The Role of Immune Cells and Cytokines in Intestinal Wound Healing



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
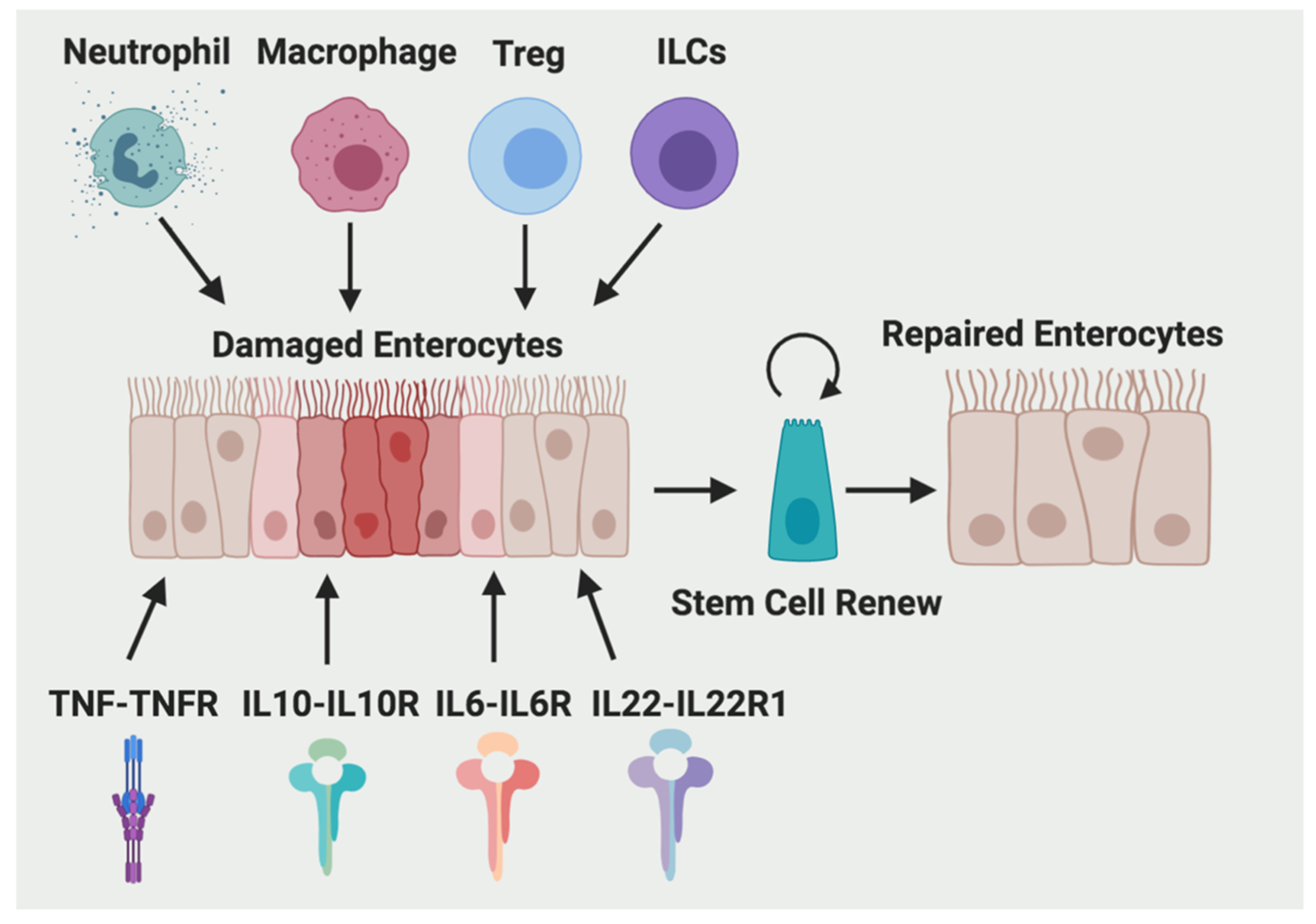
2. Immune Cells
2.1. Neutrophils
2.1.1. The Function of Neutrophils
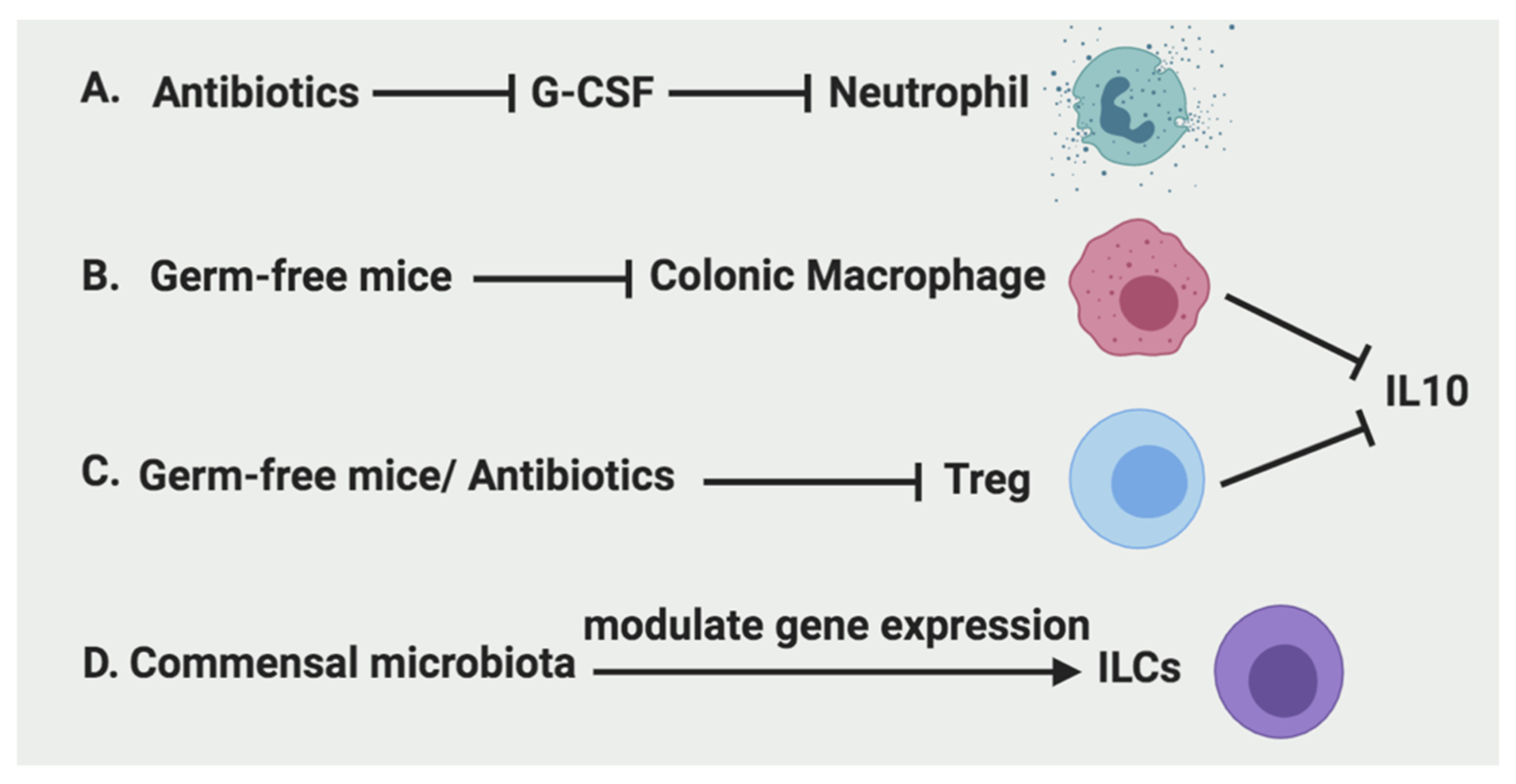
2.1.2. The Regulation of Neutrophils
2.2. Macrophages
2.2.1. The Function of Macrophages
2.2.2. The Regulation of Macrophages
2.3. Regulatory T Cells (Treg)
2.3.1. The Function of Treg
2.3.2. The Regulation of Treg
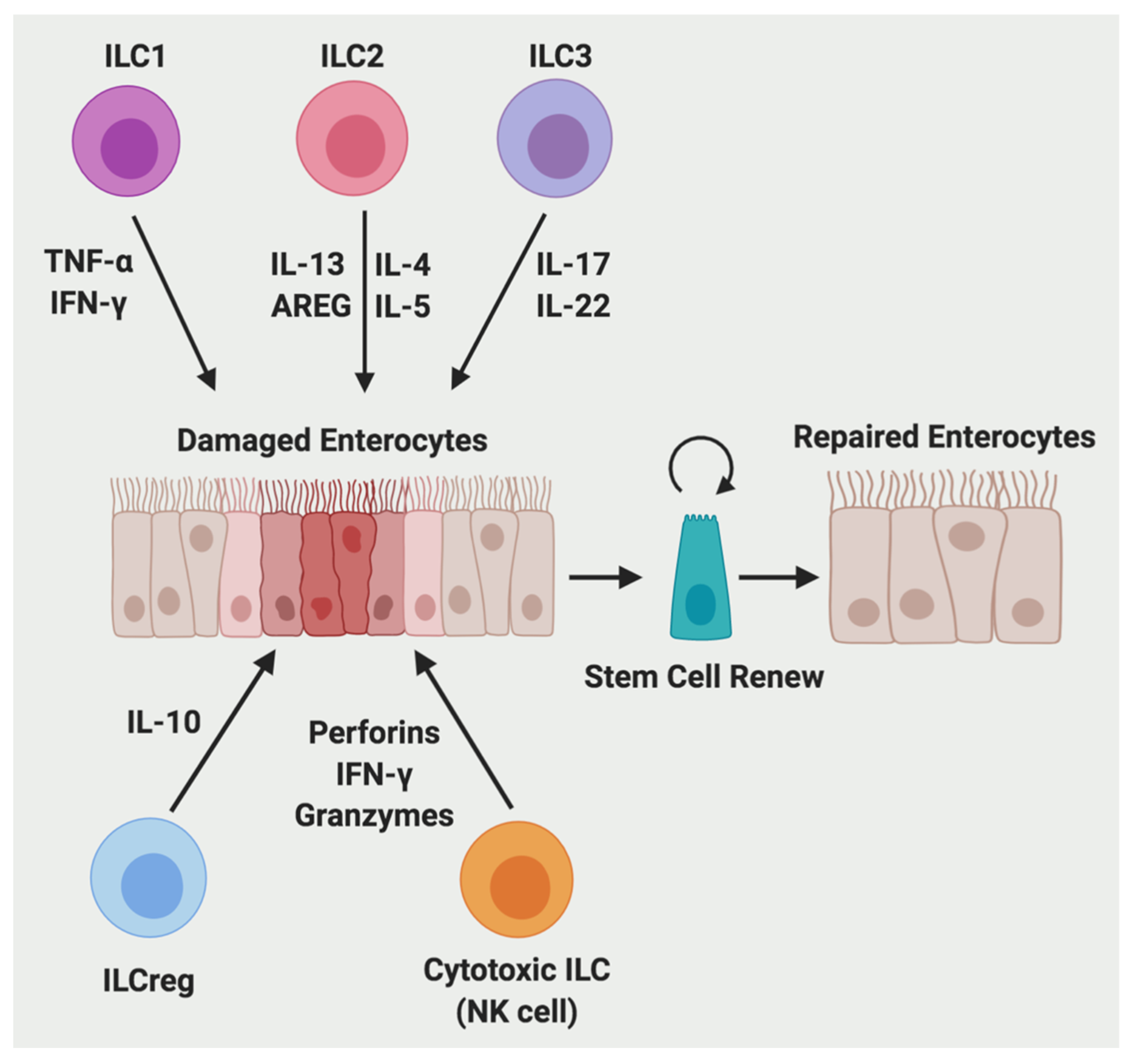
2.4. Innate Lymphoid Cells (ILCs)
2.4.1. The Function of ILCs
2.4.2. The Regulation of ILCs
3. Cytokines
3.1. IL-10
3.1.1. The Source of IL-10
3.1.2. The Function of IL-10
3.1.3. The Regulation of IL-10
3.2. TNF-α
3.2.1. The Source of TNF-α
3.2.2. The Function of TNF-α
3.2.3. The Regulation of TNF-α
3.3. IL-6
3.3.1. The Source of IL-6
3.3.2. The Function of IL-6
3.3.3. The Regulation of IL-6
3.4. IL-22
3.4.1. The Source of IL-22
3.4.2. The Function of IL-22
3.4.3. The Regulation of IL-22
4. Concluding Remarks and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Mukherjee, K.; Kavalukas, S.L.; Barbul, A. Nutritional Aspects of Gastrointestinal Wound Healing. Adv. Wound Care 2016, 5, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, M.; Konno, S. Wound healing of intestinal epithelial cells. World J. Gastroenterol. 2011, 17, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Rankin, L.C.; Artis, D. Beyond Host Defense: Emerging Functions of the Immune System in Regulating Complex Tissue Physiology. Cell 2018, 173, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Young, J.W. Human Dendritic Cells: Potent Antigen-Presenting Cells at the Crossroads of Innate and Adaptive Immunity. J. Immunol. 2005, 175, 1373–1381. [Google Scholar] [CrossRef]
- Brazil, J.C.; Quiros, M.; Nusrat, A.; Parkos, C.A. Innate immune cell–epithelial crosstalk during wound repair. J. Clin. Investig. 2019, 129, 2983–2993. [Google Scholar] [CrossRef]
- Kim, M.-H.; Liu, W.; Borjesson, D.L.; Curry, F.-R.E.; Miller, L.S.; Cheung, A.L.; Liu, F.-T.; Isseroff, R.R.; Simon, S.I. Dynamics of neutrophil infiltration during cutaneous wound healing and infection using fluorescence imaging. J. Investig. Dermatol. 2008, 128, 1812–1820. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Lahoz-Beneytez, J.; Elemans, M.; Zhang, Y.; Ahmed, R.; Salam, A.; Block, M.; Niederalt, C.; Asquith, B.; Macallan, D. Human neutrophil kinetics: Modeling of stable isotope labeling data supports short blood neutrophil half-lives. Blood 2016, 127, 3431–3438. [Google Scholar] [CrossRef]
- Colotta, F.; Re, F.; Polentarutti, N.; Sozzani, S.; Mantovani, A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 1992, 80, 2012–2020. [Google Scholar] [CrossRef]
- Williams, I.R.; Parkos, C.A. Colonic Neutrophils in Inflammatory Bowel Disease: Double-Edged Swords of the Innate Immune System with Protective and Destructive Capacity. Gastroenterology 2007, 133, 2049–2052. [Google Scholar] [CrossRef]
- Bressenot, A.; Salleron, J.; Bastien, C.; Danese, S.; Boulagnon-Rombi, C.; Peyrin-Biroulet, L. Comparing histological activity indexes in UC. Gut 2015, 64, 1412–1418. [Google Scholar] [CrossRef] [PubMed]
- Geboes, K.; Riddell, R.; Ost, A.; Jensfelt, B.; Persson, T.; Löfberg, R. A reproducible grading scale for histological assessment of inflammation in ulcerative colitis. Gut 2000, 47, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Butin-Israeli, V.; Houser, M.C.; Feng, M.; Thorp, E.B.; Nusrat, A.; Parkos, C.A.; Sumagin, R. Deposition of microparticles by neutrophils onto inflamed epithelium: A new mechanism to disrupt epithelial intercellular adhesions and promote transepithelial migration. FASEB J. 2016, 30, 4007–4020. [Google Scholar] [CrossRef] [PubMed]
- Butin-Israeli, V.; Bui, T.M.; Wiesolek, H.L.; Mascarenhas, L.; Lee, J.J.; Mehl, L.C.; Knutson, K.R.; Adam, S.A.; Goldman, R.D.; Beyder, A.; et al. Neutrophil-induced genomic instability impedes resolution of inflammation and wound healing. J. Clin. Investig. 2019, 129, 712–726. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.C.; Schmalsteig, F.C.; Finegold, M.J.; Hughes, B.J.; Rothlein, R.; Miller, L.J.; Kohl, S.; Tosi, M.F.; Jacobs, R.L.; Waldrop, T.C.; et al. The Severe and Moderate Phenotypes of Heritable Mac-1, LFA-1 Deficiency: Their Quantitative Definition and Relation to Leukocyte Dysfunction and Clinical Features. J. Infect. Dis. 1985, 152, 668–689. [Google Scholar] [CrossRef] [PubMed]
