Abstract
There are three human enzymes with HMG-CoA lyase activity that are able to synthesize ketone bodies in different subcellular compartments. The mitochondrial HMG-CoA lyase was the first to be described, and catalyzes the cleavage of 3-hydroxy-3-methylglutaryl CoA to acetoacetate and acetyl-CoA, the common final step in ketogenesis and leucine catabolism. This protein is mainly expressed in the liver and its function is metabolic, since it produces ketone bodies as energetic fuels when glucose levels are low. Another isoform is encoded by the same gene for the mitochondrial HMG-CoA lyase (HMGCL), but it is located in peroxisomes. The last HMG-CoA lyase to be described is encoded by a different gene, HMGCLL1, and is located in the cytosolic side of the endoplasmic reticulum membrane. Some activity assays and tissue distribution of this enzyme have shown the brain and lung as key tissues for studying its function. Although the roles of the peroxisomal and cytosolic HMG-CoA lyases remain unknown, recent studies highlight the role of ketone bodies in metabolic remodeling, homeostasis, and signaling, providing new insights into the molecular and cellular function of these enzymes.
1. Introduction
3-Hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) lyase (HL) catalyzes the cleavage of 3-hydroxy-3-methylglutaryl CoA into acetoacetate (AcAc) and acetyl-CoA. More than 60 years have elapsed since the discovery of this enzyme in pig heart [1]. After that, HL has been functionally and structurally well characterized in vertebrates [2], bacteria [3], and plants [4]. Both plants and vertebrates have three isoenzymes of HLs located in three different cellular compartments: mitochondria, cytosol, and peroxisomes [5,6,7,8]. Therefore, various regulation mechanisms for the synthesis of ketone bodies and/or amino acid catabolism could exist.
The mitochondrial HMG-CoA lyase (mHL) was the first human HL described and its role in mammalian metabolism has been extensively studied [9]. During ketogenesis, mHL catalyzes the transformation of HMG-CoA into acetoacetate, the first type of ketone body. Later, in the 90s, the existence of another enzyme with HMG-CoA lyase activity that could produce ketone bodies in the cell was reported. It is encoded by the same gene for mHL, HMGCL, but it is located in a different subcellular compartment, the peroxisomes [8]. There is less information available about the peroxisomal isoform of HL (pHL), and its function is still barely known. More recently, in 2004, genomic databases reported a new gene, namely, HMGCLL1 with high homology to HMGCL. This gene encoded a novel isoform of HL (er-cHL), which also had lyase activity and was capable of synthesizing acetoacetate and acetyl-CoA [6,7]. Nevertheless, its subcellular location in the cytosol and endoplasmic reticulum and its activity and tissue distribution were different from mHL and pHL, suggesting a different function for this isoform [6,7].
The existence of three enzymes with HMG-CoA lyase activity and the fact that they are able to synthesize ketone bodies in different subcellular compartments is surprising. Why does the cell need the presence of acetoacetate or β-hydroxybutyrate (BHB) in the peroxisome or the cytosol? It seems that the several functions that ketone bodies play in the cells could be related to their location in different subcellular compartments. In this review, we compare for the first time, the three human isoforms of the HMG-CoA lyase from different scopes, such as metabolic, molecular biology, phylogenetic, and clinical scopes, focusing on their different features and describing their tentative roles.
2. Ketone Body Metabolism
Ketone body metabolism, including ketogenesis and ketolysis, is considered a central metabolic process during several physiological conditions, such as fasting, caloric restriction, low carbohydrate diets, high-intensive exercises, pregnancy, or neonatal periods [10]. The major ketone bodies are β-hydroxybutyrate, acetoacetate, and acetone. They are mainly synthesized in hepatic mitochondria through the ketogenesis pathway, serving as an energy carrier for extrahepatic tissues. In addition, two novel metabolic pathways for the synthesis of ketone bodies have been described in the cytosol and peroxisomes, suggesting different regulation mechanisms of synthesis, and other tentative roles beyond energy fuel [6,11]. Moreover, some crucial molecular processes are inter-related with ketone bodies, such as β-oxidation (FAO), the tricarboxylic acid cycle (TCA), or the biosynthesis of lipids, steroids, and amino acids. Furthermore, ketone body metabolism determines the balance of cellular metabolic pairs (NADH/NAD+ and AcAc/BHB), the cellular concentration of critical signaling metabolites (acetyl-CoA), and the cellular production of BHB, thus increasingly being recognized as a cellular second messenger [10].
Acetyl-CoA, the substrate for hepatic ketogenesis, derives mainly from FAO, and to a lesser extent, from the catabolism of amino acids, especially leucine. The initial ketogenesis step consists of a reversible reaction in which two molecules of acetyl-CoA form acetoacetyl-CoA and the reaction is catalyzed by acetoacetyl-CoA thiolase (ACAT1). A third acetyl-CoA molecule is then condensed to form 3-hydroxy-3-methylglytaryl-CoA (HMG-CoA) by the mitochondrial HMG-CoA synthethase (HMGCS2). After that, HMG-CoA is transformed into AcAc and acetyl-CoA by mHL. Then, the majority of acetoacetate is reduced to BHB by the mitochondrial β-hydroxybutyrate dehydrogenase (BDH1) in an NAD+/NADH coupled reaction. In some tissues, such as the lung, AcAc could be spontaneously decarboxylate into acetone.
While acetone is not further catabolized to produce ATP, both AcAc and BHB are exported from liver mitochondria into the bloodstream and consumed by extrahepatic tissues. During ketolysis, BHB is converted back to two acetyl-CoA molecules by BDH1, 3-oxoacid CoA-transferase 1 (SCOT1), and ACAT1. These acetyl-CoAs feed into TCA to yield NADH for ATP synthesis via oxidative phosphorylation (Figure 1).
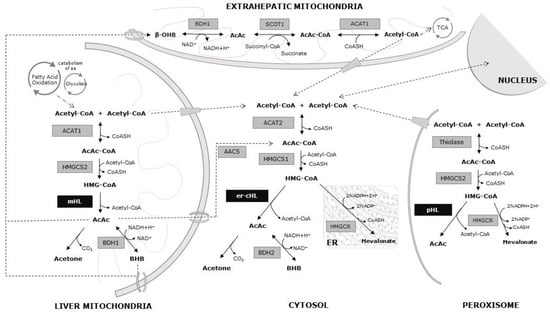
Figure 1.
Metabolic pathways of the ketone bodies. Black arrows: chemical reactions at different cellular compartments. Dotted arrows: substrate transport pathways.
