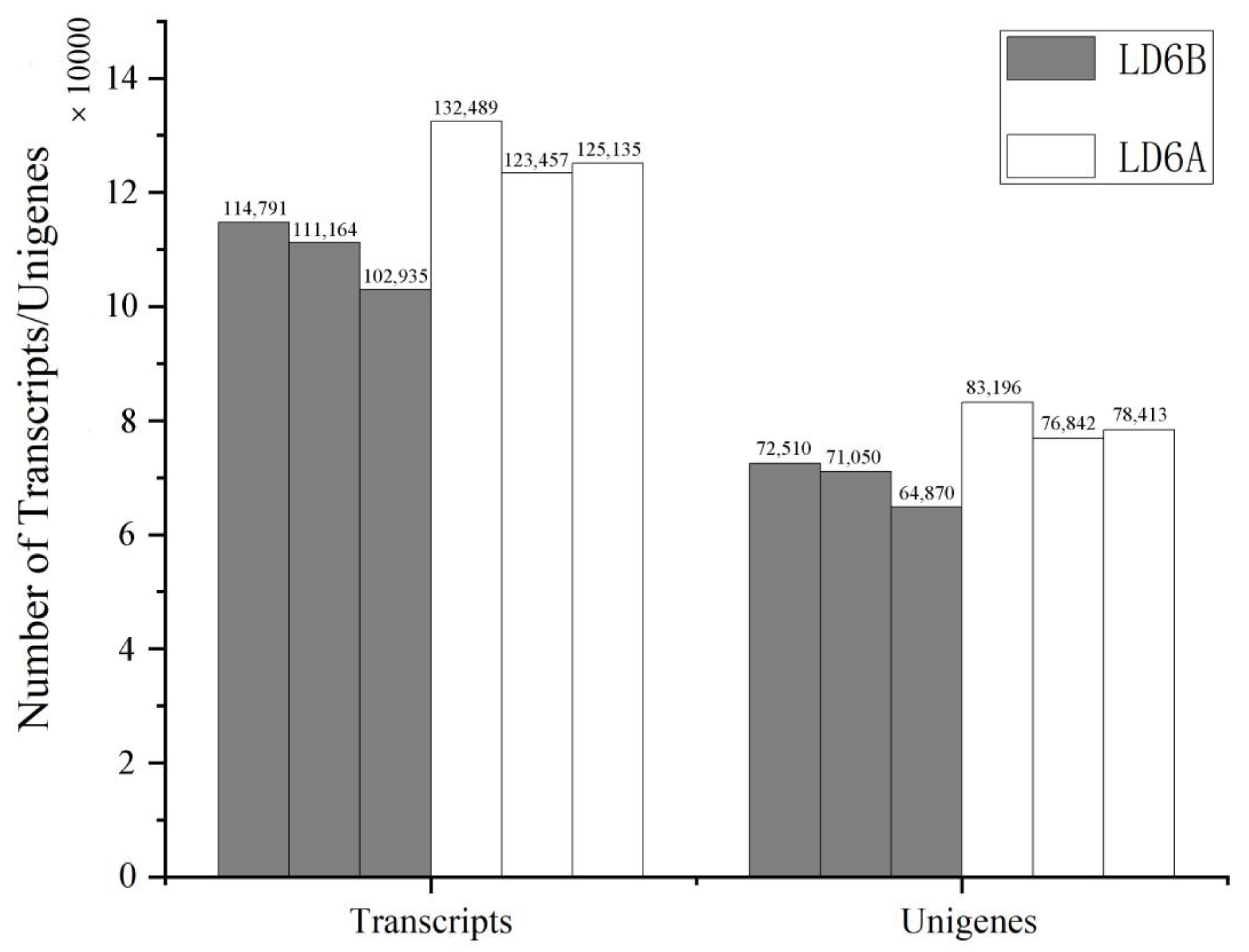
2.1. De Novo Transcriptome Analysis
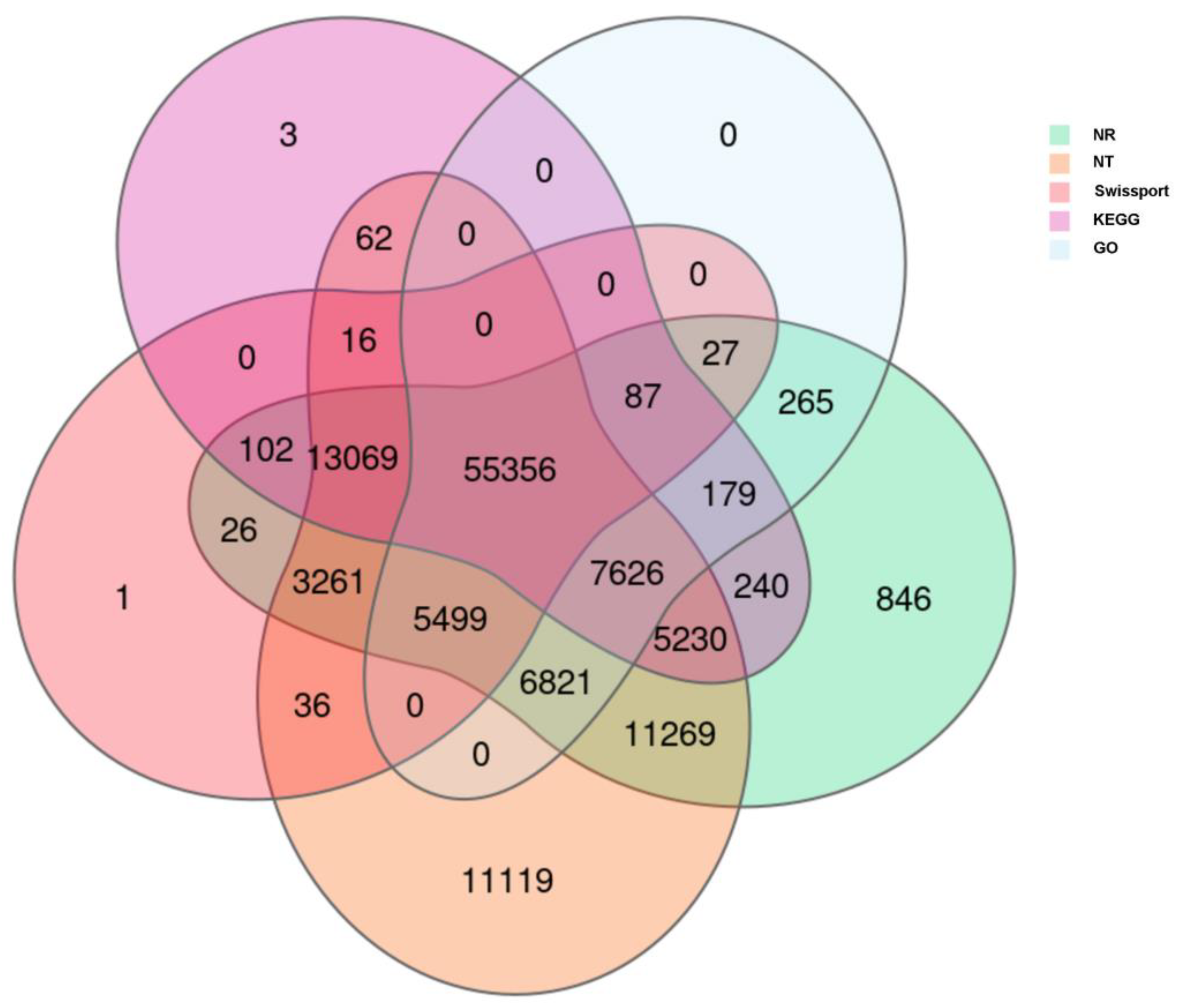
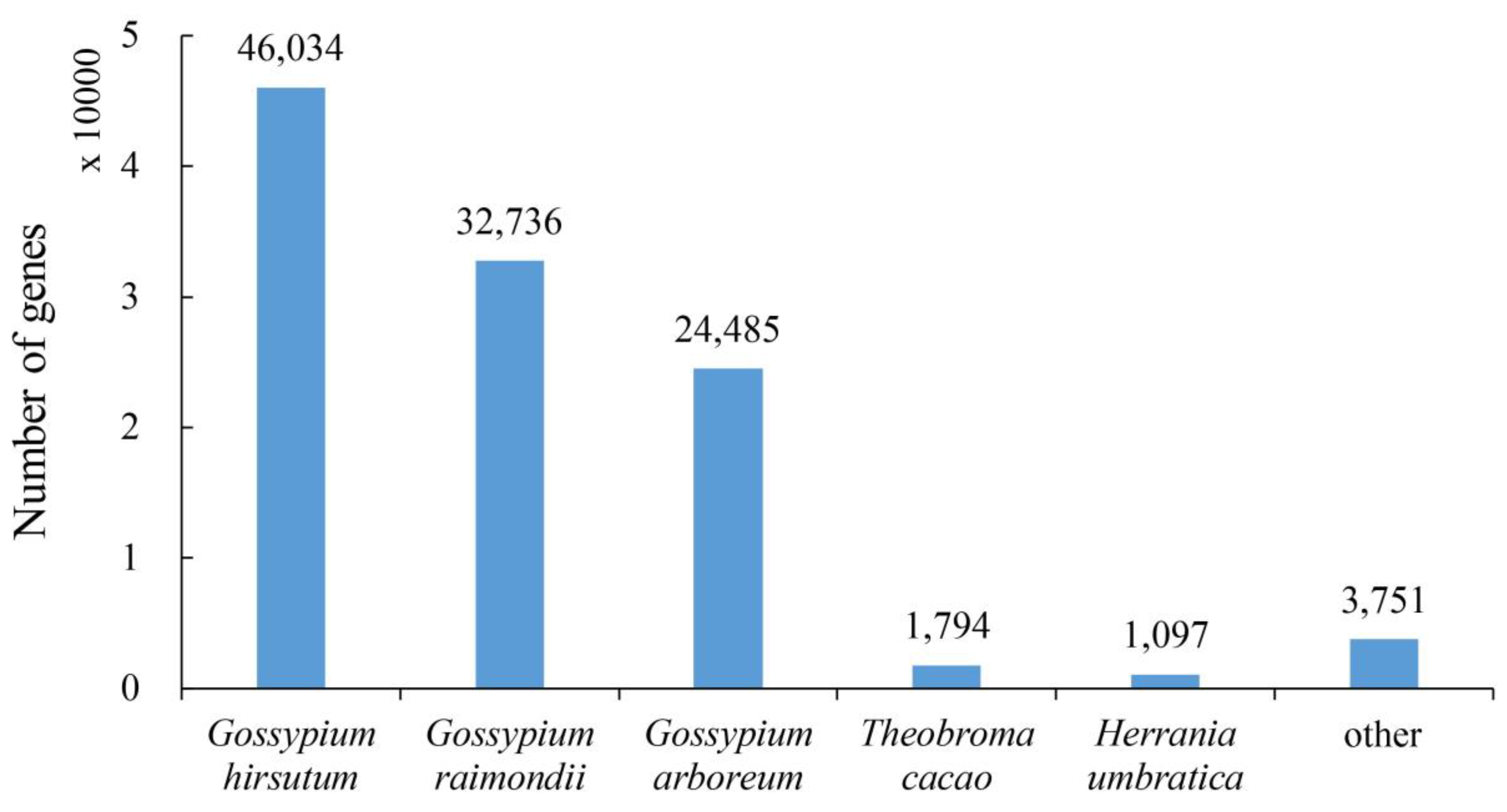
2.2. Annotation of the Assembled Cotton Genes
2.3. Annotation Difference Analysis of the Allohexaploid Progeny of Cotton
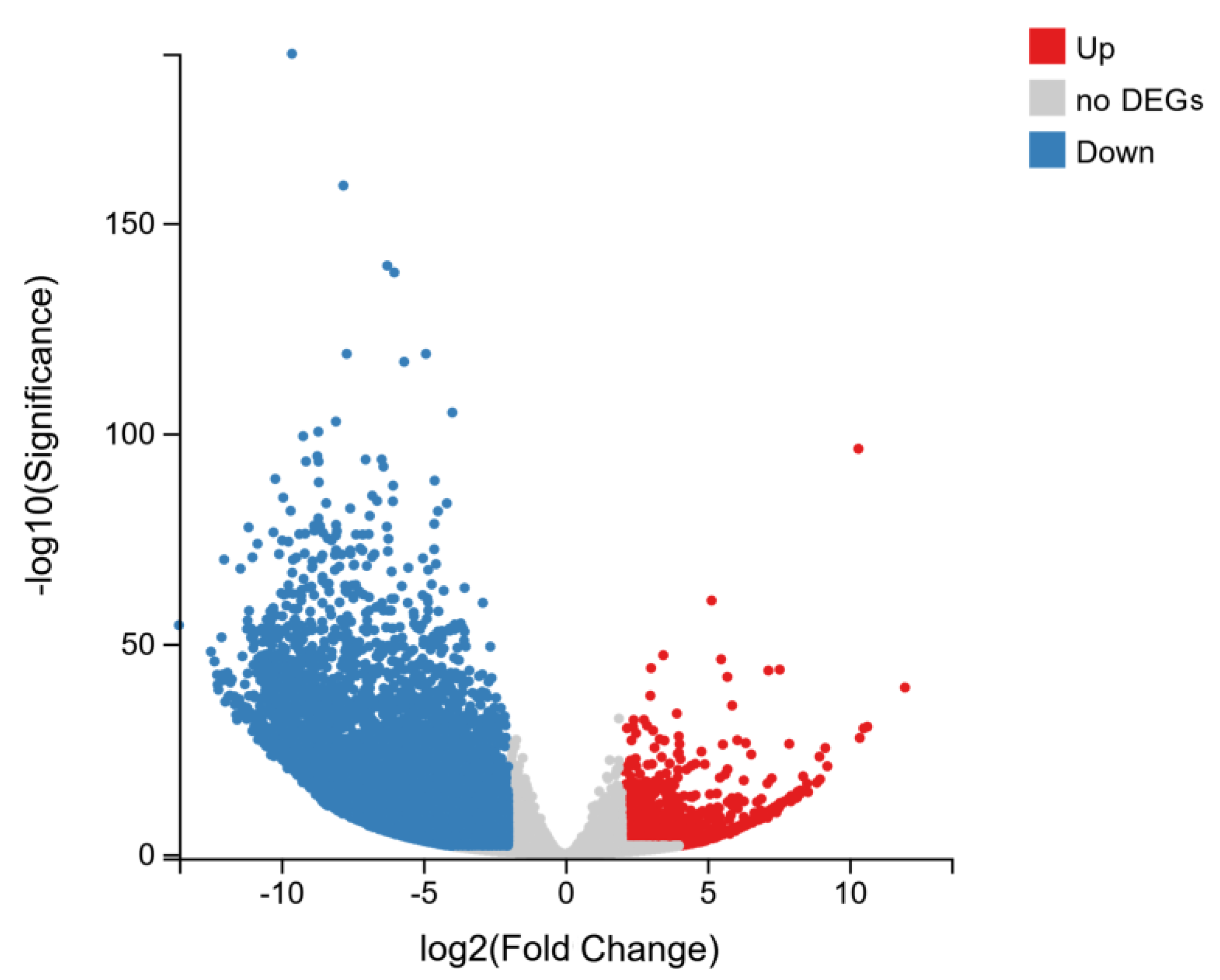
2.4. Analysis of Differentially Expressed Genes (DEGs)
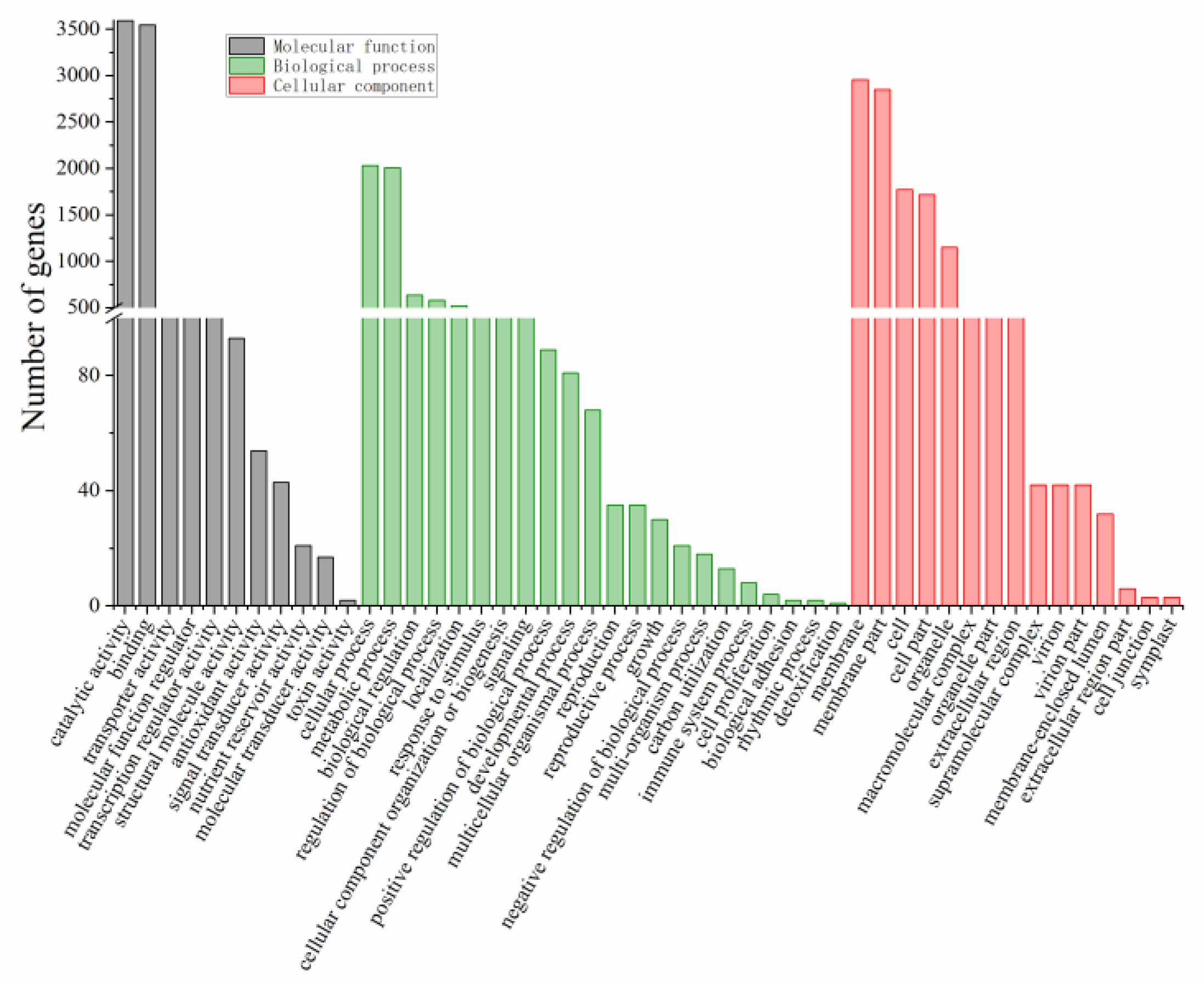
2.5. GO Annotation and Pathway Analysis of DEGs
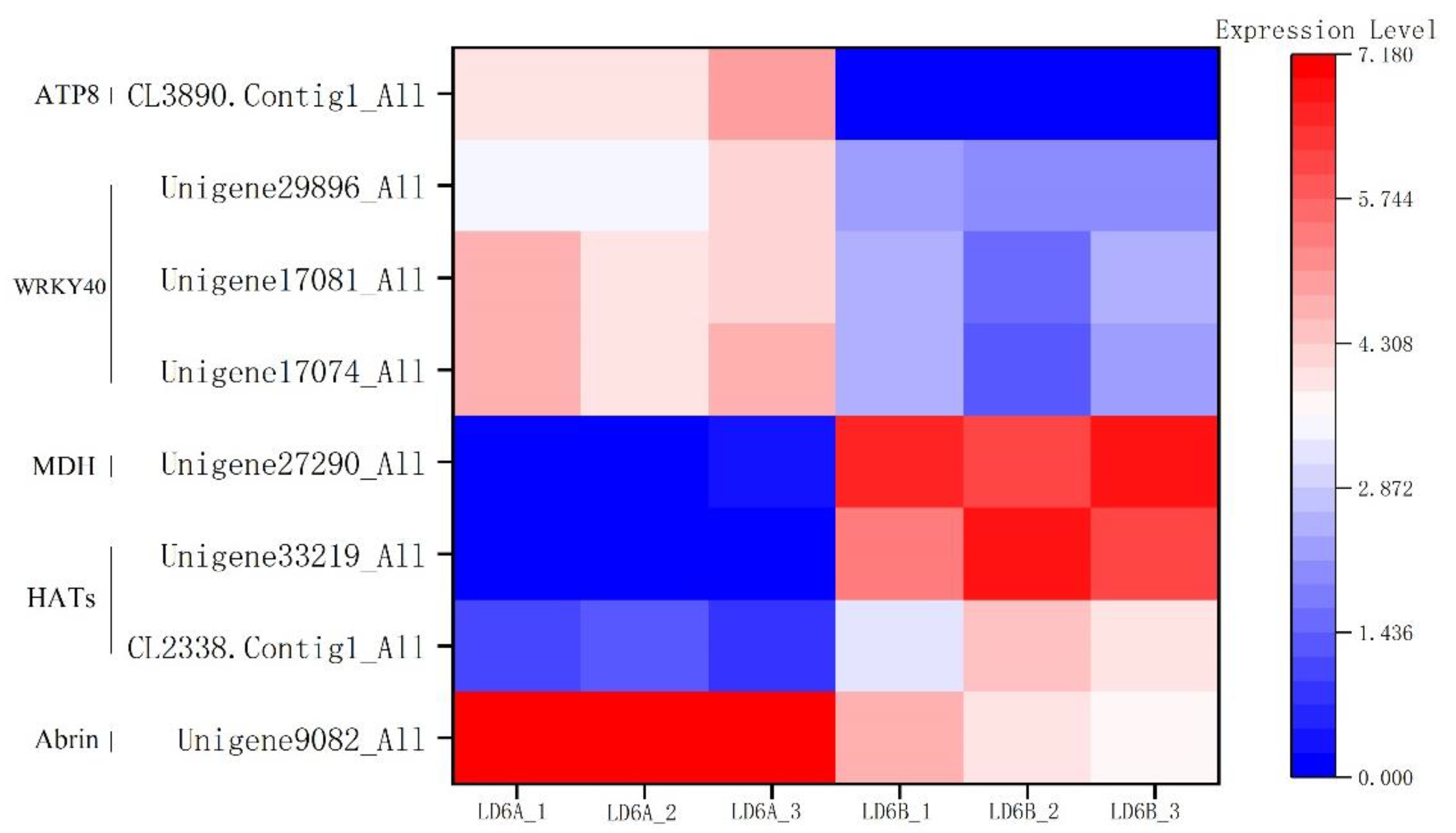
2.6. Validation of DEGs by qRT-PCR
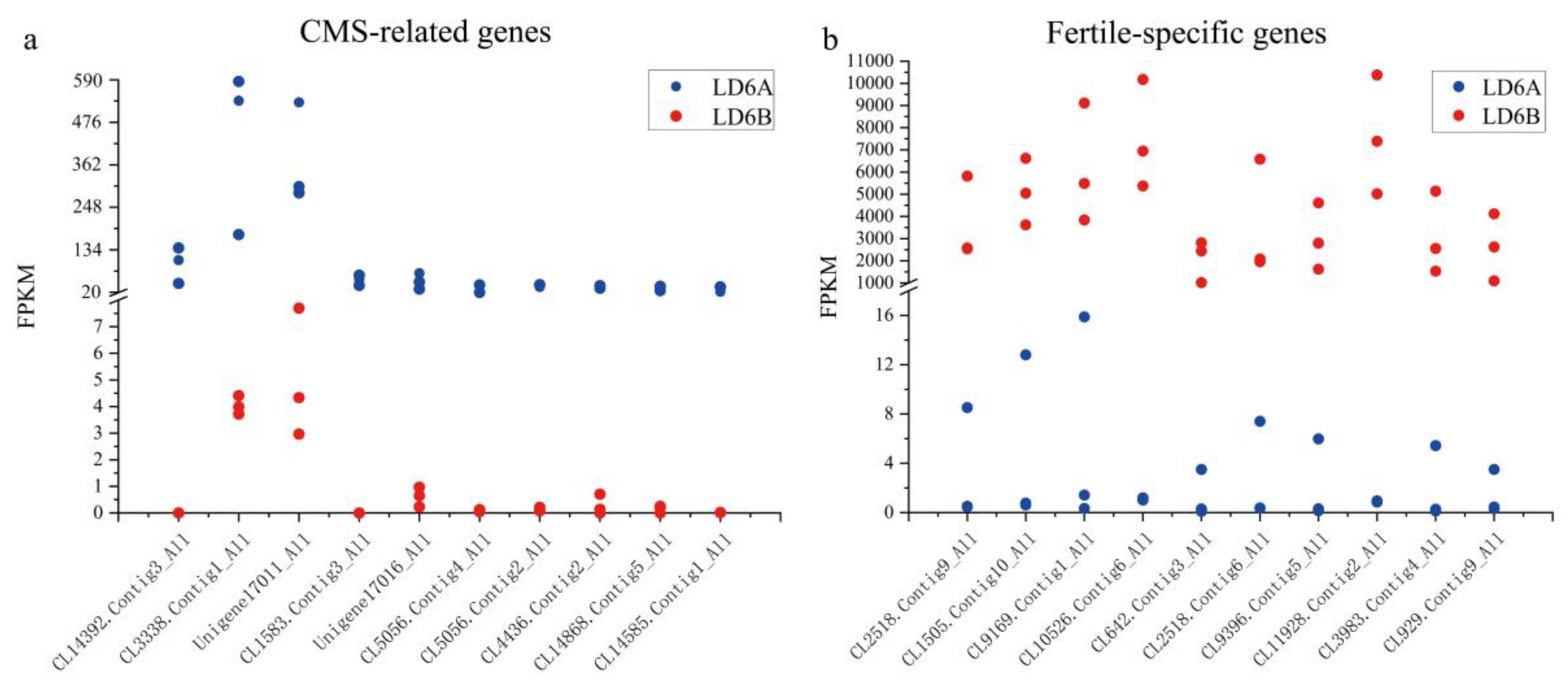
2.7. Candidate Genes Associated with CMS
2.7.1. Abrin
2.7.2. Histone Acetyltransferase (HATs)
2.7.3. TCA Cycle
2.7.4. WRKY40
2.7.5. Oxidative Phosphorylation
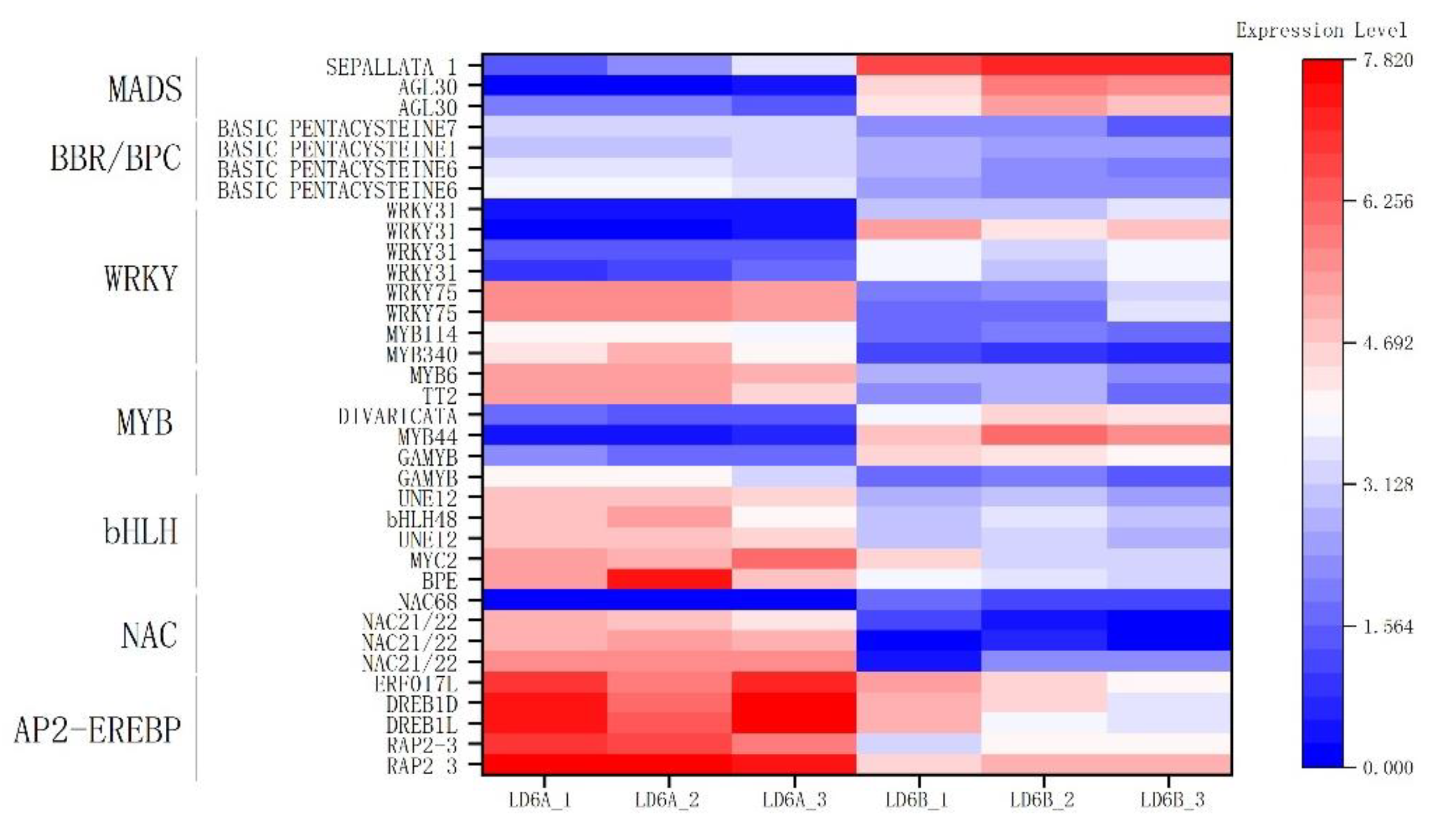
2.7.6. Transcription Factors
3. Discussion
4. Materials and Methods
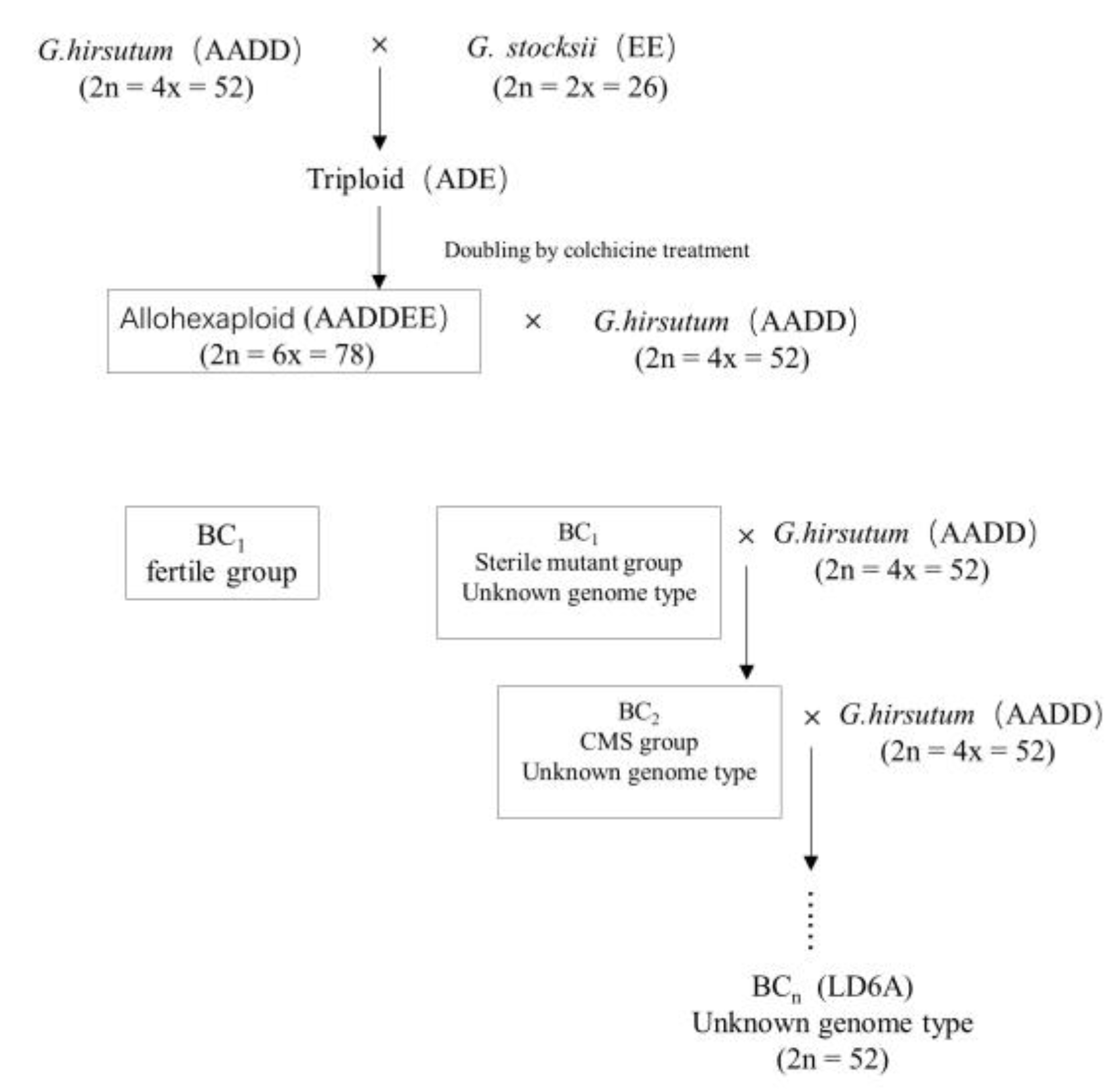
4.1. Plant Materials
4.2. RNA Extraction, cDNA Library Construction and Sequencing
4.3. De Novo Transcriptome Assembly and Gene Expression Profile
4.4. Gene Functional Annotation Analysis
4.5. Transcription Factor Analysis
4.6. Individualized Bioinformatics Analysis
4.7. Verification of Gene Expression by qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Campbell, B.T.; Saha, S.; Percy, R.; Frelichowski, J.; Jenkins, J.N.; Park, W.; Mayee, C.D.; Gotmare, V.; Dessauw, D.; Giband, M.; et al. Status of the Global Cotton Germplasm Resources. Crop Sci. 2010, 50, 1161. [Google Scholar] [CrossRef]
- Fan, S. Progresses in Research on Cotton High Yield Breeding in China. Sci. Agric. Sin. 2016, 49, 3465–3476. [Google Scholar]
- Yang, P.; Han, J.; Huang, J. Transcriptome sequencing and de novo analysis of cytoplasmic male sterility and maintenance in JA-CMS cotton. PLoS ONE 2014, 9, e112320. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Chen, P.; Khan, A.; Zhao, Y.; Chen, L.; Liu, D.; Liao, X.; Kong, X.; Zhou, R. Candidate Reference Genes Selection and Application for RT-qPCR Analysis in Kenaf with Cytoplasmic Male Sterility Background. Front. Plant Sci. 2017, 8, 1520. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wu, Y.; Zhang, M.; Zhang, J.; Stewart, J.M.; Xing, C.; Wu, J.; Jin, S. Transcriptome, cytological and biochemical analysis of cytoplasmic male sterility and maintainer line in CMS-D8 cotton. Plant Mol. Biol. 2018, 97, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Zhao, Y.; Chen, P.; Zhou, B.; Diao, Y.; Yu, M.; Huang, Z.; Zhou, R. A Comparative Analysis of the atp8 Gene between a Cytoplasmic Male Sterile Line and Its Maintainer and Further Development of a Molecular Marker Specific to Male Sterile Cytoplasm in Kenaf (Hibiscus cannabinus L.). Plant Mol. Biol. Rep. 2016, 34, 29–36. [Google Scholar] [CrossRef]
- Zheng, B.B.; Wu, X.M.; Ge, X.X.; Deng, X.X.; Grosser, J.W.; Guo, W.W. Comparative Transcript Profiling of a Male Sterile Cybrid Pummelo and Its Fertile Type Revealed Altered Gene Expression Related to Flower Development. PLoS ONE 2012, 7, 43758. [Google Scholar] [CrossRef]
- Yan, X.; Dong, C.; Yu, J.; Liu, W.; Jiang, C.; Jia, L.; Hu, Q.; Fang, X.; Wei, W. Transcriptome profile analysis of young floral buds of fertile and sterile plants from the self-pollinated offspring of the hybrid between novel restorer line NR1 and Nsa CMS line in Brassica napus. BMC Genom. 2013, 14, 26. [Google Scholar] [CrossRef]
- Liu, C.; Ma, N.; Wang, P.Y.; Fu, N.; Shen, H.L. Transcriptome Sequencing and De Novo Analysis of a Cytoplasmic Male Sterile Line and Its Near-Isogenic Restorer Line in Chili Pepper (Capsicum annuum L.). PLoS ONE 2013, 8, e65209. [Google Scholar] [CrossRef]
- Yu, Z.; Haage, K.; Streit, V.E.; Gierl, A.; Ruiz, R.A. A large number of tetraploid Arabidopsis thaliana lines, generated by a rapid strategy, reveal high stability of neo-tetraploids during consecutive generations. Theor. Appl. Genet. 2009, 118, 1107–1119. [Google Scholar] [CrossRef]
- Levings, C.S., III; Pring, D.R. Restriction Endonuclease Analysis of Mitochondrial DNA from Normal and Texas Cytoplasmic Male-Sterile Maize. Science 1976, 193, 158–160. [Google Scholar] [CrossRef]
- Dewey, R.E.; Timothy, D.H.; Levings, C.S. A mitochondrial protein associated with cytoplasmic male sterility in the T cytoplasm of maize. Proc. Natl. Acad. Sci. USA 1987, 84, 5374–5378. [Google Scholar] [CrossRef]
- Handa, H.; Gualberto, J.M.; Grienenberger, J.-M. Characterization of the mitochondrial orf B gene and its derivative, orf 224, a chimeric open reading frame specific to one mitochondrial genome of the “Polima” male-sterile cytoplasm in rape seed (Brassica napus L.). Curr. Genet. 1995, 6, 546–552. [Google Scholar] [CrossRef]
