Molecular Characterization of XX Maleness



Abstract
1. Introduction
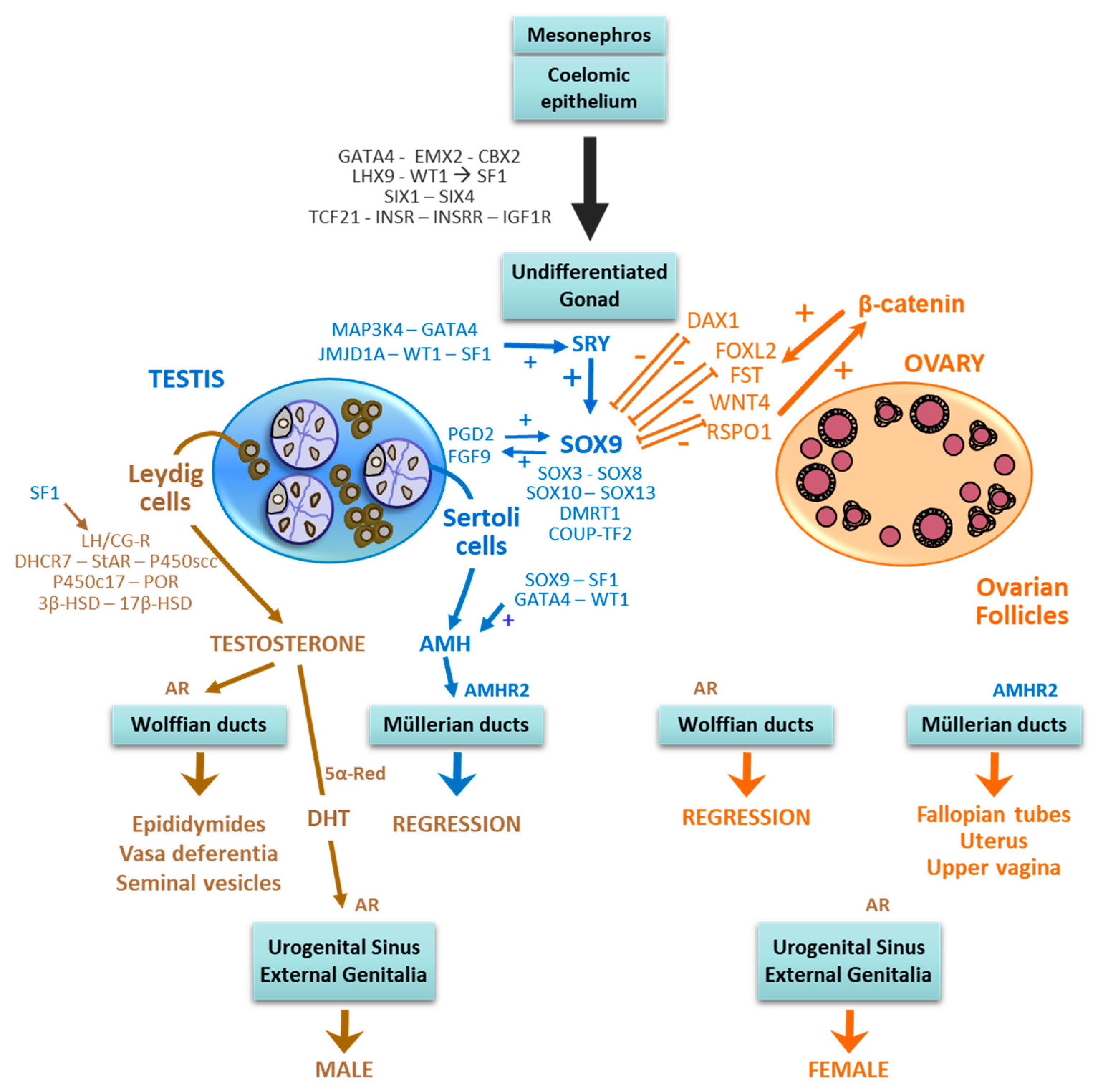
2. Molecular Mechanisms Underlying Foetal Sex Differentiation
2.1. The Sexually Undifferentiated Stage
2.2. Testicular Differentiation
2.3. Ovarian Differentiation
2.4. The Differentiation of the Internal and External Genitalia
3. Pathogenesis of XX Maleness
4. Clinical Aspects
4.1. Males
4.2. Ambiguous Genitalia
5. Genetic aspects
5.1. SRY-Positive
5.2. SRY-Negative with Increased Expression of Pro-Testicular Genes
5.2.1. SOX9
5.2.2. SOX3
5.2.3. SOX10
5.2.4. FGF9
5.2.5. DMRT1
5.3. SRY-Negative with Insufficient Expression of Pro-Ovarian Genes
5.3.1. WNT4
5.3.2. RSPO1
5.3.3. FOXL2
5.4. SRY-Negative with Mixed or Unknown Pathogenic Mechanisms
5.4.1. NR5A1 (SF1)
5.4.2. WT1
5.4.3. NR2F2 (COUP-TF2)
6. Concluding Remarks and Unresolved Questions
Author Contributions
Funding
Conflicts of Interest
References
- de la Chapelle, A.; Hortling, H.; Niemi, M.; Wennstroem, J. XX sex chromosomes in a human male. First case. Acta Med. Scand. 1964, 175, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Turcu, A.F.; Auchus, R.J. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol. Metab. Clin. N. Am. 2015, 44, 275–296. [Google Scholar] [CrossRef] [PubMed]
- Hakim, C.; Padmanabhan, V.; Vyas, A.K. Gestational Hyperandrogenism in Developmental Programming. Endocrinology 2017, 158, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Belgorosky, A.; Guercio, G.; Pepe, C.; Saraco, N.; Rivarola, M.A. Genetic and clinical spectrum of aromatase deficiency in infancy, childhood and adolescence. Horm. Res. 2009, 72, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Rey, R.A.; Grinspon, R.P. Normal male sexual differentiation and aetiology of disorders of sex development. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Baetens, D.; Verdin, H.; De Baere, E.; Cools, M. Update on the genetics of differences of sex development (DSD). Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101271. [Google Scholar] [CrossRef]
- Birk, O.S.; Casiano, D.E.; Wassif, C.A.; Cogliati, T.; Zhao, L.; Zhao, Y.; Grinberg, A.; Huang, S.; Kreidberg, J.A.; Parker, K.L.; et al. The LIM homeobox gene Lhx9 is essential for mouse gonad formation. Nature 2000, 403, 909–913. [Google Scholar] [CrossRef]
- Wilhelm, D.; Englert, C. The Wilms tumor suppressor WT1 regulates early gonad development by activation of Sf1. Genes Dev. 2002, 16, 1839–1851. [Google Scholar] [CrossRef]
- Rey, R.; Josso, N.; Racine, C. Sexual Differentiation. In Endotext; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2016. [Google Scholar]
- Hastie, N.D. Wilms’ tumour 1 (WT1) in development, homeostasis and disease. Development 2017, 144, 2862–2872. [Google Scholar] [CrossRef]
- Suntharalingham, J.P.; Buonocore, F.; Duncan, A.J.; Achermann, J.C. DAX-1 (NR0B1) and steroidogenic factor-1 (SF-1, NR5A1) in human disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 607–619. [Google Scholar] [CrossRef]
- Luo, X.; Ikeda, Y.; Parker, K.L. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell 1994, 77, 481–490. [Google Scholar] [CrossRef]
- Lin, Y.T.; Capel, B. Cell fate commitment during mammalian sex determination. Curr. Opin. Genet. Dev. 2015, 32, 144–152. [Google Scholar] [CrossRef]
- Larney, C.; Bailey, T.L.; Koopman, P. Switching on sex: Transcriptional regulation of the testis-determining gene Sry. Development 2014, 141, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Bullejos, M.; Koopman, P. Spatially dynamic expression of Sry in mouse genital ridges. Dev. Dyn. 2001, 221, 201–205. [Google Scholar] [CrossRef]
- Nagamine, C.M.; Morohashi, K.; Carlisle, C.; Chang, D.K. Sex reversal caused by Mus musculus domesticus Y chromosomes linked to variant expression of the testis-determining gene Sry. Dev. Biol. 1999, 216, 182–194. [Google Scholar] [CrossRef]
- Bullejos, M.; Koopman, P. Delayed Sry and Sox9 expression in developing mouse gonads underlies B6-YDOM sex reversal. Dev. Biol. 2005, 278, 473–481. [Google Scholar] [CrossRef]
- Palmer, S.J.; Burgoyne, P.S. In situ analysis of fetal, prepuberal and adult XX----XY chimaeric mouse testes: Sertoli cells are predominantly, but not exclusively, XY. Development 1991, 112, 265–268. [Google Scholar]
- Munger, S.C.; Natarajan, A.; Looger, L.L.; Ohler, U.; Capel, B. Fine time course expression analysis identifies cascades of activation and repression and maps a putative regulator of mammalian sex determination. PLoS Genet. 2013, 9, e1003630. [Google Scholar] [CrossRef]
- Quinn, A.; Koopman, P. The molecular genetics of sex determination and sex reversal in mammals. Semin. Reprod. Med. 2012, 30, 351–363. [Google Scholar] [CrossRef]
- Colvin, J.S.; Green, R.P.; Schmahl, J.; Capel, B.; Ornitz, D.M. Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell 2001, 104, 875–889. [Google Scholar] [CrossRef]