Additionally, acetyl-CoA and AcAc can be exported from the mitochondrial matrix to cytosol, being the precursors of multiple anabolic reactions related to fatty acid, steroid, and amino acid synthesis. An extramitochondrial variant of HMG-CoA synthetase (HMGS1) employs one acetyl-CoA molecule to convert AcAc-CoA into HMG-CoA in the cytosol. It is well known that HMG-CoA in the cytosol is metabolized by hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR) within the mevalonate pathway. It must be stressed that er-cHL is also able to cleave HMG-CoA into acetyl-CoA and AcAc, supporting a novel pathway for AcAc and BHB production in animal tissues (Figure 1).
This metabolic pathway is also completed in the peroxisomes of cells [8]. The acetyl-CoA from the cytosol can get through the peroxisomal membrane, where the thiolase condenses it to form acetoacetyl-CoA (Figure 1).
These metabolic pathways suggest a finely tuned regulation of ketone bodies synthesis. Two mechanisms are particularly relevant in this regulation, the substrate availability and the expression and activity of the enzyme HMGCS2 [12]. Although significant progress has been made in deciphering how ketogenic rates are regulated, researchers tend to focus on HMGCS2, which is classically considered to be the rate-limiting enzyme of ketogenesis. Therefore, more information is not available about the direct regulation of HMG-CoA lyase enzymes affecting the final pool of ketone bodies. An alternative splicing mechanism has been proposed for the regulation of the active forms of HMGCL and HMGCS2 genes in a tissue-specific manner [13]. Alternative inactive splicing isoforms from HMGCL and HMGCS2 genes are expressed at high levels in tissues that do not produce ketone bodies, such as heart, skeletal muscle, and brain, whereas the expression of the complete active isoform is low in these tissues. On the other hand, the expression level pattern of these isoforms is the opposite in tissues that produce ketone bodies at high rates [13]. Moreover, recent studies show that mitochondrial and peroxisomal HLs may inactivate cytosolic HL due to specific physiological requirements in Arabidopsis thaliana [4].
3. Roles of Ketone Bodies: More Than Energy Fuel
It is widely known that the main role of ketone bodies is bioenergetics. In normal circumstances, the energy yield from mitochondrial oxidation of AcAc or BHB is around 22 ATP molecules. This metabolic pathway is particularly relevant for some tissues, such as brain, where ketolysis accounts for between 60% and 70% of the energy supply during glucose deprivation [14]. The use of ketone bodies as an alternative energy fuel by the brain is an example of the importance of these metabolites, because the brain is able to use glucose and ketone bodies only to obtain energy [14]. Moreover, ketone bodies are used as a substrate for the synthesis of lipids in the brain [15], so their role as lipid precursors is imperative in the growth and development of brain [16].
Emerging studies have recently shown the potential importance of ketone bodies not only in cell metabolism, but also in metabolic remodeling, homeostasis, and signaling [10].
3.1. Metabolic Remodeling
Apart from physiological conditions, ketone bodies are essential elements in cellular metabolic remodeling during diseases and disorders. This is true for heart, where several studies show that ketone utilization is increased in both animal models and human failing hearts [17,18], although there are still discrepancies as to whether this increase in BHB oxidation improves either cardiac work or efficiency in failing hearts [19,20]. In addition, utilization of ketones has been shown to be upregulated in diabetes mellitus [21] and an increase in AcAC utilization has been recently observed in the diabetic rat heart [21]. In brain, ketone bodies can act as neuroprotective molecules. In fact, it is widely believed that the antioxidant effects of BHB and AcAc play a vital role in the prevention of neurodegeneration. The capability of BHB to reduce the production of reactive oxygen species (ROS) and prevent neuronal death was demonstrated in several models [22,23], but its mode of action is unclear. Several hypotheses point that ketone bodies can activate glutathione peroxidase and decrease ROS production [24]. Others have shown that ketone bodies reduce the production of ROS by improving mitochondrial respiration and bypassing the complex I dysfunction [25]. The ketogenic diet has been studied as a treatment in patients with complex I deficiency, and although there are a few published studies, the results show a clinical improvement of the patients [26,27]. Besides that, it has long been known that BHB is able to activate the hydroxycarboxylic acid receptor 2 (HCA2) on adipocytes in order to reduce lipolysis [28]. Interestingly, HCA2 expression is not confined to adipocytes, but is also present on neutrophils, macrophages, and brain. Therefore, BHB is able to activate a neuroprotective subset of macrophages via HCA2 receptors [29]. In any case, based on these protective effects, the ketogenic diet is used in the treatment of several diseases, such as Parkinson’s disease and Alzheimer’s disease [30].
3.2. Physiological Homeostasis
There is clear evidence that ketone bodies play a major role in the maintenance of physiological homeostasis and it is believed that they alter pathways in the aging process [31]. Although the mechanism underlying the anti-aging effects of ketone bodies has not been fully elucidated, recent studies provided strong evidences supporting their anti-inflammatory [32] and anti-oxidative effects [33,34]. Thus, prevention of senescence by BHB has been demonstrated in vascular cells [35]. Ketone bodies can modulate inflammatory and immune cell function. Mechanistically, BHB is able to inhibit NLRP3 (NACHT, LRR and PYD domains-containing protein 3) inflammasomes [32]. Furthermore, a ketone body shuttle between hepatocytes and local macrophages has been demonstrated. It should be pointed out that AcAc and BHB have segregated roles, and that AcAc is the one that protects the liver against fibrosis [36]. Among the most recent discoveries, the stemness regulation in intestinal stem cells by the production of endogenous ketone bodies is remarkable [37].
3.3. Signaling
In the last years, several studies have been published supporting the idea that the ketone bodies, and more specifically, the BHB, are signaling metabolites. In this sense, BHB could modify proteins at a post-transcriptional level, in a reaction named β-hydroxybutyrylation (kbhb). This was initially described in histone proteins, and, since then, a total of 44 histone kbhb sites have been identified in both human and mouse cells, suggesting that a variety of biological events could be regulated by ketone bodies [38]. Besides histone post-transcriptional modifications, a BHB-mediated kbhb has been described in a transcriptional factor, p53, as a novel mechanism by which ketone bodies might be involved in gene expression regulation [39]. Furthermore, since ketone bodies contribute to compartmentalized pools of acetyl-CoA, they could be involved in histone lysine acetylation dynamics [40]. Additionally, ketone bodies could act as signaling metabolites by inhibiting the activity of class I histone deacetylases (HDACs) [33]. All these findings suggest that BHB could be a key transcription and epigenetic regulator.
All these data suggest that ketone bodies and their metabolism are more important to cells than it was originally thought, and this fact could explain the existence of more than one HMG-CoA lyase enzyme in different subcellular compartments.