- Song, J.; Hedgcoth, C. A chimeric gene (orf256) is expressed as protein only in cytoplasmic male-sterile lines of wheat. Plant Mol. Biol. 1994, 26, 535–539. [Google Scholar] [CrossRef]
- Moneger, F.; Smart, C.J.; Leaver, C.J. Nuclear restoration of cytoplasmic male sterility in sunflower is associated with the tissue-specific regulation of a novel mitochondrial gene. EMBO 1994, 1, 8–17. [Google Scholar] [CrossRef]
- Chase, C.D. Cytoplasmic male sterility: A window to the world of plant mitochondrial—nuclear interactions. Trends. Genet. 2007, 23, 81–90. [Google Scholar] [CrossRef]
- Kong, X.; Liu, D.; Liao, X.; Zheng, J.; Diao, Y.; Liu, Y.; Zhou, R. Comparative Analysis of the Cytology and Transcriptomes of the Cytoplasmic Male Sterility Line H276A and Its Maintainer Line H276B of Cotton (Gossypium barbadense L.). Int. J. Mol. Sci. 2017, 18, 2240. [Google Scholar] [CrossRef]
- Chen, G.; Ye, X.; Zhang, S.; Zhu, S.; Yuan, L.; Hou, J.; Wang, C. Comparative Transcriptome Analysis between Fertile and CMS Flower Buds in Wucai (Brassica campestris L.). BMC Genom. 2018, 19, 908. [Google Scholar] [CrossRef]
- Mei, S.; Liu, T.; Wang, Z. Comparative Transcriptome Profile of the Cytoplasmic Male Sterile and Fertile Floral Buds of Radish (Raphanus sativus L.). Int. J. Mol. Sci. 2016, 17, 42. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, R.; Ou, H.; Gui, Y.; Wei, J.; Zhou, H.; Tan, H.; Li, Y. Comprehensive transcriptome analysis reveals genes in response to water deficit in the leaves of Saccharum narenga (Nees ex Steud.) hack. BMC Plant Biol. 2018, 18, 250. [Google Scholar] [CrossRef]
- Han, J.; Zhou, B.; Shan, W.; Yu, L.; Wu, W.; Wang, K. A and D genomes spatial separation at somatic metaphase in tetraploid cotton: Evidence for genomic disposition in a polyploid plant. Plant J. 2015, 84, 1167–1177. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S.; et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef]
- Li, F.; Fan, G.; Wang, K.; Sun, F.; Yuan, Y.; Song, G.; Li, Q.; Ma, Z.; Lu, C.; Zou, C.; et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 2014, 46, 567–572. [Google Scholar] [CrossRef]
- Wang, M.; Tu, L.; Yuan, D.; Zhu, D.; Shen, C.; Li, J.; Liu, F.; Pei, L.; Wang, P.; Zhao, G.; et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2019, 51, 224–229. [Google Scholar] [CrossRef]