- Morais da Silva, S.; Hacker, A.; Harley, V.; Goodfellow, P.; Swain, A.; Lovell-Badge, R. Sox9 expression during gonadal development implies a conserved role for the gene in testis differentiation in mammals and birds. Nat. Genet. 1996, 14, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Josso, N.; Lamarre, I.; Picard, J.Y.; Berta, P.; Davies, N.; Morichon, N.; Peschanski, M.; Jeny, R. Anti-Müllerian hormone in early human development. Early Hum. Dev. 1993, 33, 91–99. [Google Scholar] [CrossRef]
- Josso, N.; Rey, R.A.; Picard, J.Y. Anti-müllerian hormone: A valuable addition to the toolbox of the pediatric endocrinologist. Int. J. Endocrinol. 2013, 2013, 674105. [Google Scholar] [CrossRef] [PubMed]
- Werner, R.; Merz, H.; Birnbaum, W.; Marshall, L.; Schroder, T.; Reiz, B.; Kavran, J.M.; Baumer, T.; Capetian, P.; Hiort, O. 46,XY Gonadal Dysgenesis due to a Homozygous Mutation in Desert Hedgehog (DHH) Identified by Exome Sequencing. J. Clin. Endocrinol. Metab. 2015, 100, E1022–E1029. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.H.; Whoriskey, W.; Capel, B. Desert Hedgehog/Patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev. 2002, 16, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Canto, P.; Soderlund, D.; Reyes, E.; Mendez, J.P. Mutations in the desert hedgehog (DHH) gene in patients with 46,XY complete pure gonadal dysgenesis. J. Clin. Endocrinol. Metab. 2004, 89, 4480–4483. [Google Scholar] [CrossRef]
- Bendsen, E.; Byskov, A.G.; Laursen, S.B.; Larsen, H.P.; Andersen, C.Y.; Westergaard, L.G. Number of germ cells and somatic cells in human fetal testes during the first weeks after sex differentiation. Hum. Reprod. 2003, 18, 13–18. [Google Scholar] [CrossRef]
- Cupp, A.S.; Uzumcu, M.; Skinner, M.K. Chemotactic Role of Neurotropin 3 in the Embryonic Testis That Facilitates Male Sex Determination. Biol. Reprod. 2003, 68, 2033–2037. [Google Scholar] [CrossRef]
- Cupp, A.S.; Tessarollo, L.; Skinner, M.K. Testis Developmental Phenotypes in Neurotropin Receptor trkA and trkC Null Mutations: Role in Formation of Seminiferous Cords and Germ Cell Survival. Biol. Reprod. 2002, 66, 1838–1845. [Google Scholar] [CrossRef][Green Version]
- Brennan, J.; Karl, J.; Capel, B. Divergent Vascular Mechanisms Downstream of Sry Establish the Arterial System in the XY Gonad. Dev. Biol. 2002, 244, 418–428. [Google Scholar] [CrossRef]
- Svingen, T.; Koopman, P. Building the mammalian testis: Origins, differentiation, and assembly of the component cell populations. Genes Dev. 2013, 27, 2409–2426. [Google Scholar] [CrossRef] [PubMed]
- Sekido, R.; Lovell-Badge, R. Genetic control of testis development. Sex Dev. 2013, 7, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Mayerhofer, A.; Lahr, G.; Seidl, K.; Eusterschulte, B.; Christoph, A.; Gratzl, M. The neural cell adhesion molecule (NCAM) provides clues to the development of testicular Leydig cells. J. Androl. 1996, 17, 223–230. [Google Scholar] [PubMed]
- DeFalco, T.; Takahashi, S.; Capel, B. Two distinct origins for Leydig cell progenitors in the fetal testis. Dev. Biol. 2011, 352, 14–26. [Google Scholar] [CrossRef]
- Swain, A. Sex Determination: Time for Meiosis? The Gonad Decides. Curr. Biol. 2006, 16, R507–R509. [Google Scholar] [CrossRef]
- Hu, L.; Monteiro, A.; Johnston, H.; King, P.; O’Shaughnessy, P.J. Expression of Cyp21a1 and Cyp11b1 in the fetal mouse testis. Reproduction 2007, 134, 585–591. [Google Scholar] [CrossRef]
- Brennan, J.; Tilmann, C.; Capel, B. Pdgfr-α mediates testis cord organization and fetal Leydig cell development in the XY gonad. Genes Dev. 2003, 17, 800–810. [Google Scholar] [CrossRef]
- Word, R.A.; George, F.W.; Wilson, J.D.; Carr, B.R. Testosterone synthesis and adenylate cyclase activity in the early human fetal testis appear to be independent of human chorionic gonadotropin control. J. Clin. Endocrinol. Metab. 1989, 69, 204–208. [Google Scholar] [CrossRef]
- Jonas, K.C.; Oduwole, O.O.; Peltoketo, H.; Rulli, S.B.; Huhtaniemi, I.T. Mouse models of altered gonadotrophin action: Insight into male reproductive disorders. Reproduction 2014, 148, R63–R70. [Google Scholar] [CrossRef][Green Version]
- Kremer, H.; Kraaij, R.; Toledo, S.P.; Post, M.; Fridman, J.B.; Hayashida, C.Y.; van Reen, M.; Milgrom, E.; Ropers, H.H.; Mariman, E. Male pseudohermaphroditism due to a homozygous missense mutation of the luteinizing hormone receptor gene. Nat. Genet. 1995, 9, 160–164. [Google Scholar] [CrossRef]
- Colvin, J.S.; White, A.C.; Pratt, S.J.; Ornitz, D.M. Lung hypoplasia and neonatal death in Fgf9-null mice identify this gene as an essential regulator of lung mesenchyme. Development 2001, 128, 2095–2106. [Google Scholar] [PubMed]
- Clark, A.M.; Garland, K.K.; Russell, L.D. Desert hedgehog (Dhh) gene is required in the mouse testis for formation of adult-type Leydig cells and normal development of peritubular cells and seminiferous tubules. Biol. Reprod. 2000, 63, 1825–1838. [Google Scholar] [CrossRef] [PubMed]