4. Gene Structure and Protein Features of HMG-CoA Lyases
The mHL, pHL, and er-cHL lyases show properties of isoenzymes encoded by two distinct functional genes. Whereas mitochondrial and peroxisomal enzymes are encoded by the HMGCL gene (NT_004610.17), located at chromosome 1p36.1–p35, er-cHL is encoded by the HMGCLL1 gene (NT_007592.15) at chromosome 6p12.1. These genes show a high degree of sequence homology and similar exon/intron structure (Figure 2A). While both the genes have an open reading frame of about 1000 nt in length, the gene sizes are extremely different: HMGCL consists of 24 kb and HMGCLL1 consists of 145 kb. This variance in intron length could indicate differences in gene expression regulation [41,42]. It has been suggested that intron-poor structures are commonly found in genes involved in rapid biological responses [41]. In contrast, intron size is highly conserved in genes associated with embryonic development, since transcriptional delay is functionally significant [42].
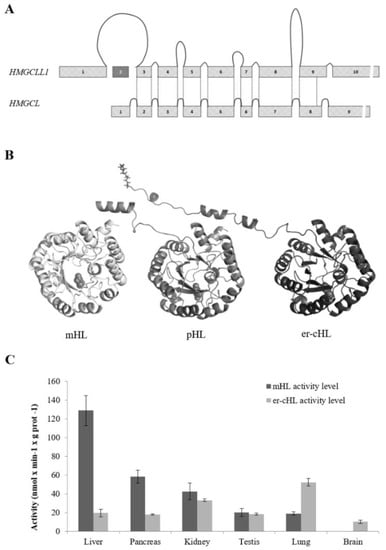
Figure 2.
Gene and protein structure, and enzymatic activity comparison of mHL, pHL, and er-cHL isoenzymes. (A) HMGCLL1 and HMGCL gene structures. The size of the exons and introns of both genes is represented in scale. The lines represent the regions that encode the equivalent protein structure in both genes. (B) 3D structure model for human mHL, pHL, and er-cHL. The three proteins share a TIM-barrel structure. The mHL protein cleaves its mitochondrial leader peptide, which is preserved in the pHL protein. The myristic acid is represented in the er-cHL protein structure at its amino-terminal end. (C) Comparison of activity levels of er-cHL and mHL proteins in different adult human tissues. This research was originally published in the Journal of Lipid Research. Arnedo M. et al. Characterization of a Novel HMG-CoA Lyase Enzyme with a Dual Location in Endoplasmic Reticulum and Cytosol. J. Lipid Res. 53 (10), 2046–2056 [6].
At least two physiological splicing variants have been reported for the HMGCL gene [13]—one bearing a deletion of exons 5 and 6 (HMGCLΔ5,6) and the other with a deletion of exons 5, 6, and 7 (HMGCLΔ5,6,7)—both lacking enzymatic activity. On the other hand, six splicing isoforms have been described in databases and experimentally by Arnedo et al. (2012) for the HMGCLL1 gene, although their possible function, in the tissues where they were found, remains unstudied.
Nevertheless, the main HMGCLL1 isoform (NM_001042406.1) encodes a protein with a structure very similar to mHL and pHL (Figure 2B). All of them have a TIM-barrel structure with an auxiliary domain composed of a β-sheet and two α-helices [2,43]. A homodimer structure was confirmed in the three isoenzymes, even in pHL [44], which had been previously described as a monomer [11]. The interaction between the monomers is located at the β9 sheet and the α11 and α12 helices, which are not components of the barrel.
The deduced amino acid sequences of mHL and er-cHL are 83% identical. The main feature of HL isoenzymes is the HMGL-like domain responsible for their lyase activity, which is located between amino acids 33 and 306 in mHL and 78 and 345 in er-cHL. The most critical amino acids for the catalytic activity of the enzymes (Arg41, Glu72, Tyr167, and Cys266 in mHL and Arg56, Glu87, Tyr182, and Cys281 in er-cHL) as well as the residues that bind to the divalent cation Mg+2, which is essential for the lyase activity (Asp42, Asn275, His233, and His235 in mHL and Asp57, Asn288, His248, and His250 in er-cHL), are well known. Furthermore, their location has been the same in the mHL secondary structure elements along evolution [45].
The three isoenzymes of HL can be distinguished by their N-terminal sequence. The mHL enzyme contains a mitochondrial leader peptide that extends from the first to the 27th amino acid. In the pHL enzyme, the C-terminal tripeptide (CKL) leads the protein to peroxisomes. The er-cHL protein has an N-myristoylation motif (MGNVPSA) at its N-terminal sequence that is likely related to its association with the cellular membranes [7]. Besides that, subcellular localization of HLs has been experimentally demonstrated. Several assays of subcellular activity [5] and fluorescence co-localization studies [6] confirmed the mitochondrial subcellular location for mHL. Peroxisomal localization of pHL was established by subcellular fractionation and enzymatic activity measures in samples obtained from mouse liver [8]. Whereas er-cHL localization was confirmed in the cytosolic side of the endoplasmic reticulum membrane, the interaction between the protein and the membrane is weak and the enzyme can be found dissociated as well [6].
Additionally, the three proteins were expressed in a bacterial model in order to study their lyase enzymatic activity. Although there were variations among the different assays, lyase enzymatic activity was confirmed in mHL, pHL, and er-cHL [6,7,45,46,47,48,49].
Finally, an HL enzyme has been described in Arabidopsis thaliana, where the At2g26800 gene is able to encode three different isoforms of the HMG-CoA lyase [4]. The alignment of the diverse isoforms with human mHL showed a high similarity, where all the key amino acids for coordination with Mg+2 or interaction with the CoA ligand are conserved. The structural model of the different HLs of A. thaliana also revealed a great homology with the human lyases [4].
5. Phylogenetic Evolution of the HMG-CoA Lyases
The strong structural similarity of mHL and er-cHL suggests that the genes encoding them have a common origin. By using MUSCLE [50] to perform a multiple sequence alignment and refining the alignment using GBLOCKS [51], a maximum likelihood phylogenetic tree can be built [52,53] which helps us to understand the phylogenetic evolution of these enzymes.
As commented above, the HLs have a TIM-barrel structure, which is common in enzymes that catalyze metabolic reactions in the body. In fact, they belong phylogenetically to one of the oldest protein families: the beta/alpha barrel scaffold. Initially, this family of proteins was arranged in the form of a hemibarrel. The fusion of two structurally equivalent genes would result in the appearance of the complete barrel. From here, the next evolutionary steps would take place by gene duplication. Each time this occurred, two new enzymes would emerge, which over time would specialize in different functions [54].
Following this hypothesis, it was plausible to speculate that HMGCL and HMGCLL1 appeared by gene duplication. The phylogenetic comparison showed that the oldest gene was HMGCL (Figure 3). Enzymes of analogous structures have been found in bacteria, specifically Alphaproteobacteria, such as Rhodobacter, Rhizobium, Sphingomonas, Rhodospirillum, and Caulobacter. It is likely that the symbiont bacteria of eukaryotes carried the HMGCL gene. The absence of this gene in mitochondrial DNA would be explained by the usual migration of this genetic material to the nuclear DNA of eukaryotic cells.