- Wang, S.; Chen, J.; Zhang, W.; Hu, Y.; Chang, L.; Fang, L.; Wang, Q.; Lv, F.; Wu, H.; Si, Z.; et al. Sequence-based ultra-dense genetic and physical maps reveal structural variations of allopolyploid cotton genomes. Genome Biol. 2015, 16, 108. [Google Scholar] [CrossRef]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J.; et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef]
- Wang, M.; Wang, P.; Lin, M.; Ye, Z.; Li, G.; Tu, L.; Shen, C.; Li, J.; Yang, Q.; Zhang, X. Evolutionary dynamics of 3D genome architecture following polyploidization in cotton. Nat. Plants 2018, 4, 90–97. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.; Fang, L.; Zhang, Z.; Ma, W.; Niu, Y.; Ju, L.; Deng, J.; Zhao, T.; Lian, J.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef]
- Fu, D.; Xiao, M.; Hayward, A.; Fu, Y.; Liu, G.; Jiang, G.; Zhang, H. Utilization of crop heterosis: A review. Euphytica 2014, 197, 161–173. [Google Scholar] [CrossRef]
- Wang, Z.; Qin, G.; Zhao, T.C. Histone Deacetylase 4 (HDAC4): Mechanism of Regulations and Biological Functions. Epigenomics 2014, 6, 139–150. [Google Scholar] [CrossRef]
- Servet, C.; Conde E Silva, N.; Zhou, D. Histone Acetyltransferase AtGCN5/HAG1 Is a Versatile Regulator of Developmental and Inducible Gene Expression in Arabidopsis. Mol. Plant 2010, 3, 670–677. [Google Scholar] [CrossRef]
- Zheng, H.; Zhang, F.; Wang, S.; Su, Y.; Ji, X.; Jiang, P.; Chen, R.; Hou, S.; Ding, Y. MLK1 and MLK2 Coordinate RGA and CCA1 Activity to Regulate Hypocotyl Elongation inArabidopsis thaliana. Plant Cell 2018, 30, 67–82. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Muhammad, I.; Zhang, B.; Tang, K.; Liu, J. Molecular Identification and Characterization of Cotton Malate Dehydrogenase Gene Familly; Tsinghua University: Beijing, China, 2017. [Google Scholar]
- Mohan, A.; Muthukrishnan, S.; Hunt, M.C.; Barstow, T.J.; Houser, T.A. Kinetics of Myoglobin Redox Form Stabilization by Malate Dehydrogenase. J. Agric. Food Chem. 2010, 58, 6994–7000. [Google Scholar] [CrossRef]
- Wang, Z.A.; Li, Q.; Ge, X.Y.; Yang, C.L.; Luo, X.L.; Zhang, A.H.; Xiao, J.L.; Tian, Y.C.; Xia, G.X.; Chen, X.Y. The mitochondrial malate dehydrogenase 1 gene GhmMDH1 is involved in plant and root growth under phosphorus deficiency conditions in cotton. Sci. Rep. 2015, 5, 10343. [Google Scholar] [CrossRef]
- Kong, X.; Liu, D.; Zheng, J.; Khan, A.; Li, B.; Diao, Y.; Zhou, R. RNA editing analysis of ATP synthase genes in the cotton cytoplasmic male sterile line H276A. Biol. Res. 2019, 52, 6. [Google Scholar] [CrossRef]