- Pannetier, M.; Chassot, A.A.; Chaboissier, M.C.; Pailhoux, E. Involvement of FOXL2 and RSPO1 in Ovarian Determination, Development, and Maintenance in Mammals. Sex. Dev. 2016, 10, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Chassot, A.A.; Gillot, I.; Chaboissier, M.C. R-spondin1, WNT4, and the CTNNB1 signaling pathway: Strict control over ovarian differentiation. Reproduction 2014, 148, R97–R110. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, S.; Megiorni, F.; Lin, L.; Mazzilli, M.C.; Gerrelli, D.; Majore, S.; Grammatico, P.; Achermann, J.C. Human RSPO1/R-spondin1 is expressed during early ovary development and augments beta-catenin signaling. PLoS ONE 2011, 6, e16366. [Google Scholar] [CrossRef] [PubMed]
- Ungewitter, E.K.; Yao, H.H. How to make a gonad: Cellular mechanisms governing formation of the testes and ovaries. Sex Dev. 2013, 7, 7–20. [Google Scholar] [CrossRef]
- Biason-Lauber, A.; Chaboissier, M.C. Ovarian development and disease: The known and the unexpected. Semin. Cell Dev. Biol. 2015, 45, 59–67. [Google Scholar] [CrossRef]
- Suzuki, H.; Kanai-Azuma, M.; Kanai, Y. From Sex Determination to Initial Folliculogenesis in Mammalian Ovaries: Morphogenetic Waves along the Anteroposterior and Dorsoventral Axes. Sex. Dev. 2015, 9, 190–204. [Google Scholar] [CrossRef]
- Carré, G.A.; Greenfield, A. Characterising novel pathways in testis determination using mouse genetics. Sex. Dev. 2014, 8, 199–207. [Google Scholar] [CrossRef]
- Heikkila, M.; Prunskaite, R.; Naillat, F.; Itaranta, P.; Vuoristo, J.; Leppaluoto, J.; Peltoketo, H.; Vainio, S. The partial female to male sex reversal in Wnt-4-deficient females involves induced expression of testosterone biosynthetic genes and testosterone production, and depends on androgen action. Endocrinology 2005, 146, 4016–4023. [Google Scholar] [CrossRef]
- Rastetter, R.H.; Bernard, P.; Palmer, J.S.; Chassot, A.A.; Chen, H.; Western, P.S.; Ramsay, R.G.; Chaboissier, M.C.; Wilhelm, D. Marker genes identify three somatic cell types in the fetal mouse ovary. Dev. Biol. 2014, 394, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Uhlenhaut, N.H.; Jakob, S.; Anlag, K.; Eisenberger, T.; Sekido, R.; Kress, J.; Treier, A.C.; Klugmann, C.; Klasen, C.; Holter, N.I.; et al. Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell 2009, 139, 1130–1142. [Google Scholar] [CrossRef] [PubMed]
- Edson, M.A.; Nagaraja, A.K.; Matzuk, M.M. The mammalian ovary from genesis to revelation. Endocr. Rev. 2009, 30, 624–712. [Google Scholar] [CrossRef] [PubMed]
- Hummitzsch, K.; Irving-Rodgers, H.F.; Hatzirodos, N.; Bonner, W.; Sabatier, L.; Reinhardt, D.P.; Sado, Y.; Ninomiya, Y.; Wilhelm, D.; Rodgers, R.J. A new model of development of the mammalian ovary and follicles. PLoS ONE 2013, 8, e55578. [Google Scholar] [CrossRef]
- Jost, A. Problems of fetal endocrinology: The gonadal and hypophyseal hormones. Recent Prog. Horm. Res. 1953, 8, 379–418. [Google Scholar]
- Mendonça, B.B.; Costa, E.M.; Belgorosky, A.; Rivarola, M.A.; Domenice, S. 46,XY DSD due to impaired androgen production. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 243–262. [Google Scholar] [CrossRef]
- Freire, A.V.; Grinspon, R.P.; Rey, R.A. Importance of Serum Testicular Protein Hormone Measurement in the Assessment of Disorders of Sex Development. Sex. Dev. 2018, 12, 30–40. [Google Scholar] [CrossRef]
- Josso, N. Anti-Mullerian hormone: A look back and ahead. Reproduction 2019. [Google Scholar] [CrossRef]
- Imbeaud, S.; Faure, E.; Lamarre, I.; Mattei, M.G.; di, C.N.; Tizard, R.; Carre-Eusebe, D.; Belville, C.; Tragethon, L.; Tonkin, C.; et al. Insensitivity to anti-mullerian hormone due to a mutation in the human anti-mullerian hormone receptor. Nat. Genet. 1995, 11, 382–388. [Google Scholar] [CrossRef]
- Moses, M.M.; Behringer, R.R. A gene regulatory network for Mullerian duct regression. Env. Epigenet 2019, 5, dvz017. [Google Scholar] [CrossRef]
- Auchus, R.J.; Miller, W.L. Defects in androgen biosynthesis causing 46,XY disorders of sexual development. Semin. Reprod. Med. 2012, 30, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.A.; Houk, C.P.; Ahmed, S.F.; Hughes, I.A. In collaboration with the participants in the International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. Consensus Statement on Management of Intersex Disorders. Pediatrics 2006, 118, e488–e500. [Google Scholar] [PubMed]
- de la Chapelle, A. Analytic review: Nature and origin of males with XX sex chromosomes. Am. J. Hum. Genet. 1972, 24, 71–105. [Google Scholar] [PubMed]
- Vorona, E.; Zitzmann, M.; Gromoll, J.; Schuring, A.N.; Nieschlag, E. Clinical, endocrinological, and epigenetic features of the 46,XX male syndrome, compared with 47,XXY Klinefelter patients. J. Clin. Endocrinol. Metab. 2007, 92, 3458–3465. [Google Scholar] [CrossRef]
- Aksglæde, L.; Skakkebæk, N.E.; Juul, A. Abnormal sex chromosome constitution and longitudinal growth: Serum levels of insulin-like growth factor (IGF)-I, IGF binding protein-3, luteinizing hormone, and testosterone in 109 males with 47,XXY, 47,XYY, or sex-determining region of the Y chromosome (SRY)-positive 46,XX karyotypes. J. Clin. Endocrinol. Metab. 2008, 93, 169–176. [Google Scholar] [CrossRef]