- Kühl, A.A.; Kakirman, H.; Janotta, M.; Dreher, S.; Cremer, P.; Pawlowski, N.N.; Loddenkemper, C.; Heimesaat, M.M.; Grollich, K.; Zeitz, M.; et al. Aggravation of Different Types of Experimental Colitis by Depletion or Adhesion Blockade of Neutrophils. Gastroenterology 2007, 133, 1882–1892. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- McNamee, E.N. Neutrophil-derived microRNAs put the (DNA) breaks on intestinal mucosal healing. J. Clin. Investig. 2019, 129, 499–502. [Google Scholar] [CrossRef]
- Campbell, E.L.; Bruyninckx, W.J.; Kelly, C.J.; Glover, L.E.; McNamee, E.N.; Bowers, B.E.; Bayless, A.J.; Scully, M.; Saeedi, B.J.; Golden-Mason, L.; et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 2014, 40, 66–77. [Google Scholar] [CrossRef]
- Furuta, G.T.; Turner, J.R.; Taylor, C.T.; Hershberg, R.M.; Comerford, K.; Narravula, S.; Podolsky, D.K.; Colgan, S.P. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J. Exp. Med. 2001, 193, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kirpich, I.; Liu, Y.; Ma, Z.; Barve, S.; McClain, C.J.; Feng, W. Lactobacillus rhamnosus GG Treatment Potentiates Intestinal Hypoxia-Inducible Factor, Promotes Intestinal Integrity and Ameliorates Alcohol-Induced Liver Injury. Am. J. Pathol. 2011, 179, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Yang, W.; Huang, X.; Cao, A.T.; Bilotta, A.J.; Xiao, Y.; Sun, M.; Chen, L.; Ma, C.; Liu, X.; et al. Neutrophils Promote Amphiregulin Production in Intestinal Epithelial Cells through TGF-β and Contribute to Intestinal Homeostasis. J. Immunol. 2018, 201, 2492–2501. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, H.S.; Liu, Y.; Menkiti, O.R.; Mei, J.; Dai, N.; O’Leary, C.E.; Oliver, P.M.; Kolls, J.K.; Weiser, J.N.; Worthen, G.S. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat. Med. 2014, 20, 524–530. [Google Scholar] [CrossRef]
- Xue, X.; Ramakrishnan, S.; Anderson, E.; Taylor, M.; Zimmermann, E.M.; Spence, J.R.; Huang, S.; Greenson, J.K.; Shah, Y.M. Endothelial PAS Domain Protein 1 Activates the Inflammatory Response in the Intestinal Epithelium to Promote Colitis in Mice. Gastroenterology 2013, 145, 831–841. [Google Scholar] [CrossRef]
- Xue, X.; Shah, Y.M. Hypoxia-inducible factor-2α is essential in activating the COX2/mPGES-1/PGE 2 signaling axis in colon cancer. Carcinogenesis 2013, 34, 163–169. [Google Scholar] [CrossRef]
- Xue, X.; Taylor, M.; Anderson, E.; Hao, C.; Qu, A.; Greenson, J.K.; Zimmermann, E.M.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-Inducible Factor-2α Activation Promotes Colorectal Cancer Progression by Dysregulating Iron Homeostasis. Cancer Res. 2012, 72, 2285–2293. [Google Scholar] [CrossRef]
- Triner, D.; Xue, X.; Schwartz, A.J.; Jung, I.; Colacino, J.A.; Shah, Y.M. Epithelial Hypoxia-Inducible Factor 2α Facilitates the Progression of Colon Tumors through Recruiting Neutrophils. Mol. Cell. Biol. 2017, 37, e00481-16. [Google Scholar] [CrossRef]
- Schuerer-Maly, C.C.; Eckmann, L.; Kagnoff, M.F.; Falco, M.T.; Maly, F.E. Colonic epithelial cell lines as a source of interleukin-8: Stimulation by inflammatory cytokines and bacterial lipopolysaccharide. Immunology 1994, 81, 85–91. [Google Scholar]
- Sumagin, R.; Robin, A.Z.; Nusrat, A.; Parkos, C.A. Transmigrated neutrophils in the intestinal lumen engage ICAM-1 to regulate the epithelial barrier and neutrophil recruitment. Mucosal Immunol. 2014, 7, 905–915. [Google Scholar] [CrossRef]
- Sumagin, R.; Brazil, J.C.; Nava, P.; Nishio, H.; Alam, A.; Luissint, A.C.; Weber, D.A.; Neish, A.S.; Nusrat, A.; Parkos, C.A. Neutrophil interactions with epithelial-expressed ICAM-1 enhances intestinal mucosal wound healing. Mucosal Immunol. 2016, 9, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.H.; Kljavin, N.M.; Ota, N.; Leonard, J.; Roose-Girma, M.; Diehl, L.; Ouyang, W.; Ghilardi, N. Opposing consequences of IL-23 signaling mediated by innate and adaptive cells in chemically induced colitis in mice. Mucosal Immunol. 2012, 5, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Bain, C.C.; Bravo-Blas, A.; Scott, C.L.; Perdiguero, E.G.; Geissmann, F.; Henri, S.; Malissen, B.; Osborne, L.C.; Artis, D.; Mowat, A.M. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 2014, 15, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.N.; Houston, S.A.; Wemyss, K.; Bridgeman, H.M.; Barbera, T.A.; Zangerle-Murray, T.; Strangward, P.; Ridley, A.J.L.; Wang, P.; Tamoutounour, S.; et al. Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4 expression. J. Exp. Med. 2018, 215, 1507–1518. [Google Scholar] [CrossRef]