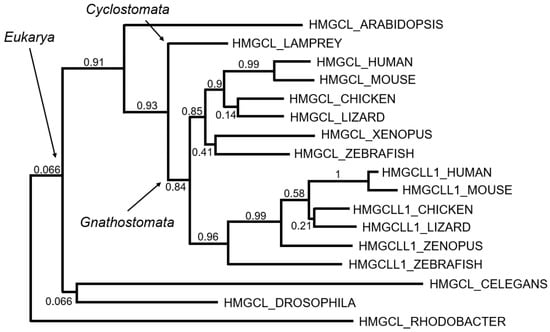
Figure 3.
Tentative evolution of the HMG-CoA lyases. Maximum likelihood phylogenetic tree based on multiple sequence alignment is shown in Figure S1. Bootstrap values (0, lowest to 1, highest) are indicated as measurements of branch confidence scores.
Assuming the idea that the first enzyme was the mitochondrial lyase, we could wonder when the gene duplication that led to the appearance of the HMGCLL1 gene occurred. Sequence homology suggests that HMGCLL1 appeared for the first time in vertebrates with mandibles, Gnathostomata, about 420 million years ago (Figure 3). In this period, there was a division between Agnatha (lamprey and related) and Gnathostomata by complete genomic duplication [55,56]. Humans come from this last branch and it is believed that this was the origin of many of the genes that appeared duplicated in the human genome.
Moreover, a convergent-like evolution has been recently described in plants. The HL isoforms have been found at the same localizations (mitochondria, peroxisome, and cytosol) as higher organisms. The subcellular location in these three cell compartments can be explained for the alternative splicing of the HMGCL gene, as it has been described in the case of Arabidopsis thaliana [4].
6. Tissue-Specific Expression of HMG-CoA Lyases
Although mHL function has been deeply studied, relatively very little is known about er-cHL and pHL. The different tissue distribution of the three human lyase enzymes could provide insights into their specific function.
The widespread adoption of RNA-seq and the completion of large projects, such as ENCODE [57] and GTEx [58], give us a valuable opportunity to study the tissue-specific expression of HMGCL and HMGCLL1. As it would be expected, the liver is the tissue that shows the highest mRNA expression level of HMGCL, since it is the main ketogenic tissue. Interestingly, HMGCLL1 is preferentially expressed in brain and lung tissues [59]. Furthermore, the analysis of HMGCLL1 expression using the web-based application http://evodevoapp.kaessmannlab.org [58] reinforced these data and suggested an important role of HMGCLL1 during fetal brain development.
Besides public datasets, tissue-specific distribution of mHL and er-cHL has been confirmed not only at the mRNA level, but at the protein and enzymatic activity levels as well. Thus, the liver is the tissue that shows the highest mHL activity, as well as the highest protein and mRNA expression levels [5]. A high activity level was also found in pancreas and kidney. Interestingly, mHL activity was not detected in the brain mitochondrial fraction, but a significant activity of er-cHL was reported in the brain, despite the difficulties in studying the human brain immediately after death due to the fast degradation of proteins. The highest er-cHL activity was found in lung tissues [6] (Figure 2C). Overall, gene expression data found in public databases were experimentally confirmed. Unfortunately, there is very less information about the tissue distribution of the pHL isoform and its function in the peroxisomes is yet to be extensively studied.
Together, these observations underscore the importance of tissue-specific expression of mHL and er-cHL, possibly reflecting a response to different signals as well as different roles in the synthesis of ketone bodies (see Section 3, above).
7. HMG-CoA Lyase Related Diseases
To date, only the deficiency of the mHL protein has been reported to cause a genetic disease, 3-hydroxy-3-methylglutaric aciduria (OMIM 246450) [60]. The absence of this enzyme causes ketone body production failure and the accumulation of toxic acid metabolites of leucine catabolism that may disrupt normal organ function. For this reason, it is included within alterations of fatty acid metabolism and within organic acidurias.
Since the description of the deficiency in 1976, more than 150 patients have been reported in the literature [47,61]. It happens in all ethnic groups throughout the world, but with a greater incidence in countries of the Mediterranean area, Spain and Portugal [47] and in Saudi Arabia [62]. It is considered a rare disease of autosomal recessive nature with a very low incidence (<1/100,000 among live newborns). Despite its low frequency, it is recommended that hospitals with neonate units should have the capacity to diagnose it early, because its treatment is effective and simple [63].
Although there is a considerable clinical heterogeneity, in most cases, the disease appears in the first year of life, and in about 50% of patients, the disease appears in the neonatal period, which highlights the importance of ketone bodies in the brain metabolism of newborns. In rare cases, it can also be found in adolescents or adults [64,65,66].
The onset of symptoms is initiated by fasting, infection, dietary protein load, or the stress of birth. Affected patients commonly present acute clinical manifestations, such as metabolic acidosis and hypoketotic hypoglycemia, accompanied by vomiting, dehydration, hypotonia, and lethargy. Other chronic manifestations of the disease are less frequent and include hepatomegaly, macrocephaly, and less frequently microcephaly, seizures, dilated cardiomyopathy, arrhythmia, and acute pancreatitis. As already discussed, the finding of higher enzymatic activity in pancreas [5] justifies that this organ may be injured by the accumulation of toxic metabolites.
Abnormalities in cerebral white matter and basal ganglia have also been reported, although the pathogenesis of brain damage in this disease is poorly known. As in pancreas, it has been proposed that the acid compounds accumulating in 3-hydroxy-3-methylglutaric aciduria induce a pro-inflammatory response in astrocytes that may possibly be involved in the neuropathology of this disease [67].
This deficiency is a life-threatening condition and may cause irreversible coma and/or death; however, prognosis is good for the patients and the symptoms often improve around puberty. While the overall physical development of the patients appeared normal, the cognitive outcome was highly variable [61], perhaps due to vulnerability and affectation of the brain. Besides, pregnancy is considered especially stressful due to constant changes in fetal growth and subsequent metabolic demand. Several cases of metabolic acidosis and hypoglycemia during pregnancy, delivery, and postpartum period have been described that can be fatal if not treated appropriately [68].
Patients with HMG-CoA lyase deficiency show a diagnostic urinary organic acid pattern, with high levels of 3-hydroxy-3-methylglutaric acid, 3-hydroxy-isovaleric acid, 3-methylglutaric acid, and 3-methylglutaconic acid. A confirmation diagnosis requires direct enzyme activity testing or a genetic study of mutations in the gene HMGCL. So far, several mutations have been reported in this gene to cause a decrease in the HMG-CoA lyase activity [61,69,70,71,72,73]. At present, despite the considerable progress made in the genetic diagnosis of the deficiency, thanks to the use of next-generation sequencing gene panels, genotype–phenotype correlations are difficult to establish.