- Rey, R.A. Mini-puberty and true puberty: Differences in testicular function. Ann. Endocrinol. (Paris) 2014, 75, 58–63. [Google Scholar] [CrossRef]
- Boucekkine, C.; Toublanc, J.E.; Abbas, N.; Chaabouni, S.; Ouahid, S.; Semrouni, M.; Jaubert, F.; Toublanc, M.; McElreavey, K.; Vilain, E. Clinical and anatomical spectrum in XX sex reversed patients. Relationship to the presence of Y specific DNA-sequences. Clin. Endocrinol. 1994, 40, 733–742. [Google Scholar] [CrossRef]
- Rey, R.A.; Belville, C.; Nihoul-Fékété, C.; Michel-Calemard, L.; Forest, M.G.; Lahlou, N.; Jaubert, F.; Mowszowicz, I.; David, M.; Saka, N.; et al. Evaluation of gonadal function in 107 intersex patients by means of serum antimüllerian hormone measurement. J. Clin. Endocrinol. Metab. 1999, 84, 627–631. [Google Scholar] [CrossRef]
- Huang, W.J.; Yen, P.H. Genetics of spermatogenic failure. Sex. Dev. 2008, 2, 251–259. [Google Scholar] [CrossRef]
- Aksglæde, L.; Jorgensen, N.; Skakkebæk, N.E.; Juul, A. Low semen volume in 47 adolescents and adults with 47,XXY Klinefelter or 46,XX male syndrome. Int. J. Androl. 2009, 32, 376–384. [Google Scholar] [CrossRef]
- Maciel-Guerra, A.T.; de Mello, M.P.; Coeli, F.B.; Ribeiro, M.L.; Miranda, M.L.; Marques-de-Faria, A.P.; Baptista, M.T.; Moraes, S.G.; Guerra-Junior, G. XX Maleness and XX true hermaphroditism in SRY-negative monozygotic twins: Additional evidence for a common origin. J. Clin. Endocrinol. Metab. 2008, 93, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.A.; Nordenstrom, A.; Houk, C.P.; Ahmed, S.F.; Auchus, R.; Baratz, A.; Baratz Dalke, K.; Liao, L.M.; Lin-Su, K.; Looijenga 3rd, L.H.; et al. Global Disorders of Sex Development Update since 2006: Perceptions, Approach and Care. Horm. Res. Paediatr. 2016, 85, 158–180. [Google Scholar] [CrossRef] [PubMed]
- Guercio, G.; Rey, R.A. Fertility issues in the management of patients with disorders of sex development. Endocr. Dev. 2014, 27, 87–98. [Google Scholar] [CrossRef]
- Dutta, D.; Shivaprasad, K.S.; Das, R.N.; Ghosh, S.; Chatterjee, U.; Chowdhury, S.; Dasgupta, R. Ovotesticular disorder of sexual development due to 47,XYY/46,XY/45,X mixed gonadal dysgenesis in a phenotypic male presenting as cyclical haematuria: Clinical presentation and assessment of long-term outcomes. Andrologia 2014, 46, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Cools, M.; Looijenga, L.H.; Wolffenbuttel, K.P.; T’Sjoen, G. Managing the risk of germ cell tumourigenesis in disorders of sex development patients. Endocr. Dev. 2014, 27, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Verkauskas, G.; Jaubert, F.; Lortat-Jacob, S.; Malan, V.; Thibaud, E.; Nihoul-Fekete, C. The long-term followup of 33 cases of true hermaphroditism: A 40-year experience with conservative gonadal surgery. J. Urol. 2007, 177, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, R.; Ramdial, P.K. The gonads of 111 South African patients with ovotesticular disorder of sex differentiation. J. Pediatr. Surg. 2009, 44, 556–560. [Google Scholar] [CrossRef]
- Matsui, F.; Shimada, K.; Matsumoto, F.; Itesako, T.; Nara, K.; Ida, S.; Nakayama, M. Long-term outcome of ovotesticular disorder of sex development: A single center experience. Int. J. Urol. 2011, 18, 231–236. [Google Scholar] [CrossRef]
- Sinclair, A.H.; Berta, P.; Palmer, M.S.; Hawkins, J.R.; Griffiths, B.L.; Smith, M.J.; Foster, J.W.; Frischauf, A.M.; Lovell-Badge, R.; Goodfellow, P.N. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 1990, 346, 240–244. [Google Scholar] [CrossRef]
- McElreavey, K.; Rappaport, R.; Vilain, E.; Abbas, N.; Richaud, F.; Lortat-Jacob, S.; Berger, R.; Le Coniat, M.; Boucekkine, C.; Kucheria, K.; et al. A minority of 46,XX true hermaphrodites are positive for the Y-DNA sequence including SRY. Hum. Genet. 1992, 90, 121–125. [Google Scholar] [CrossRef]
- McElreavey, K.; Vilain, E.; Abbas, N.; Herskowitz, I.; Fellous, M. A regulatory cascade hypothesis for mammalian sex determination: SRY represses a negative regulator of male development. Proc. Natl. Acad. Sci. USA 1993, 90, 3368–3372. [Google Scholar] [CrossRef] [PubMed]
- Ortenberg, J.; Oddoux, C.; Craver, R.; McElreavey, K.; Salas-Cortes, L.; Guillen-Navarro, E.; Ostrer, H.; Sarafoglou, K.; Clarke, V.; Yee, H. SRY gene expression in the ovotestes of XX true hermaphrodites. J. Urol. 2002, 167, 1828–1831. [Google Scholar] [CrossRef]
- Lefebvre, V.; Dumitriu, B.; Penzo-Mendez, A.; Han, Y.; Pallavi, B. Control of cell fate and differentiation by Sry-related high-mobility-group box (Sox) transcription factors. Int. J. Biochem. Cell Biol. 2007, 39, 2195–2214. [Google Scholar] [CrossRef] [PubMed]
- Gonen, N.; Lovell-Badge, R. The regulation of Sox9 expression in the gonad. Curr. Top. Dev. Biol. 2019, 134, 223–252. [Google Scholar] [CrossRef]
- Eggers, S.; Ohnesorg, T.; Sinclair, A. Genetic regulation of mammalian gonad development. Nat. Rev. Endocrinol. 2014, 10, 673–683. [Google Scholar] [CrossRef]