- De Schepper, S.; Verheijden, S.; Aguilera-Lizarraga, J.; Viola, M.F.; Boesmans, W.; Stakenborg, N.; Voytyuk, I.; Schmidt, I.; Boeckx, B.; Dierckx de Casterlé, I.; et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell 2018, 175, 400–415. [Google Scholar] [CrossRef]
- Bain, C.C.; Scott, C.L.; Uronen-Hansson, H.; Gudjonsson, S.; Jansson, O.; Grip, O.; Guilliams, M.; Malissen, B.; Agace, W.W.; Mowat, A.M. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013, 6, 498–510. [Google Scholar] [CrossRef]
- Isidro, R.A.; Appleyard, C.B. Colonic macrophage polarization in homeostasis, inflammation, and cancer. Am. J. Physiol.-Gastrointest. Liver Physiol. 2016, 311, G59–G73. [Google Scholar] [CrossRef]
- Rivollier, A.; He, J.; Kole, A.; Valatas, V.; Kelsall, B.L. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J. Exp. Med. 2012, 209, 139–155. [Google Scholar] [CrossRef]
- Baillie, J.K.; Arner, E.; Daub, C.; De Hoon, M.; Itoh, M.; Kawaji, H.; Lassmann, T.; Carninci, P.; Forrest, A.R.R.; Hayashizaki, Y.; et al. Analysis of the human monocyte-derived macrophage transcriptome and response to lipopolysaccharide provides new insights into genetic aetiology of inflammatory bowel disease. PLoS Genet. 2017, 13, e1006641. [Google Scholar] [CrossRef]
- Smythies, L.E.; Sellers, M.; Clements, R.H.; Mosteller-Barnum, M.; Meng, G.; Benjamin, W.H.; Orenstein, J.M.; Smith, P.D. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J. Clin. Investig. 2005, 115, 66–75. [Google Scholar] [CrossRef]
- Bain, C.C.; Mowat, A.M. Macrophages in intestinal homeostasis and inflammation. Immunol. Rev. 2014, 260, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef]
- Pull, S.L.; Doherty, J.M.; Mills, J.C.; Gordon, J.I.; Stappenbeck, T.S. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc. Natl. Acad. Sci. USA 2005, 102, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Seno, H.; Miyoshi, H.; Brown, S.L.; Geske, M.J.; Colonna, M.; Stappenbeck, T.S. Efficient colonic mucosal wound repair requires Trem2 signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 256–261. [Google Scholar] [CrossRef]
- Saha, S.; Aranda, E.; Hayakawa, Y.; Bhanja, P.; Atay, S.; Brodin, N.P.; Li, J.; Asfaha, S.; Liu, L.; Tailor, Y.; et al. Macrophage-derived extracellular vesicle-packaged WNTs rescue intestinal stem cells and enhance survival after radiation injury. Nat. Commun. 2016, 7, 13096. [Google Scholar] [CrossRef]
- Cosín-Roger, J.; Ortiz-Masiá, D.; Calatayud, S.; Hernández, C.; Esplugues, J.V.; Barrachina, M.D. The activation of Wnt signaling by a STAT6-dependent macrophage phenotype promotes mucosal repair in murine IBD. Mucosal Immunol. 2016, 9, 986–998. [Google Scholar] [CrossRef]
- Ochi, T.; Feng, Y.; Kitamoto, S.; Nagao-Kitamoto, H.; Kuffa, P.; Atarashi, K.; Honda, K.; Teitelbaum, D.H.; Kamada, N. Diet-dependent, microbiota-independent regulation of IL-10-producing lamina propria macrophages in the small intestine. Sci. Rep. 2016, 6, 27634. [Google Scholar] [CrossRef]
- Morhardt, T.L.; Hayashi, A.; Ochi, T.; Quirós, M.; Kitamoto, S.; Nagao-Kitamoto, H.; Kuffa, P.; Atarashi, K.; Honda, K.; Kao, J.Y.; et al. IL-10 produced by macrophages regulates epithelial integrity in the small intestine. Sci. Rep. 2019, 9, 1223. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, Z. Inflammatory bowel disease related innate immunity and adaptive immunity. Am. J. Transl. Res. 2016, 8, 2490–2497. [Google Scholar]
- Geremia, A.; Biancheri, P.; Allan, P.; Corazza, G.R.; Di Sabatino, A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 3–10. [Google Scholar] [CrossRef]