- Narayanan, S.; Surolia, A.; Karande, A.A. Ribosome-inactivating protein and apoptosis: Abrin causes cell death via mitochondrial pathway in Jurkat cells. Biochem. J. 2004, 377, 233–240. [Google Scholar] [CrossRef]
- Levings, C.R. Thoughts on Cytoplasmic Male Sterility in cms-T Maize. Plant Cell 1993, 5, 1285–1290. [Google Scholar] [CrossRef]
- Huang, J.; Chen, D.; Yan, H.; Xie, F.; Yu, Y.; Zhang, L.; Sun, M.; Peng, X. Acetylglutamate kinase is required for both gametophyte function and embryo development in Arabidopsis thaliana. J. Integr. Plant Biol. 2017, 59, 642–656. [Google Scholar] [CrossRef]
- Sharoni, A.M.; Nuruzzaman, M.; Satoh, K.; Shimizu, T.; Kondoh, H.; Sasaya, T.; Choi, I.; Omura, T.; Kikuchi, S. Gene Structures, Classification and Expression Models of the AP2/EREBP Transcription Factor Family in Rice. Plant Cell Physiol. 2011, 52, 344–360. [Google Scholar] [CrossRef]
- Olsen, A.N.; Ernst, H.A.; Leggio, L.L.; Skriver, K. NAC transcription factors: Structurally distinct, functionally diverse. Trends. Plant Sci. 2005, 10, 79–87. [Google Scholar] [CrossRef]
- Zhou, Q.; Anderson, D.J. The bHLH Transcription Factors OLIG2 and OLIG1 Couple Neuronal and Glial Subtype Specification. Cell 2002, 109, 61–73. [Google Scholar] [CrossRef]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends. Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Chen, L.; Song, Y.; Li, S.; Zhang, L.; Zou, C.; Yu, D. The role of WRKY transcription factors in plant abiotic stresses. Biochim. Biophys. Acta 2012, 1819, 120–128. [Google Scholar] [CrossRef]
- Shao, S.; Li, B.; Zhang, Z.; Zhou, Y.; Jiang, J.; Li, X. Expression of a cotton MADS-box gene is regulated in anther development and in response to phytohormone signaling. J. Genet. Genom. 2010, 37, 805–816. [Google Scholar] [CrossRef]
- Prewitt, S.F.; Ayre, B.G.; McGarry, R.C. Cotton CENTRORADIALIS/TERMINAL FLOWER 1/SELF-PRUNING genes functionally diverged to differentially impact plant architecture. J. Exp. Bot. 2018, 69, 5403–5417. [Google Scholar] [CrossRef]
- Livak, K.; Schmittgen, T. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-∆∆Ct Method. Methods 2000, 25, 402–408. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | LD6B | LD6A | |
|---|---|---|---|
| Total raw reads | 73.88 Mb | 73.88 Mb | |
| Total trinity transcripts | 709,971 | ||