- de Santa Barbara, P.; Bonneaud, N.; Boizet, B.; Desclozeaux, M.; Moniot, B.; Sudbeck, P.; Scherer, G.; Poulat, F.; Berta, P. Direct interaction of SRY-related protein SOX9 and steroidogenic factor 1 regulates transcription of the human anti-Müllerian hormone gene. Mol. Cell. Biol. 1998, 18, 6653–6665. [Google Scholar] [CrossRef]
- Arango, N.A.; Lovell-Badge, R.; Behringer, R.R. Targeted mutagenesis of the endogenous mouse Mis gene promoter: In vivo definition of genetic pathways of vertebrate sexual development. Cell 1999, 99, 409–419. [Google Scholar] [CrossRef]
- Wagner, T.; Wirth, J.; Meyer, J.; Zabel, B.; Held, M.; Zimmer, J.; Pasantes, J.; Bricarelli, F.D.; Keutel, J.; Hustert, E. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell 1994, 79, 1111–1120. [Google Scholar] [CrossRef]
- Foster, J.W.; Dominguez-Steglich, M.A.; Guioli, S.; Kowk, G.; Weller, P.A.; Stevanoviç, M.; Weissenbach, J.; Mansour, S.; Young, I.D.; Goodfellow, P.N. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature 1994, 372, 525–530. [Google Scholar] [CrossRef]
- Vidal, V.P.; Chaboissier, M.C.; de Rooij, D.G.; Schedl, A. Sox9 induces testis development in XX transgenic mice. Nat. Genet. 2001, 28, 216–217. [Google Scholar] [CrossRef]
- Huang, B.; Wang, S.B.; Ning, Y.; Lamb, A.N.; Bartley, J. Autosomal XX sex reversal caused by duplication of SOX9. Am. J. Med. Genet. 1999, 87, 349–353. [Google Scholar] [CrossRef]
- Lee, G.M.; Ko, J.M.; Shin, C.H.; Yang, S.W. A Korean boy with 46,XX testicular disorder of sex development caused by SOX9 duplication. Ann. Pediatr. Endocrinol. Metab. 2014, 19, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.J.; Willatt, L.; Homfray, T.; Woods, C.G. A SOX9 Duplication and Familial 46,XX Developmental Testicular Disorder. N. Engl. J. Med. 2011, 364, 91–93. [Google Scholar] [CrossRef]
- Vetro, A.; Ciccone, R.; Giorda, R.; Patricelli, M.G.; Della, M.E.; Forlino, A.; Zuffardi, O. XX males SRY negative: A confirmed cause of infertility. J. Med. Genet. 2011, 48, 710–712. [Google Scholar] [CrossRef] [PubMed]
- Vetro, A.; Dehghani, M.R.; Kraoua, L.; Giorda, R.; Beri, S.; Cardarelli, L.; Merico, M.; Manolakos, E.; Parada-Bustamante, A.; Castro, A.; et al. Testis development in the absence of SRY: Chromosomal rearrangements at SOX9 and SOX3. Eur. J. Hum. Genet. 2015, 23, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.J.; Sock, E.; Buchberger, A.; Just, W.; Denzer, F.; Hoepffner, W.; German, J.; Cole, T.; Mann, J.; Seguin, J.H.; et al. Copy number variation of two separate regulatory regions upstream of SOX9 causes isolated 46,XY or 46,XX disorder of sex development. J. Med. Genet. 2015, 52, 240–247. [Google Scholar] [CrossRef]
- Refai, O.; Friedman, A.; Terry, L.; Jewett, T.; Pearlman, A.; Perle, M.A.; Ostrer, H. De novo 12;17 translocation upstream of SOX9 resulting in 46,XX testicular disorder of sex development. Am. J. Med. Genet. A 2010, 152, 422–426. [Google Scholar] [CrossRef]
- Benko, S.; Gordon, C.T.; Mallet, D.; Sreenivasan, R.; Thauvin-Robinet, C.; Brendehaug, A.; Thomas, S.; Bruland, O.; David, M.; Nicolino, M.; et al. Disruption of a long distance regulatory region upstream of SOX9 in isolated disorders of sex development. J. Med. Genet. 2011, 48, 825–830. [Google Scholar] [CrossRef]
- Croft, B.; Ohnesorg, T.; Hewitt, J.; Bowles, J.; Quinn, A.; Tan, J.; Corbin, V.; Pelosi, E.; van den Bergen, J.; Sreenivasan, R.; et al. Human sex reversal is caused by duplication or deletion of core enhancers upstream of SOX9. Nat. Commun. 2018, 9, 5319. [Google Scholar] [CrossRef]
- Hyon, C.; Chantot-Bastaraud, S.; Harbuz, R.; Bhouri, R.; Perrot, N.; Peycelon, M.; Sibony, M.; Rojo, S.; Piguel, X.; Bilan, F.; et al. Refining the regulatory region upstream of SOX9 associated with 46,XX testicular disorders of Sex Development (DSD). Am. J. Med. Genet. A 2015, 167, 1851–1858. [Google Scholar] [CrossRef]
- López-Hernández, B.; Méndez, J.P.; Coral-Vázquez, R.M.; Benitez-Granados, J.; Zenteno, J.C.; Villegas-Ruiz, V.; Calzada-León, R.; Soderlund, D.; Canto, P. Duplication of SOX9 associated with 46,XX ovotesticular disorder of sex development. Reprod. Biomed. Online 2018, 37, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Shankara Narayana, N.; Kean, A.M.; Ewans, L.; Ohnesorg, T.; Ayers, K.L.; Watson, G.; Vasilaras, A.; Sinclair, A.H.; Twigg, S.M.; Handelsman, D.J. Painful ovulation in a 46,XX SRY -ve adult male with SOX9 duplication. Endocrinol. Diabetes Metab. Case Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ohnesorg, T.; van den Bergen, J.A.; Belluoccio, D.; Shankara-Narayana, N.; Kean, A.M.; Vasilaras, A.; Ewans, L.; Ayers, K.L.; Sinclair, A.H. A duplication in a patient with 46,XX ovo-testicular disorder of sex development refines the SOX9 testis-specific regulatory region to 24 kb. Clin. Genet. 2017, 92, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Ji, X.; Xing, Y.; Chen, Y.W.; Tao, J. A rare case of 46, XX SRY-negative male with approximately 74-kb duplication in a region upstream of SOX9. Eur. J. Med. Genet. 2013, 56, 695–698. [Google Scholar] [CrossRef]