- Roncarolo, M.G.; Gregori, S.; Bacchetta, R.; Battaglia, M.; Gagliani, N. The Biology of T Regulatory Type 1 Cells and Their Therapeutic Application in Immune-Mediated Diseases. Immunity 2018, 49, 1004–1019. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.A.R.; Rodrigues, B.L.; Ayrizono, M.D.L.S.; Leal, R.F. The Immunological Basis of Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2016, 2016, 2097274. [Google Scholar] [CrossRef]
- Betto, T.; Amano, H.; Ito, Y.; Eshima, K.; Yoshida, T.; Matsui, Y.; Yamane, S.; Inoue, T.; Otaka, F.; Kobayashi, K.; et al. Vascular endothelial growth factor receptor 1 tyrosine kinase signaling facilitates healing of DSS-induced colitis by accumulation of Tregs in ulcer area. Biomed. Pharmacother. 2019, 111, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Vent-Schmidt, J.; McGeough, M.D.; Wong, M.; Hoffman, H.M.; Steiner, T.S.; Levings, M.K. Tr1 Cells, but Not Foxp3+ Regulatory T Cells, Suppress NLRP3 Inflammasome Activation via an IL-10–Dependent Mechanism. J. Immunol. 2015, 195, 488–497. [Google Scholar] [CrossRef]
- Cook, L.; Stahl, M.; Han, X.; Nazli, A.; MacDonald, K.N.; Wong, M.Q.; Tsai, K.; Dizzell, S.; Jacobson, K.; Bressler, B.; et al. Suppressive and Gut Reparative Functions of Human Type 1 T-regulatory Cells. Gastroenterology 2019. [Google Scholar] [CrossRef]
- Desreumaux, P.; Foussat, A.; Allez, M.; Beaugerie, L.; Hébuterne, X.; Bouhnik, Y.; Nachury, M.; Brun, V.; Bastian, H.; Belmonte, N.; et al. Safety and Efficacy of Antigen-Specific Regulatory T-Cell Therapy for Patients with Refractory Crohn’s Disease. Gastroenterology 2012, 143, 1207–1217. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Kim, K.S.; Hong, S.-W.; Han, D.; Yi, J.; Jung, J.; Yang, B.-G.; Lee, J.Y.; Lee, M.; Surh, C.D. Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 2016, 351, 858–863. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Cherrier, D.E.; Serafini, N.; Di Santo, J.P. Innate Lymphoid Cell Development: A T Cell Perspective. Immunity 2018, 48, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Walker, J.A.; Jolin, H.E.; Drynan, L.F.; Hams, E.; Camelo, A.; Barlow, J.L.; Neill, D.R.; Panova, V.; Koch, U.; et al. Transcription factor RORα is critical for nuocyte development. Nat. Immunol. 2012, 13, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate production of TH2 cytokines by adipose tissue-associated c-Kit+Sca-1+ lymphoid cells. Nature 2010, 463, 540–544. [Google Scholar] [CrossRef]
- Cupedo, T.; Crellin, N.K.; Papazian, N.; Rombouts, E.J.; Weijer, K.; Grogan, J.L.; Fibbe, W.E.; Cornelissen, J.J.; Spits, H. Human fetal lymphoid tissue–inducer cells are interleukin 17–producing precursors to RORC+ CD127+ natural killer–like cells. Nat. Immunol. 2009, 10, 66–74. [Google Scholar] [CrossRef]
- Wang, S.; Xia, P.; Chen, Y.; Qu, Y.; Xiong, Z.; Ye, B.; Du, Y.; Tian, Y.; Yin, Z.; Xu, Z.; et al. Regulatory Innate Lymphoid Cells Control Innate Intestinal Inflammation. Cell 2017, 171, 201–216. [Google Scholar] [CrossRef]
- Robinette, M.L.; Fuchs, A.; Cortez, V.S.; Lee, J.S.; Wang, Y.; Durum, S.K.; Gilfillan, S.; Colonna, M.; Shaw, L.; Yu, B.; et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat. Immunol. 2015, 16, 306–317. [Google Scholar] [CrossRef]
- Cerwenka, A.; Lanier, L.L. Natural killer cells, viruses and cancer. Nat. Rev. Immunol. 2001, 1, 41–49. [Google Scholar] [CrossRef]
- Pokrovskii, M.; Hall, J.A.; Ochayon, D.E.; Yi, R.; Chaimowitz, N.S.; Seelamneni, H.; Carriero, N.; Watters, A.; Waggoner, S.N.; Littman, D.R.; et al. Characterization of Transcriptional Regulatory Networks that Promote and Restrict Identities and Functions of Intestinal Innate Lymphoid Cells. Immunity 2019, 51, 185–197. [Google Scholar] [CrossRef]
- Sonnenberg, G.F.; Artis, D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat. Med. 2015, 21, 698–708. [Google Scholar] [CrossRef]
- Fuchs, A.; Vermi, W.; Lee, J.S.; Lonardi, S.; Gilfillan, S.; Newberry, R.D.; Cella, M.; Colonna, M. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12-and IL-15-responsive IFN-γ-producing cells. Immunity 2013, 38, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Forkel, M.; van Tol, S.; Höög, C.; Michaëlsson, J.; Almer, S.; Mjösberg, J. Distinct Alterations in the Composition of Mucosal Innate Lymphoid Cells in Newly Diagnosed and Established Crohn’s Disease and Ulcerative Colitis. J. Crohn’s Colitis 2018, 13, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Zhu, X.; Wu, J.; He, L.; Lu, T.; Wang, Y.; Liu, B.; Ye, B.; Sun, L.; Fan, D.; et al. IL-13 secreted by ILC2s promotes the self-renewal of intestinal stem cells through circular RNA circPan3. Nat. Immunol. 2019, 20, 183–194. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.W.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef] [PubMed]