Although no disease has been reported to be related to pHL or er-cHL deficiency, a decrease in HMGCLL1 gene expression level has been found in 18 cancer types [74]. Moreover, using the gene expression profiling interactive analysis (GEPIA) [75] that interrogated the TCGA pan-cancer and GTEx data repositories, a significant decrease in HMGLL1 expression was observed in many cancer types, such as glioblastoma, lung adenocarcinoma, and testicular tumors. In contrast, HMGCL expression appears to be upregulated in several cancer tissues compared to corresponding normal tissues. This opposite behavior is highly surprising and further studies are required to characterize the roles of HMGCL and HMGCLL1 expression in cancer cells. Connections between ketone bodies and cancer are under active investigation, and beyond the realm of fuel metabolism, the latest studies highlight the imperative roles of ketone bodies in tumor cell signaling [39,76,77].
Recently, a study reported that a specific HMGCLL1 haplotype acts as a genetic biomarker for intrinsic sensitivity to tyrosine kinase inhibitor therapy in chronic myeloid leukemia (CML) patients [78]. The study suggested that inhibition of the HMGCLL1 gene could be a potential therapeutic strategy to improve the response to imatinib therapy in CML patients [78].
Apart from its possible implication in cancer, a mutation in the HMGLL1 gene has been reported in a patient with hypopituitarism [79], although its relevance in this disease has not been deeply studied given the fact that the patient also carried another mutation in the HESX1 gene, which has been thoroughly studied in patients with hypopituitarism.
8. Conclusions
For many years, ketone body metabolism has been mainly associated with energy fuel and lipid metabolism. During the last 10 years, an increasing burst of studies on the functions of ketone bodies have been published. These studies provide insights into the key role of ketone bodies in metabolic remodeling, signaling, and homeostasis. Therefore, it has been proposed that ketone body synthesis pathways in cytosol and peroxisomes are crucial issues with attractive conceptual potential that requires a more thorough research. In this context, HMG-CoA lyase isoenzymes could be exciting tools to clarify the potential importance of ketone body synthesis in cytosol or peroxisomes, and in poorly explored tissues, such as the brain or lung. Addressing these questions promises to offer an exciting task for the years to come.
Supplementary Materials
The following are available online at https://www.mdpi.com/1422-0067/20/24/6124/s1, Figure S1: Multiple sequence alignment of representative sequences from the HMGCL (mHL) and HMGCLL1 (er-cHL) family of proteins. Sequences were obtained from Uniprot (www.uniprot.org) and correspond to organisms: Human, Mouse, Chicken, Anole lizard, Xenopus, Zebra fish, Sea lamprey, Drosophila, C. elegans, Arabidopsis and Rhodobacter.
Author Contributions
M.A. and A.L.-P.: manuscript writing, collection, and assembly of data; C.L.-C., M.G.-S., and R.A.-P.: literature review, collection, and assembly of data; P.G.-P.: computational simulation. G.B.-L., B.P., and J.P.: conception and design, manuscript editing, and approval of the manuscript.
Funding
This work was supported by: Diputación General de Aragón-FEDER: European Social Fund (Grupo de Referencia B32_17R, to Juan Pié.), as well as grants from the Ministerio de Ciencia, Innovación y Universidades/Agencia Estatal de Investigación RTC-2017-6494-1 and RTI2018-094434-B-I00 (MCIU/AEI/FEDER, UE) to Paulino Gómez-Puertas. Beatriz Puisac and Juan Pié are members of CIBERER-GCV02 and María Arnedo, Ana Latorre-Pellicer, Cristina Lucia-Campos, Marta Gil-Salvador, Rebeca Antoñanzas-Peréz, Beatriz Puisac and Juan Pié are members of ISS-Aragon at the School of Medicine, University of Zaragoza.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| HMG-CoA | 3-Hydroxy-3-methylglutaryl coenzyme A |
| HL | HMG-CoA lyase |
| mHL | Mitochondrial HMG-CoA lyase |
| pHL | Peroxisomal HMG-CoA lyase |
| er-cHL | HMG-CoA lyase-like 1 |
| BHB | β-Hydroxybutyrate |
| AcAc | Acetoacetate |
| FAO | β-Oxidation |
| TCA | Tricarboxylic acid cycle |
| ACAT1 | Acetoacetyl-CoA thiolase |
| HMGCS1 | HMG-CoA synthetase 1 |
| HMGCS2 | HMG-CoA synthetase 2 |
| SCOT1 | 3-Oxoacid-CoA 1 |
| BDH1 | β-Hydroxybutyrate dehydrogenase |
| HMGCR | HMG-CoA reductase |
| ROS | Reactive oxygen species |
| HCA2 | Hydroxycarboxylic acid receptor 2 |
| Kbhb | β-Hydroxybutyrylation |
| HDAC | Class I histone deacetylase |
| CML | Chronic myeloid leukemia |
References
- Bachhawat, B.K.; Robinson, W.G.; Coon, M.J. The enzymatic cleavage of beta-hydroxy-beta-methylglutaryl coenzyme A to acetoacetate and acetyl coenzyme A. J. Biol. Chem. 1955, 216, 727–736. [Google Scholar] [PubMed]
- Fu, Z.; Runquist, J.A.; Forouhar, F.; Hussain, M.; Hunt, J.F.; Miziorko, H.M.; Kim, J.J. Crystal structure of human 3-hydroxy-3-methylglutaryl-CoA Lyase: Insights into catalysis and the molecular basis for hydroxymethylglutaric aciduria. J. Biol. Chem. 2006, 281, 7526–7532. [Google Scholar] [CrossRef] [PubMed]
- Forouhar, F.; Hussain, M.; Farid, R.; Benach, J.; Abashidze, M.; Edstrom, W.C.; Vorobiev, S.M.; Xiao, R.; Acton, T.B.; Fu, Z.; et al. Crystal structures of two bacterial 3-hydroxy-3-methylglutaryl-CoA lyases suggest a common catalytic mechanism among a family of TIM barrel metalloenzymes cleaving carbon-carbon bonds. J. Biol. Chem. 2006, 281, 7533–7545. [Google Scholar] [CrossRef] [PubMed]