| N50 (transcripts) | 1541 | ||
| Total assembled bases (transcripts) | 680,840,189 | ||
| Total trinity genes | 128,901 | ||
| N50 (genes) | 2036 | ||
| Average length (genes) | 1434 | ||
| Total assembled bases (genes) | 184,861,957 | ||
| GC (%) | 40.66 |
| Type | ID | Term | Number | Rich Ratio | p Value |
|---|---|---|---|---|---|
| Biological process | GO:0045490 | pectin catabolic process | 89 | 0.028507367 | 1.52 × 10−39 |
| GO:0042545 | cell wall modification | 61 | 0.019538757 | 4.79 × 10−26 | |
| GO:0005975 | carbohydrate metabolic process | 198 | 0.063420884 | 3.23 × 10−16 | |
| GO:0071555 | cell wall organization | 108 | 0.034593209 | 3.49 × 10−10 | |
| GO:0016042 | lipid catabolic process | 57 | 0.018257527 | 3.77 × 10−9 | |
| GO:0051017 | actin filament bundle assembly | 13 | 0.004163997 | 1.60 × 10−8 | |
| Cellular component | GO:0016021 | integral component of membrane | 2742 | 0.637377964 | 2.30 × 10−29 |
| GO:0005576 | extracellular region | 144 | 0.033472803 | 3.17 × 10−23 | |
| GO:0005618 | cell wall | 92 | 0.021385402 | 5.67 × 10−10 | |
| Molecular function | GO:0004857 | enzyme inhibitor activity | 69 | 0.011551984 | 1.53 × 10−28 |
| GO:0030599 | pectinesterase activity | 61 | 0.010212623 | 5.75 × 10−26 | |
| GO:0045330 | aspartyl esterase activity | 61 | 0.010212623 | 5.75 × 10−26 | |
| GO:0004650 | polygalacturonase activity | 54 | 0.009040683 | 1.65 × 10−20 | |
| GO:0030570 | pectate lyase activity | 32 | 0.005357442 | 6.03 × 10−17 | |
| GO:0005096 | GTPase activator activity | 59 | 0.009877783 | 2.81 × 10−14 | |
| GO:0003779 | actin binding | 63 | 0.010547464 | 5.97 × 10−11 | |
| GO:0045735 | nutrient reservoir activity | 21 | 0.003515821 | 7.83 × 10−11 | |
| GO:0004575 | sucrose alpha-glucosidase activity | 14 | 0.002343881 | 1.11 × 10−9 | |
| GO:0015299 | solute: proton antiporter activity | 34 | 0.005692282 | 2.59 × 10−9 | |
| GO:0020037 | heme binding | 132 | 0.022099448 | 7.05 × 10−8 |
| Pathway | Pathway ID | Genes with Pathway Annotation | p Value | Q Value | |
|---|---|---|---|---|---|
| DEGs (2934) | All Genes (19,296) | ||||
| Biosynthesis of other secondary metabolites | |||||
| Flavone and flavonol biosynthesis | ko00944 | 8 | 28 | 5.749022 × 10−3 | 2.874511 × 10−2 |
| Stilbenoid, diarylheptanoid and gingerol biosynthesis | ko00945 | 27 | 150 | 2.693941 × 10−3 | 1.454728 × 10−2 |