- Sawa, S.; Lochner, M.; Satoh-Takayama, N.; Dulauroy, S.; Bérard, M.; Kleinschek, M.; Cua, D.; Di Santo, J.P.; Eberl, G. RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat. Immunol. 2011, 12, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, J.G.; Woo, V.; Viladomiu, M.; Putzel, G.; Lima, S.; Diehl, G.E.; Marderstein, A.R.; Gandara, J.; Perez, A.R.; Withers, D.R.; et al. Microbiota-Induced TNF-like Ligand 1A Drives Group 3 Innate Lymphoid Cell-Mediated Barrier Protection and Intestinal T Cell Activation during Colitis. Immunity 2018, 49, 1077–1089. [Google Scholar] [CrossRef]
- Longman, R.S.; Diehl, G.E.; Victorio, D.A.; Huh, J.R.; Galan, C.; Miraldi, E.R.; Swaminath, A.; Bonneau, R.; Scherl, E.J.; Littman, D.R. CX3CR1+ mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J. Exp. Med. 2014, 211, 1571–1583. [Google Scholar] [CrossRef]
- Hanash, A.M.; Dudakov, J.A.; Hua, G.; O’Connor, M.H.; Young, L.F.; Singer, N.V.; West, M.L.; Jenq, R.R.; Holland, A.M.; Kappel, L.W.; et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity 2012, 37, 339–350. [Google Scholar] [CrossRef]
- Lindemans, C.A.; Calafiore, M.; Mertelsmann, A.M.; O’Connor, M.H.; Dudakov, J.A.; Jenq, R.R.; Velardi, E.; Young, L.F.; Smith, O.M.; Lawrence, G.; et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 2015, 528, 560–564. [Google Scholar] [CrossRef]
- Gronke, K.; Hernández, P.P.; Zimmermann, J.; Klose, C.S.N.; Kofoed-Branzk, M.; Guendel, F.; Witkowski, M.; Tizian, C.; Amann, L.; Schumacher, F.; et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature 2019, 566, 249–253. [Google Scholar] [CrossRef]
- Sonnenberg, G.F.; Monticelli, L.A.; Elloso, M.M.; Fouser, L.A.; Artis, D. CD4+ lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity 2011, 34, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Domingo, P.; Romera-Hernandez, M.; Karrich, J.J.; Cornelissen, F.; Papazian, N.; Lindenbergh-Kortleve, D.J.; Butler, J.A.; Boon, L.; Coles, M.C.; Samsom, J.N.; et al. Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. J. Exp. Med. 2015, 212, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Steel, A.W.; Mela, C.M.; Lindsay, J.O.; Gazzard, B.G.; Goodier, M.R. Increased proportion of CD16+ NK cells in the colonic lamina propria of inflammatory bowel disease patients, but not after azathioprine treatment. Aliment. Pharmacol. Ther. 2011, 33, 115–126. [Google Scholar] [CrossRef]
- Yusung, S.; McGovern, D.; Lin, L.; Hommes, D.; Lagishetty, V.; Braun, J. NK cells are biologic and biochemical targets of 6-mercaptopurine in Crohn’s disease patients. Clin. Immunol. 2017, 175, 82–90. [Google Scholar] [CrossRef]
- Lin, L.; Ma, C.; Wei, B.; Aziz, N.; Rajalingam, R.; Yusung, S.; Erlich, H.A.; Trachtenberg, E.A.; Targan, S.R.; McGovern, D.P.B.; et al. Human NK Cells Licensed by Killer Ig Receptor Genes Have an Altered Cytokine Program That Modifies CD4+ T Cell Function. J. Immunol. 2014, 193, 940–949. [Google Scholar] [CrossRef]
- Gury-BenAri, M.; Thaiss, C.A.; Serafini, N.; Winter, D.R.; Giladi, A.; Lara-Astiaso, D.; Levy, M.; Salame, T.M.; Weiner, A.; David, E.; et al. The Spectrum and Regulatory Landscape of Intestinal Innate Lymphoid Cells Are Shaped by the Microbiome. Cell 2016, 166, 1231–1246. [Google Scholar] [CrossRef]
- Li, S.; Bostick, J.W.; Ye, J.; Qiu, J.; Zhang, B.; Urban, J.F.; Avram, D.; Zhou, L. Aryl Hydrocarbon Receptor Signaling Cell Intrinsically Inhibits Intestinal Group 2 Innate Lymphoid Cell Function. Immunity 2018, 49, 915–928. [Google Scholar] [CrossRef]