- Hemmerlin, A.; Huchelmann, A.; Tritsch, D.; Schaller, H.; Bach, T.J. The specific molecular architecture of plant 3-hydroxy-3-methylglutaryl-CoA lyase. J. Biol. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Puisac, B.; Arnedo, M.; Casale, C.H.; Ribate, M.P.; Castiella, T.; Ramos, F.J.; Ribes, A.; Perez-Cerda, C.; Casals, N.; Hegardt, F.G.; et al. Differential HMG-CoA lyase expression in human tissues provides clues about 3-hydroxy-3-methylglutaric aciduria. J. Inherit. Metab. Dis. 2010, 33, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Arnedo, M.; Menao, S.; Puisac, B.; Teresa-Rodrigo, M.E.; Gil-Rodriguez, M.C.; Lopez-Vinas, E.; Gomez-Puertas, P.; Casals, N.; Casale, C.H.; Hegardt, F.G.; et al. Characterization of a novel HMG-CoA lyase enzyme with a dual location in endoplasmic reticulum and cytosol. J. Lipid Res. 2012, 53, 2046–2056. [Google Scholar] [CrossRef]
- Montgomery, C.; Pei, Z.; Watkins, P.A.; Miziorko, H.M. Identification and characterization of an extramitochondrial human 3-hydroxy-3-methylglutaryl-CoA lyase. J. Biol. Chem. 2012, 287, 33227–33236. [Google Scholar] [CrossRef]
- Ashmarina, L.I.; Rusnak, N.; Miziorko, H.M.; Mitchell, G.A. 3-Hydroxy-3-methylglutaryl-CoA lyase is present in mouse and human liver peroxisomes. J. Biol. Chem. 1994, 269, 31929–31932. [Google Scholar]
- Robinson, A.M.; Williamson, D.H. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Ashmarina, L.I.; Robert, M.F.; Elsliger, M.A.; Mitchell, G.A. Characterization of the hydroxymethylglutaryl-CoA lyase precursor, a protein targeted to peroxisomes and mitochondria. Biochem. J. 1996, 315 (Pt 1), 71–75. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. beta-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef] [PubMed]
- Puisac, B.; Ramos, M.; Arnedo, M.; Menao, S.; Gil-Rodriguez, M.C.; Teresa-Rodrigo, M.E.; Pie, A.; de Karam, J.C.; Wesselink, J.J.; Gimenez, I.; et al. Characterization of splice variants of the genes encoding human mitochondrial HMG-CoA lyase and HMG-CoA synthase, the main enzymes of the ketogenesis pathway. Mol. Biol. Rep. 2012, 39, 4777–4785. [Google Scholar] [CrossRef] [PubMed]
- Cahill, G.F., Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Nehlig, A. Brain uptake and metabolism of ketone bodies in animal models. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 265–275. [Google Scholar] [CrossRef]
- Morris, A.A. Cerebral ketone body metabolism. J. Inherit. Metab. Dis. 2005, 28, 109–121. [Google Scholar] [CrossRef]
- Bedi, K.C., Jr.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B.; et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 2016, 133, 706–716. [Google Scholar] [CrossRef]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Kruger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016, 133, 698–705. [Google Scholar] [CrossRef]
- Horton, J.L.; Davidson, M.T.; Kurishima, C.; Vega, R.B.; Powers, J.C.; Matsuura, T.R.; Petucci, C.; Lewandowski, E.D.; Crawford, P.A.; Muoio, D.M.; et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. Jci Insight 2019, 4. [Google Scholar] [CrossRef]
- Ho, K.L.; Zhang, L.; Wagg, C.; Al Batran, R.; Gopal, K.; Levasseur, J.; Leone, T.; Dyck, J.R.B.; Ussher, J.R.; Muoio, D.M.; et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc. Res. 2019, 115, 1606–1616. [Google Scholar] [CrossRef]
- Abdurrachim, D.; Woo, C.C.; Teo, X.Q.; Chan, W.X.; Radda, G.K.; Lee, P.T.H. A new hyperpolarized (13)C ketone body probe reveals an increase in acetoacetate utilization in the diabetic rat heart. Sci. Rep. 2019, 9, 5532. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.; Canfield, J.; Copes, N.; Rehan, M.; Lipps, D.; Bradshaw, P.C. D-beta-hydroxybutyrate extends lifespan in C. elegans. Aging 2014, 6, 621–644. [Google Scholar] [CrossRef] [PubMed]
- Julio-Amilpas, A.; Montiel, T.; Soto-Tinoco, E.; Geronimo-Olvera, C.; Massieu, L. Protection of hypoglycemia-induced neuronal death by beta-hydroxybutyrate involves the preservation of energy levels and decreased production of reactive oxygen species. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2015, 35, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.R.; Ribeiro, L.C.; Hagenn, M.; Siqueira, I.R.; Araujo, E.; Torres, I.L.; Gottfried, C.; Netto, C.A.; Goncalves, C.A. Ketogenic diet increases glutathione peroxidase activity in rat hippocampus. Neurochem. Res. 2003, 28, 1793–1797. [Google Scholar] [CrossRef]
- Achanta, L.B.; Rae, C.D. beta-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef]
- Steriade, C.; Andrade, D.M.; Faghfoury, H.; Tarnopolsky, M.A.; Tai, P. Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) may respond to adjunctive ketogenic diet. Pediatr. Neurol. 2014, 50, 498–502. [Google Scholar] [CrossRef]
- Frey, S.; Geffroy, G.; Desquiret-Dumas, V.; Gueguen, N.; Bris, C.; Belal, S.; Amati-Bonneau, P.; Chevrollier, A.; Barth, M.; Henrion, D.; et al. The addition of ketone bodies alleviates mitochondrial dysfunction by restoring complex I assembly in a MELAS cellular model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 284–291. [Google Scholar] [CrossRef]
- Ahmed, K.; Tunaru, S.; Offermanns, S. GPR109A, GPR109B and GPR81, a family of hydroxy-carboxylic acid receptors. Trends Pharmacol. Sci. 2009, 30, 557–562. [Google Scholar] [CrossRef]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Muller-Fielitz, H.; Pokorna, B.; Vollbrandt, T.; Stolting, I.; Nadrowitz, R.; et al. The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef]