| Phenylpropanoid biosynthesis | ko00940 | 209 | 1613 | 2.371287 × 10−4 | 1.684862 × 10−3 |
| Carbohydrate metabolism | |||||
| Glycolysis/gluconeogenesis | ko00010 | 119 | 902 | 2.607198 × 10−3 | 1.454728 × 10−2 |
| Inositol phosphate metabolism | ko00562 | 98 | 709 | 1.457824 × 10−3 | 8.945738 × 10−3 |
| Starch and sucrose metabolism | ko00500 | 262 | 2022 | 4.067709 × 10−5 | 3.922434 × 10−4 |
| Galactose metabolism | ko00052 | 134 | 896 | 5.676684 × 10−6 | 8.515026 × 10−5 |
| Amino sugar and nucleotide sugar metabolism | ko00520 | 230 | 1532 | 2.096337 × 10−9 | 7.075137 × 10−8 |
| Pentose and glucuronate interconversions | ko00040 | 224 | 993 | 2.589022 × 10−30 | 3.495180 × 10−28 |
| Digestive system | |||||
| Cholesterol metabolism | ko04979 | 26 | 122 | 2.363962 × 10−4 | 1.684862 × 10−3 |
| Energy metabolism | |||||
| Carbon fixation in photosynthetic organisms | ko00710 | 71 | 485 | 1.431960 × 10−3 | 8.945738 × 10−3 |
| Oxidative phosphorylation | ko00190 | 131 | 930 | 1.187092 × 10−4 | 1.001609 × 10−3 |
| Photosynthesis | ko00195 | 39 | 189 | 1.710027 × 10−5 | 2.098670 × 10−4 |
| Lipid metabolism | |||||
| Glycerophospholipid metabolism | ko00564 | 134 | 992 | 5.903167 × 10−4 | 3.984638 × 10−3 |
| Linoleic acid metabolism | ko00591 | 30 | 143 | 1.108358 × 10−4 | 9.975222 × 10−4 |
| Ether lipid metabolism | ko00565 | 61 | 351 | 3.005129 × 10−5 | 3.120711 × 10−4 |
| Arachidonic acid metabolism | ko00590 | 37 | 178 | 2.341729 × 10−5 | 2.634445 × 10−4 |
| Glycerolipid metabolism | ko00561 | 122 | 814 | 1.313738 × 10−5 | 1.773546 × 10−4 |
| Steroid biosynthesis | ko00100 | 51 | 216 | 1.062244 × 10−8 | 2.390049 × 10−7 |
| Cutin, suberine and wax biosynthesis | ko00073 | 59 | 260 | 3.884301 × 10−9 | 1.048761 × 10−7 |
| Metabolism of other amino acids | |||||
| Cyan amino acid metabolism | ko00460 | 136 | 986 | 2.254576 × 10−4 | 1.684862 × 10−3 |
| Diterpenoid biosynthesis | ko00904 | 36 | 236 | 1.022848 × 10−2 | 4.931589 × 10−2 |
| Monoterpenoid biosynthesis | ko00902 | 24 | 135 | 5.264254 × 10−3 | 2.733363 × 10−2 |
| Signal transduction | |||||
| Phosphatidylinositol signaling system | ko04070 | 104 | 770 | 2.261160 × 10−3 | 1.327203 × 10−2 |
| Transcription | |||||
| RNA polymerase | ko03020 | 166 | 795 | 3.529257 × 10−19 | 1.588166 × 10−17 |
| Transport and catabolism | |||||
| Endocytosis | ko04144 | 285 | 2152 | 3.517584 × 10−6 | 5.935923 × 10−5 |