- Kim, M.H.; Taparowsky, E.J.; Kim, C.H. Retinoic Acid Differentially Regulates the Migration of Innate Lymphoid Cell Subsets to the Gut. Immunity 2015, 43, 107–119. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Ueda, Y.; Kayama, H.; Jeon, S.G.; Kusu, T.; Isaka, Y.; Rakugi, H.; Yamamoto, M.; Takeda, K. Commensal microbiota induce LPS hyporesponsiveness in colonic macrophages via the production of IL-10. Int. Immunol. 2010, 22, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Quiros, M.; Nishio, H.; Neumann, P.A.; Siuda, D.; Brazil, J.C.; Azcutia, V.; Hilgarth, R.; O’Leary, M.N.; Garcia-Hernandez, V.; Leoni, G.; et al. Macrophage-derived IL-10 mediates mucosal repair by epithelial WISP-1 signaling. J. Clin. Investig. 2017, 127, 3510–3520. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Layunta, E.; Grasa, L.; Pardo, J.; García, S.; Alcalde, A.I.; Mesonero, J.E. Toll-like receptors 2 and 4 modulate intestinal IL-10 differently in ileum and colon. United Eur. Gastroenterol. J. 2018, 6, 446–453. [Google Scholar] [CrossRef]
- Sun, M.; Wu, W.; Chen, L.; Yang, W.; Huang, X.; Ma, C.; Chen, F.; Xiao, Y.; Zhao, Y.; Ma, C.; et al. Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nat. Commun. 2018, 9, 3555. [Google Scholar] [CrossRef] [PubMed]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef]
- Pennica, D.; Nedwin, G.E.; Hayflick, J.S.; Seeburg, P.H.; Derynck, R.; Palladino, M.A.; Kohr, W.J.; Aggarwal, B.B.; Goeddel, D. V Human tumour necrosis factor: Precursor structure, expression and homology to lymphotoxin. Nature 1984, 312, 724–729. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Bradford, E.M.; Ryu, S.H.; Singh, A.P.; Lee, G.; Goretsky, T.; Sinh, P.; Williams, D.B.; Cloud, A.L.; Gounaris, E.; Patel, V.; et al. Epithelial TNF Receptor Signaling Promotes Mucosal Repair in Inflammatory Bowel Disease. J. Immunol. 2017, 199, 1886–1897. [Google Scholar] [CrossRef]
- Roulis, M.; Armaka, M.; Manoloukos, M.; Apostolaki, M.; Kollias, G. Intestinal epithelial cells as producers but not targets of chronic TNF suffice to cause murine Crohn-like pathology. Proc. Natl. Acad. Sci. USA 2011, 108, 5396–5401. [Google Scholar] [CrossRef]
- Robak, T.; Gla̧dalska, A.; Stȩpień, H. The tumour necrosis factor family of receptors/ligands in the serum of patients with rheumatoid arthritis. Eur. Cytokine Netw. 1998, 9, 145–154. [Google Scholar]
- Waage, A.; Halstensen, A.; Espevik, T. Association between tumour necrosis factor in serum and fatal outcome in patients with meningococcal disease. Lancet 1987, 329, 355–357. [Google Scholar] [CrossRef]
- Kwiatkowski, D.; Sambou, I.; Twumasi, P.; Greenwood, B.M.; Hill, A.V.S.; Manogue, K.R.; Cerami, A.; Castracane, J.; Brewster, D.R. TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet 1990, 336, 1201–1204. [Google Scholar] [CrossRef]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Petito, V.; Graziani, C.; Lopetuso, L.R.; Fossati, M.; Battaglia, A.; Arena, V.; Scannone, D.; Quaranta, G.; Quagliariello, A.; Del Chierico, F.; et al. Anti-tumor necrosis factor α therapy associates to type 17 helper T lymphocytes immunological shift and significant microbial changes in dextran sodium sulphate colitis. World J. Gastroenterol. 2019, 25, 1465–1477. [Google Scholar] [CrossRef]
- Pogrel, M.A. Compression osteosynthesis in mandibular fractures. Int. J. Oral Maxillofac. Surg. 1986, 15, 521–524. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Mizoguchi, A.; Takedatsu, H.; Cario, E.; De Jong, Y.P.; Ooi, C.J.; Xavier, R.J.; Terhorst, C.; Podolsky, D.K.; Bhan, A.K. Role of tumor necrosis factor receptor 2 (TNFR2) in colonic epithelial hyperplasia and chronic intestinal inflammation in mice. Gastroenterology 2002, 122, 134–144. [Google Scholar] [CrossRef]
- Punit, S.; Dubé, P.E.; Liu, C.Y.; Girish, N.; Washington, M.K.; Polk, D.B. Tumor Necrosis Factor Receptor 2 Restricts the Pathogenicity of CD8(+) T Cells in Mice with Colitis. Gastroenterology 2015, 149, 993–1005. [Google Scholar] [CrossRef]
- Chen, L.-W.; Egan, L.; Li, Z.-W.; Greten, F.R.; Kagnoff, M.F.; Karin, M. The two faces of IKK and NF-κB inhibition: Prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat. Med. 2003, 9, 575–581. [Google Scholar] [CrossRef]
- Falvo, J.V.; Tsytsykova, A.V.; Goldfeld, A.E. Transcriptional control of the TNF gene. TNF Pathophysiol. 2010, 11, 27–60. [Google Scholar]
- Drouet, C.; Shakhov, A.N.; Jongeneel, C. V Enhancers and transcription factors controlling the inducibility of the tumor necrosis factor-alpha promoter in primary macrophages. J. Immunol. 1991, 147, 1694–1700. [Google Scholar]