- Yang, H.; Shan, W.; Zhu, F.; Wu, J.; Wang, Q. Ketone Bodies in Neurological Diseases: Focus on Neuroprotection and Underlying Mechanisms. Front. Neurol. 2019, 10, 585. [Google Scholar] [CrossRef]
- Roberts, M.N.; Wallace, M.A.; Tomilov, A.A.; Zhou, Z.; Marcotte, G.R.; Tran, D.; Perez, G.; Gutierrez-Casado, E.; Koike, S.; Knotts, T.A.; et al. A Ketogenic Diet Extends Longevity and Healthspan in Adult Mice. Cell Metab. 2017, 26, 539–546. [Google Scholar] [CrossRef]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Board, M.; Lopez, C.; van den Bos, C.; Callaghan, R.; Clarke, K.; Carr, C. Acetoacetate is a more efficient energy-yielding substrate for human mesenchymal stem cells than glucose and generates fewer reactive oxygen species. Int. J. Biochem. Cell Biol. 2017, 88, 75–83. [Google Scholar] [CrossRef]
- Han, Y.M.; Bedarida, T.; Ding, Y.; Somba, B.K.; Lu, Q.; Wang, Q.; Song, P.; Zou, M.H. beta-Hydroxybutyrate Prevents Vascular Senescence through hnRNP A1-Mediated Upregulation of Oct4. Mol. Cell 2018, 71, 1064–1078 e1065. [Google Scholar] [CrossRef]
- Puchalska, P.; Martin, S.E.; Huang, X.; Lengfeld, J.E.; Daniel, B.; Graham, M.J.; Han, X.; Nagy, L.; Patti, G.J.; Crawford, P.A. Hepatocyte-Macrophage Acetoacetate Shuttle Protects against Tissue Fibrosis. Cell Metab. 2019, 29, 383–398. [Google Scholar] [CrossRef]
- Cheng, C.W.; Biton, M.; Haber, A.L.; Gunduz, N.; Eng, G.; Gaynor, L.T.; Tripathi, S.; Calibasi-Kocal, G.; Rickelt, S.; Butty, V.L.; et al. Ketone Body Signaling Mediates Intestinal Stem Cell Homeostasis and Adaptation to Diet. Cell 2019, 178, 1115–1131. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine beta-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef]
- Liu, K.; Li, F.; Sun, Q.; Lin, N.; Han, H.; You, K.; Tian, F.; Mao, Z.; Li, T.; Tong, T.; et al. p53 beta-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 2019, 10, 243. [Google Scholar] [CrossRef]
- Ruan, H.B.; Crawford, P.A. Ketone bodies as epigenetic modifiers. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 260–266. [Google Scholar] [CrossRef]
- Jeffares, D.C.; Penkett, C.J.; Bahler, J. Rapidly regulated genes are intron poor. Trends Genet. 2008, 24, 375–378. [Google Scholar] [CrossRef]
- Seoighe, C.; Korir, P.K. Evidence for intron length conservation in a set of mammalian genes associated with embryonic development. BMC Bioinform. 2011, 12, S16. [Google Scholar] [CrossRef]
- Casals, N.; Gomez-Puertas, P.; Pie, J.; Mir, C.; Roca, R.; Puisac, B.; Aledo, R.; Clotet, J.; Menao, S.; Serra, D.; et al. Structural (betaalpha)8 TIM barrel model of 3-hydroxy-3-methylglutaryl-coenzyme A lyase. J. Biol. Chem. 2003, 278, 29016–29023. [Google Scholar] [CrossRef]
- Tuinstra, R.L.; Burgner, J.W., II; Miziorko, H.M. Investigation of the oligomeric status of the peroxisomal isoform of human 3-hydroxy-3-methylglutaryl-CoA lyase. Arch. Biochem. Biophys. 2002, 408, 286–294. [Google Scholar] [CrossRef]
- Tuinstra, R.L.; Miziorko, H.M. Investigation of conserved acidic residues in 3-hydroxy-3-methylglutaryl-CoA lyase: Implications for human disease and for functional roles in a family of related proteins. J. Biol. Chem. 2003, 278, 37092–37098. [Google Scholar] [CrossRef]
- Roberts, J.R.; Narasimhan, C.; Hruz, P.W.; Mitchell, G.A.; Miziorko, H.M. 3-Hydroxy-3-methylglutaryl-CoA lyase: Expression and isolation of the recombinant human enzyme and investigation of a mechanism for regulation of enzyme activity. J. Biol. Chem. 1994, 269, 17841–17846. [Google Scholar]
- Menao, S.; Lopez-Vinas, E.; Mir, C.; Puisac, B.; Gratacos, E.; Arnedo, M.; Carrasco, P.; Moreno, S.; Ramos, M.; Gil, M.C.; et al. Ten novel HMGCL mutations in 24 patients of different origin with 3-hydroxy-3-methyl-glutaric aciduria. Hum. Mutat. 2009, 30, E520–E529. [Google Scholar] [CrossRef]
- Montgomery, C.; Miziorko, H.M. Influence of multiple cysteines on human 3-hydroxy-3-methylglutaryl-CoA lyase activity and formation of inter-subunit adducts. Arch. Biochem. Biophys. 2011, 511, 48–55. [Google Scholar] [CrossRef]
- Wanders, R.J.; Schutgens, R.B.; Zoeters, P.H. 3-Hydroxy-3-methylglutaryl-CoA lyase in human skin fibroblasts: Study of its properties and deficient activity in 3-hydroxy-3-methylglutaric aciduria patients using a simple spectrophotometric method. Clin. Chim. Acta 1988, 171, 95–101. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Lang, D.; Thoma, R.; Henn-Sax, M.; Sterner, R.; Wilmanns, M. Structural evidence for evolution of the beta/alpha barrel scaffold by gene duplication and fusion. Science 2000, 289, 1546–1550. [Google Scholar] [CrossRef]
- Smith, J.J.; Keinath, M.C. The sea lamprey meiotic map improves resolution of ancient vertebrate genome duplications. Genome Res. 2015, 25, 1081–1090. [Google Scholar] [CrossRef]
- Smith, J.J.; Timoshevskaya, N.; Ye, C.; Holt, C.; Keinath, M.C.; Parker, H.J.; Cook, M.E.; Hess, J.E.; Narum, S.R.; Lamanna, F.; et al. The sea lamprey germline genome provides insights into programmed genome rearrangement and vertebrate evolution. Nat. Genet. 2018, 50, 270–277. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Cardoso-Moreira, M.; Halbert, J.; Valloton, D.; Velten, B.; Chen, C.; Shao, Y.; Liechti, A.; Ascencao, K.; Rummel, C.; Ovchinnikova, S.; et al. Gene expression across mammalian organ development. Nature 2019, 571, 505–509. [Google Scholar] [CrossRef]
- Available online: https://gtexportal.org (accessed on 31 October 2019).