- Croft, B.; Ohnesorg, T.; Sinclair, A.H. The Role of Copy Number Variants in Disorders of Sex Development. Sex. Dev. 2018, 12, 19–29. [Google Scholar] [CrossRef]
- Symon, A.; Harley, V. SOX9: A genomic view of tissue specific expression and action. Int. J. Biochem. Cell Biol. 2017, 87, 18–22. [Google Scholar] [CrossRef]
- Weiss, J.; Meeks, J.J.; Hurley, L.; Raverot, G.; Frassetto, A.; Jameson, J.L. Sox3 Is Required for Gonadal Function, but Not Sex Determination, in Males and Females. Mol. Cell. Biol. 2003, 23, 8084–8091. [Google Scholar] [CrossRef]
- Sutton, E.; Hughes, J.; White, S.; Sekido, R.; Tan, J.; Arboleda, V.; Rogers, N.; Knower, K.; Rowley, L.; Eyre, H.; et al. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J. Clin. Investig. 2011, 121, 328–341. [Google Scholar] [CrossRef]
- Grinspon, R.P.; Nevado, J.; Mori Alvarez, M.L.; del Rey, G.; Castera, R.; Venara, M.; Chiesa, A.; Podestá, M.; Lapunzina, P.; Rey, R.A. 46,XX ovotesticular DSD associated with a SOX3 gene duplication in a SRY-negative boy. Clin. Endocrinol. 2016, 85, 669–675. [Google Scholar] [CrossRef]
- Moalem, S.; Babul-Hirji, R.; Stavropolous, D.J.; Wherrett, D.; Bagli, D.J.; Thomas, P.; Chitayat, D. XX male sex reversal with genital abnormalities associated with a de novo SOX3 gene duplication. Am. J. Med. Genet. A 2012, 158, 1759–1764. [Google Scholar] [CrossRef]
- Mizuno, K.; Kojima, Y.; Kamisawa, H.; Moritoki, Y.; Nishio, H.; Nakane, A.; Kurokawa, S.; Kohri, K.; Hayashi, Y. Elucidation of distinctive genomic DNA structures in patients with 46,XX testicular disorders of sex development using genome wide analyses. J. Urol. 2014, 192, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Haines, B.; Hughes, J.; Corbett, M.; Shaw, M.; Innes, J.; Patel, L.; Gecz, J.; Clayton-Smith, J.; Thomas, P. Interchromosomal insertional translocation at Xq26.3 alters SOX3 expression in an individual with XX male sex reversal. J. Clin. Endocrinol. Metab. 2015, 100, E815–E820. [Google Scholar] [CrossRef] [PubMed]
- Aleck, K.A.; Argueso, L.; Stone, J.; Hackel, J.G.; Erickson, R.P. True hermaphroditism with partial duplication of chromosome 22 and without SRY. Am. J. Med. Genet. 1999, 85, 2–4. [Google Scholar] [CrossRef]
- Nicholl, R.M.; Grimsley, L.; Butler, L.; Palmer, R.W.; Rees, H.C.; Savage, M.O.; Costeloe, K. Trisomy 22 and intersex. Arch. Dis. Child. Fetal Neonatal Ed. 1994, 71, F57–F58. [Google Scholar] [CrossRef] [PubMed]
- Seeherunvong, T.; Perera, E.M.; Bao, Y.; Benke, P.J.; Benigno, A.; Donahue, R.P.; Berkovitz, G.D. 46,XX sex reversal with partial duplication of chromosome arm 22q. Am. J. Med. Genet. 2004, 127, 149–151. [Google Scholar] [CrossRef]
- Falah, N.; Posey, J.E.; Thorson, W.; Benke, P.; Tekin, M.; Tarshish, B.; Lupski, J.R.; Harel, T. 22q11.2q13 duplication including SOX10 causes sex-reversal and peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome, and Hirschsprung disease. Am. J. Med. Genet. A 2017, 173, 1066–1070. [Google Scholar] [CrossRef]
- Chiang, H.S.; Wu, Y.N.; Wu, C.C.; Hwang, J.L. Cytogenic and molecular analyses of 46,XX male syndrome with clinical comparison to other groups with testicular azoospermia of genetic origin. J. Formos. Med. Assoc. 2013, 112, 72–78. [Google Scholar] [CrossRef]
- Igarashi, M.; Mikami, H.; Katsumi, M.; Miyado, M.; Izumi, Y.; Ogata, T.; Fukami, M. SOX3 Overdosage Permits Normal Sex Development in Females with Random X Inactivation. Sex. Dev. 2015, 9, 125–129. [Google Scholar] [CrossRef]
- Pingault, V.; Bodereau, V.; Baral, V.; Marcos, S.; Watanabe, Y.; Chaoui, A.; Fouveaut, C.; Leroy, C.; Verier-Mine, O.; Francannet, C.; et al. Loss-of-function mutations in SOX10 cause Kallmann syndrome with deafness. Am. J. Hum. Genet. 2013, 92, 707–724. [Google Scholar] [CrossRef]
- Polanco, J.C.; Wilhelm, D.; Davidson, T.L.; Knight, D.; Koopman, P. Sox10 gain-of-function causes XX sex reversal in mice: Implications for human 22q-linked disorders of sex development. Hum. Mol. Genet. 2010, 19, 506–516. [Google Scholar] [CrossRef]
- Brunner, B.; Hornung, U.; Shan, Z.; Nanda, I.; Kondo, M.; Zend-Ajusch, E.; Haaf, T.; Ropers, H.H.; Shima, A.; Schmid, M.; et al. Genomic organization and expression of the doublesex-related gene cluster in vertebrates and detection of putative regulatory regions for DMRT1. Genomics 2001, 77, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, R.E.; Gearhart, M.D.; Minkina, A.; Krentz, A.D.; Bardwell, V.J.; Zarkower, D. Sexual cell-fate reprogramming in the ovary by DMRT1. Curr. Biol. 2015, 25, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Matson, C.K.; Zarkower, D. Sex and the singular DM domain: Insights into sexual regulation, evolution and plasticity. Nat. Rev. Genet. 2012, 13, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Svingen, T.; Ng, E.T.; Koopman, P. Female-to-male sex reversal in mice caused by transgenic overexpression of Dmrt1. Development 2015, 142, 1083–1088. [Google Scholar] [CrossRef] [PubMed]