- Krämer, B.; Wiegmann, K.; Krönke, M. Regulation of the Human TNF Promoter by the Transcription Factor Ets. J. Biol. Chem. 1995, 270, 6577–6583. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Garbers, C.; Hermanns, H.M.; Schaper, F.; Müller-Newen, G.; Grötzinger, J.; Rose-John, S.; Scheller, J. Plasticity and cross-talk of Interleukin 6-type cytokines. Cytokine Growth Factor Rev. 2012, 23, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Clevers, H. Reparative inflammation takes charge of tissue regeneration. Nature 2016, 529, 307–315. [Google Scholar] [CrossRef]
- Meyer, T.A.; Wang, J.; Tiao, G.M.; Ogle, C.K.; Fischer, J.E.; Hasselgren, P.-O. Sepsis and endotoxemia stimulate intestinal interleukin-6 production. Surgery 1995, 118, 336–342. [Google Scholar] [CrossRef]
- Hosokawa, T.; Kusugami, K.; Ina, K.; Ando, T.; Shinoda, M.; Imada, A.; Ohsuga, M.; Sakai, T.; Matsuura, T.; Ito, K.; et al. Interleukin-6 and soluble interleukin-6 receptor in the colonic mucosa of inflammatory bowel disease. J. Gastroenterol. Hepatol. 1999, 14, 987–996. [Google Scholar] [CrossRef]
- Parisinos, C.A.; Serghiou, S.; Katsoulis, M.; George, M.J.; Patel, R.S.; Hemingway, H.; Hingorani, A.D. Variation in Interleukin 6 Receptor Gene Associates with Risk of Crohn’s Disease and Ulcerative Colitis. Gastroenterology 2018, 155, 303–306. [Google Scholar] [CrossRef]
- Ito, H.; Takazoe, M.; Fukuda, Y.; Hibi, T.; Kusugami, K.; Andoh, A.; Matsumoto, T.; Yamamura, T.; Azuma, J.; Nishimoto, N.; et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease. Gastroenterology 2004, 126, 989–996. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.-Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef]
- Taniguchi, K.; Wu, L.-W.; Grivennikov, S.I.; de Jong, P.R.; Lian, I.; Yu, F.-X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, N.; Zheng, Y.; de Wilde, R.F.; Maitra, A.; Pan, D. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev. 2010, 24, 2383–2388. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, R.; Tsuchiya, K.; Nemoto, Y.; Akiyama, J.; Nakamura, T.; Kanai, T.; Watanabe, M. Requirement of Notch activation during regeneration of the intestinal epithelia. Am. J. Physiol. Liver Physiol. 2009, 296, G23–G35. [Google Scholar]
- Wang, Q.; Wang, J.J.; Boyce, S.; Fischer, J.E.; Hasselgren, P.-O. Endotoxemia and IL-1β Stimulate Mucosal IL-6 Production in Different Parts of the Gastrointestinal Tract. J. Surg. Res. 1998, 76, 27–31. [Google Scholar] [PubMed]
- Parikh, A.A.; Salzman, A.L.; Fischer, J.E.; Szabo, C.; Hasselgren, P.-O. Interleukin-1β and interferon-γ regulate interleukin-6 production in cultured human intestinal epithelial cells. Shock 1997, 29, 531–537. [Google Scholar] [CrossRef]
- McGee, D.W.; Beagley, K.W.; Aicher, W.K.; McGhee, J.R. Transforming growth factor-beta and IL-1 beta act in synergy to enhance IL-6 secretion by the intestinal epithelial cell line, IEC-6. J. Immunol. 1993, 151, 970–978. [Google Scholar]
- Hershko, D.D.; Robb, B.W.; Luo, G.; Hasselgren, P.-O. Multiple transcription factors regulating the IL-6 gene are activated by cAMP in cultured Caco-2 cells. Am. J. Physiol. 2002, 283, R1140–R1148. [Google Scholar]
- Dumoutier, L.; Louahed, J.; Renauld, J.-C. Cloning and Characterization of IL-10-Related T Cell-Derived Inducible Factor (IL-TIF), a Novel Cytokine Structurally Related to IL-10 and Inducible by IL-9. J. Immunol. 2000, 164, 1814–1819. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Izotova, L.S.; Mirochnitchenko, O.V.; Esterova, E.; Dickensheets, H.; Donnelly, R.P.; Pestka, S. Identification of the Functional Interleukin-22 (IL-22) Receptor Complex: The il-10r2 chain (il-10rβ) is a common chain of both the il-10 and il-22 (il-10-related t cell-derived inducible factor, il-tif) receptor complexes. J. Biol. Chem. 2001, 276, 2725–2732. [Google Scholar] [CrossRef]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.-A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Stevens, S.; Flavell, R.A. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 2008, 29, 947–957. [Google Scholar] [CrossRef]