- Gibson, K.M.; Breuer, J.; Nyhan, W.L. 3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: Review of 18 reported patients. Eur. J. Pediatr. 1988, 148, 180–186. [Google Scholar] [CrossRef]
- Grunert, S.C.; Schlatter, S.M.; Schmitt, R.N.; Gemperle-Britschgi, C.; Mrazova, L.; Balci, M.C.; Bischof, F.; Coker, M.; Das, A.M.; Demirkol, M.; et al. 3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: Clinical presentation and outcome in a series of 37 patients. Mol. Genet. Metab. 2017, 121, 206–215. [Google Scholar] [CrossRef]
- Ozand, P.T.; Devol, E.B.; Gascon, G.G. Neurometabolic diseases at a national referral center: Five years experience at the King Faisal Specialist Hospital and Research Centre. J. Child. Neurol. 1992, 7, S4–S11. [Google Scholar] [CrossRef]
- Bolhuis, P.A.; Page-Christiaens, G.C. The advisory report ‘Neonatal screening’ from the Health Council of The Netherlands. Ned. Tijdschr Geneeskd 2005, 149, 2857–2860. [Google Scholar]
- Vargas, C.R.; Sitta, A.; Schmitt, G.; Ferreira, G.C.; Cardoso, M.L.; Coelho, D.; Gibson, K.M.; Wajner, M. Incidence of 3-hydroxy-3-methylglutaryl-coenzyme A lyase (HL) deficiency in Brazil, South America. J. Inherit. Metab. Dis. 2008, 31, 511–515. [Google Scholar] [CrossRef]
- Bischof, F.; Nagele, T.; Wanders, R.J.; Trefz, F.K.; Melms, A. 3-hydroxy-3-methylglutaryl-CoA lyase deficiency in an adult with leukoencephalopathy. Ann. Neurol. 2004, 56, 727–730. [Google Scholar] [CrossRef]
- Reimao, S.; Morgado, C.; Almeida, I.T.; Silva, M.; Corte Real, H.; Campos, J. 3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: Initial presentation in a young adult. J. Inherit. Metab. Dis. 2009, 32, S49–S52. [Google Scholar] [CrossRef]
- Fernandes, C.G.; Rodrigues, M.D.N.; Seminotti, B.; Colin-Gonzalez, A.L.; Santamaria, A.; Quincozes-Santos, A.; Wajner, M. Induction of a Proinflammatory Response in Cortical Astrocytes by the Major Metabolites Accumulating in HMG-CoA Lyase Deficiency: The Role of ERK Signaling Pathway in Cytokine Release. Mol. Neurobiol. 2015, 53, 3586–3595. [Google Scholar] [CrossRef]
- Santosa, D.; Donner, M.G.; Vom Dahl, S.; Fleisch, M.; Hoehn, T.; Mayatepek, E.; Heldt, K.; Niehues, T.; Haussinger, D. Favourable Outcome in Two Pregnancies in a Patient with 3-Hydroxy-3-Methylglutaryl-CoA Lyase Deficiency. JIMD Rep. 2017, 37, 1–5. [Google Scholar]
- Pie, J.; Lopez-Vinas, E.; Puisac, B.; Menao, S.; Pie, A.; Casale, C.; Ramos, F.J.; Hegardt, F.G.; Gomez-Puertas, P.; Casals, N. Molecular genetics of HMG-CoA lyase deficiency. Mol. Genet. Metab. 2007, 92, 198–209. [Google Scholar] [CrossRef]
- Buesa, C.; Pie, J.; Barcelo, A.; Casals, N.; Mascaro, C.; Casale, C.H.; Haro, D.; Duran, M.; Smeitink, J.A.; Hegardt, F.G. Aberrantly spliced mRNAs of the 3-hydroxy-3-methylglutaryl coenzyme A lyase (HL) gene with a donor splice-site point mutation produce hereditary HL deficiency. J. Lipid Res. 1996, 37, 2420–2432. [Google Scholar]
- Casale, C.H.; Casals, N.; Pie, J.; Zapater, N.; Perez-Cerda, C.; Merinero, B.; Martinez-Pardo, M.; Garcia-Penas, J.J.; Garcia-Gonzalez, J.M.; Lama, R.; et al. A nonsense mutation in the exon 2 of the 3-hydroxy-3-methylglutaryl coenzyme A lyase (HL) gene producing three mature mRNAs is the main cause of 3-hydroxy-3-methylglutaric aciduria in European Mediterranean patients. Arch. Biochem. Biophys. 1998, 349, 129–137. [Google Scholar] [CrossRef]
- Casals, N.; Pie, J.; Casale, C.H.; Zapater, N.; Ribes, A.; Castro-Gago, M.; Rodriguez-Segade, S.; Wanders, R.J.; Hegardt, F.G. A two-base deletion in exon 6 of the 3-hydroxy-3-methylglutaryl coenzyme A lyase (HL) gene producing the skipping of exons 5 and 6 determines 3-hydroxy-3-methylglutaric aciduria. J. Lipid Res. 1997, 38, 2303–2313. [Google Scholar]
- Puisac, B.; Lopez-Vinas, E.; Moreno, S.; Mir, C.; Perez-Cerda, C.; Menao, S.; Lluch, D.; Pie, A.; Gomez-Puertas, P.; Casals, N.; et al. Skipping of exon 2 and exons 2 plus 3 of HMG-CoA lyase (HL) gene produces the loss of beta sheets 1 and 2 in the recently proposed (beta-alpha)8 TIM barrel model of HL. Biophys. Chem. 2005, 115, 241–245. [Google Scholar] [CrossRef]
- Li, M.; Sun, Q.; Wang, X. Transcriptional landscape of human cancers. Oncotarget 2017, 8, 34534–34551. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Kang, H.B.; Fan, J.; Lin, R.; Elf, S.; Ji, Q.; Zhao, L.; Jin, L.; Seo, J.H.; Shan, C.; Arbiser, J.L.; et al. Metabolic Rewiring by Oncogenic BRAF V600E Links Ketogenesis Pathway to BRAF-MEK1 Signaling. Mol. Cell 2015, 59, 345–358. [Google Scholar] [CrossRef]
- Luo, W.; Qin, L.; Li, B.; Liao, Z.; Liang, J.; Xiao, X.; Xiao, X.; Mo, Y.; Huang, G.; Zhang, Z.; et al. Inactivation of HMGCL promotes proliferation and metastasis of nasopharyngeal carcinoma by suppressing oxidative stress. Sci. Rep. 2017, 7, 11954. [Google Scholar] [CrossRef]
- Park, J.H.; Woo, Y.M.; Youm, E.M.; Hamad, N.; Won, H.H.; Naka, K.; Park, E.J.; Park, J.H.; Kim, H.J.; Kim, S.H.; et al. HMGCLL1 is a predictive biomarker for deep molecular response to imatinib therapy in chronic myeloid leukemia. Leukemia 2019, 33, 1439–1450. [Google Scholar] [CrossRef]
- Fang, Q.; Benedetti, A.F.; Ma, Q.; Gregory, L.; Li, J.Z.; Dattani, M.; Sadeghi-Nejad, A.; Arnhold, I.J.; Mendonca, B.B.; Camper, S.A.; et al. HESX1 mutations in patients with congenital hypopituitarism: Variable phenotypes with the same genotype. Clin. Endocrinol. 2016, 85, 408–414. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).