- Vainio, S.; Heikkila, M.; Kispert, A.; Chin, N.; McMahon, A.P. Female development in mammals is regulated by Wnt-4 signalling. Nature 1999, 397, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.K.; Mohammed, M.; Ching, S.T.; Delot, E.; Chen, X.N.; Dewing, P.; Swain, A.; Rao, P.N.; Elejalde, B.R.; Vilain, E. Up-regulation of WNT-4 signaling and dosage-sensitive sex reversal in humans. Am. J. Hum. Genet. 2001, 68, 1102–1109. [Google Scholar] [CrossRef]
- Biason-Lauber, A.; Konrad, D.; Navratil, F.; Schoenle, E.J. A WNT4 Mutation Associated with Mullerian-Duct Regression and Virilization in a 46,XX Woman. N. Engl. J. Med. 2004, 351, 792–798. [Google Scholar] [CrossRef]
- Biason-Lauber, A.; De Filippo, G.; Konrad, D.; Scarano, G.; Nazzaro, A.; Schoenle, E.J. WNT4 deficiency—A clinical phenotype distinct from the classic Mayer-Rokitansky-Kuster-Hauser syndrome: A case report. Hum. Reprod. 2007, 22, 224–229. [Google Scholar] [CrossRef]
- Philibert, P.; Biason-Lauber, A.; Rouzier, R.; Pienkowski, C.; Paris, F.; Konrad, D.; Schoenle, E.; Sultan, C. Identification and Functional Analysis of a New WNT4 Gene Mutation among 28 Adolescent Girls with Primary Amenorrhea and Mullerian Duct Abnormalities: A French Collaborative Study. J. Clin. Endocrinol. Metab. 2008, 93, 895–900. [Google Scholar] [CrossRef]
- Mandel, H.; Shemer, R.; Borochowitz, Z.U.; Okopnik, M.; Knopf, C.; Indelman, M.; Drugan, A.; Tiosano, D.; Gershoni-Baruch, R.; Choder, M.; et al. SERKAL syndrome: An autosomal-recessive disorder caused by a loss-of-function mutation in WNT4. Am. J. Hum. Genet. 2008, 82, 39–47. [Google Scholar] [CrossRef]
- Chassot, A.A.; Gregoire, E.P.; Magliano, M.; Lavery, R.; Chaboissier, M.C. Genetics of ovarian differentiation: Rspo1, a major player. Sex. Dev. 2008, 2, 219–227. [Google Scholar] [CrossRef]
- Chassot, A.A.; Ranc, F.; Gregoire, E.P.; Roepers-Gajadien, H.L.; Taketo, M.M.; Camerino, G.; de Rooij, D.G.; Schedl, A.; Chaboissier, M.C. Activation of beta-catenin signaling by Rspo1 controls differentiation of the mammalian ovary. Hum. Mol. Genet. 2008, 17, 1264–1277. [Google Scholar] [CrossRef] [PubMed]
- Parma, P.; Radi, O.; Vidal, V.; Chaboissier, M.C.; Dellambra, E.; Valentini, S.; Guerra, L.; Schedl, A.; Camerino, G. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat. Genet. 2006, 38, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Micali, G.; Nasca, M.R.; Innocenzi, D.; Frasin, L.A.; Radi, O.; Parma, P.; Camerino, G.; Schwartz, R.A. Association of palmoplantar keratoderma, cutaneous squamous cell carcinoma, dental anomalies, and hypogenitalism in four siblings with 46,XX karyotype: A new syndrome. J. Am. Acad. Derm. 2005, 53, S234–S239. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, S.; Megiorni, F.; De Bernardo, C.; Felici, A.; Marrocco, G.; Maggiulli, G.; Grammatico, B.; Remotti, D.; Saccucci, P.; Valentini, F.; et al. Syndromic true hermaphroditism due to an R-spondin1 (RSPO1) homozygous mutation. Hum. Mutat. 2008, 29, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Tallapaka, K.; Venugopal, V.; Dalal, A.; Aggarwal, S. Novel RSPO1 mutation causing 46,XX testicular disorder of sex development with palmoplantar keratoderma: A review of literature and expansion of clinical phenotype. Am. J. Med. Genet. A 2018, 176, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Naasse, Y.; Bakhchane, A.; Charoute, H.; Jennane, F.; Bignon-Topalovic, J.; Malki, A.; Bashamboo, A.; Barakat, A.; Rouba, H.; McElreavey, K. A Novel Homozygous Missense Mutation in the FU-CRD2 Domain of the R-spondin1 Gene Associated with Familial 46,XX DSD. Sex. Dev. 2017, 11, 269–274. [Google Scholar] [CrossRef]
- Radi, O.; Parma, P.; Imbeaud, S.; Nasca, M.R.; Uccellatore, F.; Maraschio, P.; Tiepolo, L.; Micali, G.; Camerino, G. XX sex reversal, palmoplantar keratoderma, and predisposition to squamous cell carcinoma: Genetic analysis in one family. Am. J. Med. Genet. Part A 2005, 138, 241–246. [Google Scholar] [CrossRef]
- Boulanger, L.; Pannetier, M.; Gall, L.; Allais-Bonnet, A.; Elzaiat, M.; Le Bourhis, D.; Daniel, N.; Richard, C.; Cotinot, C.; Ghyselinck, N.B.; et al. FOXL2 is a female sex-determining gene in the goat. Curr. Biol. 2014, 24, 404–408. [Google Scholar] [CrossRef]
- Ottolenghi, C.; Pelosi, E.; Tran, J.; Colombino, M.; Douglass, E.; Nedorezov, T.; Cao, A.; Forabosco, A.; Schlessinger, D. Loss of Wnt4 and Foxl2 leads to female-to-male sex reversal extending to germ cells. Hum. Mol. Genet. 2007, 16, 2795–2804. [Google Scholar] [CrossRef]
- Crisponi, L.; Deiana, M.; Loi, A.; Chiappe, F.; Uda, M.; Amati, P.; Bisceglia, L.; Zelante, L.; Nagaraja, R.; Porcu, S.; et al. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat. Genet. 2001, 27, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Domenice, S.; Machado, A.Z.; Ferreira, F.M.; Ferraz-de-Souza, B.; Lerario, A.M.; Lin, L.; Nishi, M.Y.; Gomes, N.L.; da Silva, T.E.; Silva, R.B.; et al. Wide spectrum of NR5A1-related phenotypes in 46,XY and 46,XX individuals. Birth Defects Res. C Embryo Today 2016, 108, 309–320. [Google Scholar] [CrossRef]