- Wolk, K.; Kunz, S.; Asadullah, K.; Sabat, R. Cutting Edge: Immune Cells as Sources and Targets of the IL-10 Family Members? J. Immunol. 2002, 168, 5397–5402. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Witte, E.; Hoffmann, U.; Doecke, W.-D.; Endesfelder, S.; Asadullah, K.; Sterry, W.; Volk, H.-D.; Wittig, B.M.; Sabat, R. IL-22 Induces Lipopolysaccharide-Binding Protein in Hepatocytes: A Potential Systemic Role of IL-22 in Crohn’s Disease. J. Immunol. 2007, 178, 5973–5981. [Google Scholar] [CrossRef] [PubMed]
- Schmechel, S.; Konrad, A.; Diegelmann, J.; Glas, J.; Wetzke, M.; Paschos, E.; Lohse, P.; Göke, B.; Brand, S. Linking genetic susceptibility to Crohn’s disease with Th17 cell function: IL-22 serum levels are increased in Crohn’s disease and correlate with disease activity and IL23R genotype status. Inflamm. Bowel Dis. 2007, 14, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Valdez, P.A.; Danilenko, D.M.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; de Sauvage, F.J.; et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008, 14, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Brand, S.; Beigel, F.; Olszak, T.; Zitzmann, K.; Eichhorst, S.T.; Otte, J.-M.; Diepolder, H.; Marquardt, A.; Jagla, W.; Popp, A.; et al. IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am. J. Physiol. Liver Physiol. 2006, 290, G827–G838. [Google Scholar] [CrossRef]
- Geng, H.; Bu, H.-F.; Liu, F.; Wu, L.; Pfeifer, K.; Chou, P.M.; Wang, X.; Sun, J.; Lu, L.; Pandey, A.; et al. In Inflamed Intestinal Tissues and Epithelial Cells, Interleukin 22 Signaling Increases Expression of H19 Long Noncoding RNA, Which Promotes Mucosal Regeneration. Gastroenterology 2018, 155, 144–155. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef]
- Pelczar, P.; Witkowski, M.; Perez, L.G.; Kempski, J.; Hammel, A.G.; Brockmann, L.; Kleinschmidt, D.; Wende, S.; Haueis, C.; Bedke, T.; et al. A pathogenic role for T cell–derived IL-22BP in inflammatory bowel disease. Science 2016, 354, 358–362. [Google Scholar] [CrossRef]
- Huber, S.; Gagliani, N.; Zenewicz, L.A.; Huber, F.J.; Bosurgi, L.; Hu, B.; Hedl, M.; Zhang, W.; O’Connor Jr, W.; Murphy, A.J.; et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012, 491, 259–263. [Google Scholar] [CrossRef]
- Monteleone, I.; Rizzo, A.; Sarra, M.; Sica, G.; Sileri, P.; Biancone, L.; MacDonald, T.T.; Pallone, F.; Monteleone, G. Aryl Hydrocarbon Receptor-Induced Signals Up-regulate IL-22 Production and Inhibit Inflammation in the Gastrointestinal Tract. Gastroenterology 2011, 141, 237–248. [Google Scholar] [CrossRef]
- Moriwaki, K.; Balaji, S.; McQuade, T.; Malhotra, N.; Kang, J.; Chan, F.K.-M. The Necroptosis Adaptor RIPK3 Promotes Injury-Induced Cytokine Expression and Tissue Repair. Immunity 2014, 41, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Allez, M.; Skolnick, B.E.; Wisniewska-Jarosinska, M.; Petryka, R.; Overgaard, R.V. Anti-NKG2D monoclonal antibody (NNC0142-0002) in active Crohn’s disease: A randomised controlled trial. Gut 2017, 66, 1918–1925. [Google Scholar] [CrossRef] [PubMed]
- Medina-Contreras, O.; Harusato, A.; Nishio, H.; Flannigan, K.L.; Ngo, V.; Leoni, G.; Neumann, P.-A.; Geem, D.; Lili, L.N.; Ramadas, R.A.; et al. Cutting Edge: IL-36 Receptor Promotes Resolution of Intestinal Damage. J. Immunol. 2016, 196, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Grainger, J.R.; Wohlfert, E.A.; Fuss, I.J.; Bouladoux, N.; Askenase, M.H.; Legrand, F.; Koo, L.Y.; Brenchley, J.M.; Fraser, I.D.C.; Belkaid, Y. Inflammatory monocytes regulate pathologic responses to commensals during acute gastrointestinal infection. Nat. Med. 2013, 19, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, X.; Falcon, D.M. The Role of Immune Cells and Cytokines in Intestinal Wound Healing. Int. J. Mol. Sci. 2019, 20, 6097. https://doi.org/10.3390/ijms20236097
Xue X, Falcon DM. The Role of Immune Cells and Cytokines in Intestinal Wound Healing. International Journal of Molecular Sciences. 2019; 20(23):6097. https://doi.org/10.3390/ijms20236097
Chicago/Turabian StyleXue, Xiang, and Daniel M. Falcon. 2019. "The Role of Immune Cells and Cytokines in Intestinal Wound Healing" International Journal of Molecular Sciences 20, no. 23: 6097. https://doi.org/10.3390/ijms20236097
APA StyleXue, X., & Falcon, D. M. (2019). The Role of Immune Cells and Cytokines in Intestinal Wound Healing. International Journal of Molecular Sciences, 20(23), 6097. https://doi.org/10.3390/ijms20236097