| Phagosome | ko04145 | 111 | 697 | 2.018673 × 10−6 | 3.893155 × 10−5 |
| Gene ID | Protein Identity | Fold Change | |
|---|---|---|---|
| RNA-Seq | qRT-PCR | ||
| LOC107915747 | cytochrome P450 83B1-like | 5.16 | 4.40 |
| LOC107892026 | zinc finger protein CONSTANS-LIKE 16-like | 6.06 | 5.83 |
| LOC107903815 | methyltransferase-like protein | 5.21 | 1.5 |
| LOC107893683 | mitochondrial uncoupling protein 3-like | 5.56 | 3.13 |
| LOC107911279 | transcription factor MYB114-like | 5.04 | 5.03 |
| LOC107926337 | probable calcium-binding protein CML49 | 6.69 | 0.19 |
| LOC107905948 | probable calcium-binding protein CML13 | −10.76 | −2.74 |
| LOC107941623 | pollen allergen Che a 1-like | −12.94 | −26.59 |
| LOC107942901 | pollen-specific protein-like At4g18596 | −11.48 | −8.12 |
| LOC107915309 | plasma membrane ATPase 4-like | −10.61 | −1.30 |
| LOC107908343 | V-type proton ATPase subunit G1-like (ATP6V1G1) | −10.45 | −4.71 |
| LOC107903454 | cytochrome P450 76A2-like | −10.59 | −4.71 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, J.; Kong, X.; Li, B.; Khan, A.; Li, Z.; Liu, Y.; Kang, H.; Ullah Dawar, F.; Zhou, R. Comparative Transcriptome Analysis between a Novel Allohexaploid Cotton Progeny CMS Line LD6A and Its Maintainer Line LD6B. Int. J. Mol. Sci. 2019, 20, 6127. https://doi.org/10.3390/ijms20246127
Zheng J, Kong X, Li B, Khan A, Li Z, Liu Y, Kang H, Ullah Dawar F, Zhou R. Comparative Transcriptome Analysis between a Novel Allohexaploid Cotton Progeny CMS Line LD6A and Its Maintainer Line LD6B. International Journal of Molecular Sciences. 2019; 20(24):6127. https://doi.org/10.3390/ijms20246127
Chicago/Turabian StyleZheng, Jie, Xiangjun Kong, Bin Li, Aziz Khan, Zhiling Li, Yiding Liu, Haodong Kang, Farman Ullah Dawar, and Ruiyang Zhou. 2019. "Comparative Transcriptome Analysis between a Novel Allohexaploid Cotton Progeny CMS Line LD6A and Its Maintainer Line LD6B" International Journal of Molecular Sciences 20, no. 24: 6127. https://doi.org/10.3390/ijms20246127
APA StyleZheng, J., Kong, X., Li, B., Khan, A., Li, Z., Liu, Y., Kang, H., Ullah Dawar, F., & Zhou, R. (2019). Comparative Transcriptome Analysis between a Novel Allohexaploid Cotton Progeny CMS Line LD6A and Its Maintainer Line LD6B. International Journal of Molecular Sciences, 20(24), 6127. https://doi.org/10.3390/ijms20246127