- Baetens, D.; Stoop, H.; Peelman, F.; Todeschini, A.L.; Rosseel, T.; Coppieters, F.; Veitia, R.A.; Looijenga, L.H.; De Baere, E.; Cools, M. NR5A1 is a novel disease gene for 46,XX testicular and ovotesticular disorders of sex development. Genet. Med. 2017, 19, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Bashamboo, A.; Donohoue, P.A.; Vilain, E.; Rojo, S.; Calvel, P.; Seneviratne, S.N.; Buonocore, F.; Barseghyan, H.; Bingham, N.; Rosenfeld, J.A.; et al. A recurrent p.Arg92Trp variant in steroidogenic factor-1 (NR5A1) can act as a molecular switch in human sex development. Hum. Mol. Genet. 2016, 25, 3446–3453. [Google Scholar] [CrossRef] [PubMed]
- Miyado, M.; Inui, M.; Igarashi, M.; Katoh-Fukui, Y.; Takasawa, K.; Hakoda, A.; Kanno, J.; Kashimada, K.; Miyado, K.; Tamano, M.; et al. The p.R92W variant of NR5A1/Nr5a1 induces testicular development of 46,XX gonads in humans, but not in mice: Phenotypic comparison of human patients and mutation-induced mice. Biol. Sex Differ. 2016, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Takasawa, K.; Hakoda, A.; Kanno, J.; Takada, S.; Miyado, M.; Baba, T.; Morohashi, K.I.; Tajima, T.; Hata, K.; et al. Identical NR5A1 Missense Mutations in Two Unrelated 46,XX Individuals with Testicular Tissues. Hum. Mutat. 2017, 38, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Takasawa, K.; Igarashi, M.; Ono, M.; Takemoto, A.; Takada, S.; Yamataka, A.; Ogata, T.; Morio, T.; Fukami, M.; Kashimada, K. Phenotypic Variation in 46,XX Disorders of Sex Development due to the NR5A1 p.R92W Variant: A Sibling Case Report and Literature Review. Sex. Dev. 2017, 11, 284–288. [Google Scholar] [CrossRef]
- Knarston, I.M.; Robevska, G.; van den Bergen, J.A.; Eggers, S.; Croft, B.; Yates, J.; Hersmus, R.; Looijenga, L.H.J.; Cameron, F.J.; Monhike, K.; et al. NR5A1 gene variants repress the ovarian-specific WNT signaling pathway in 46,XX disorders of sex development patients. Hum. Mutat. 2019, 40, 207–216. [Google Scholar] [CrossRef]
- Saito-Hakoda, A.; Kanno, J.; Suzuki, D.; Kawashima, S.; Kamimura, M.; Hirano, K.; Sakai, K.; Igarashi, M.; Fukami, M.; Fujiwara, I. A Follow-Up from Infancy to Puberty in a Japanese Male with SRY-Negative 46,XX Testicular Disorder of Sex Development Carrying a p.Arg92Trp Mutation in NR5A1. Sex. Dev. 2019, 13, 60–66. [Google Scholar] [CrossRef]
- Bashamboo, A.; Eozenou, C.; Rojo, S.; McElreavey, K. Anomalies in human sex determination provide unique insights into the complex genetic interactions of early gonad development. Clin. Genet. 2017, 91, 143–156. [Google Scholar] [CrossRef]
- Swartz, J.M.; Ciarlo, R.; Guo, M.H.; Abrha, A.; Weaver, B.; Diamond, D.A.; Chan, Y.M.; Hirschhorn, J.N. A 46,XX Ovotesticular Disorder of Sex Development Likely Caused by a Steroidogenic Factor-1 (NR5A1) Variant. Horm. Res. Paediatr. 2017, 87, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Niaudet, P.; Gubler, M.C. WT1 and glomerular diseases. Pediatr. Nephrol. 2006, 21, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.L.; de Paula, L.C.P.; Silva, J.M.; Silva, T.E.; Lerario, A.M.; Nishi, M.Y.; Batista, R.L.; Faria Junior, J.A.D.; Moraes, D.; Costa, E.M.F.; et al. A 46,XX testicular disorder of sex development caused by a Wilms’ tumour Factor-1 (WT1) pathogenic variant. Clin. Genet. 2019, 95, 172–176. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, L.; Cui, X.; Lin, X.; Li, Y.; Wang, Y.; Wang, Y.; Qin, Y.; Chen, D.; Han, C.; et al. Wt1 directs the lineage specification of sertoli and granulosa cells by repressing Sf1 expression. Development 2017, 144, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Al Turki, S.; Manickaraj, A.K.; Mercer, C.L.; Gerety, S.S.; Hitz, M.P.; Lindsay, S.; D’Alessandro, L.C.; Swaminathan, G.J.; Bentham, J.; Arndt, A.K.; et al. Rare variants in NR2F2 cause congenital heart defects in humans. Am. J. Hum. Genet. 2014, 94, 574–585. [Google Scholar] [CrossRef]
- Bashamboo, A.; Eozenou, C.; Jorgensen, A.; Bignon-Topalovic, J.; Siffroi, J.P.; Hyon, C.; Tar, A.; Nagy, P.; Solyom, J.; Halasz, Z.; et al. Loss of Function of the Nuclear Receptor NR2F2, Encoding COUP-TF2, Causes Testis Development and Cardiac Defects in 46,XX Children. Am. J. Hum. Genet. 2018, 102, 487–493. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Genetics | Ref. | Genitalia | Gonads | Molecular Findings |
|---|---|---|---|---|
| Duplication of SOX9 gene | [92] | Ambiguous | Scrotal Histol. NA | ~8-cM duplication dup(17)(q23.1q24.3) involving the entire SOX9 gene |
| [93] | Male | Scrotal Histol. NA | Duplication extends at least 5 kb upstream and downstream of SOX9 | |
| Duplication/Triplication of SOX9 regulatory sequences | [94] | Male | Scrotal Testes | 178-kb duplication 600 kb upstream of SOX9 |
| [95] | Male | Scrotal Testes | 96-kb triplication 514 kb upstream of SOX9 | |
| [96] | Pt 1: Male Pt 2: Ambiguous Pt 3: Male | Pt 1: Histol. NA Pt 2: Scrotal. Bilateral ovotestes Pt 3: Histol. NA | Pt 1: 364-kb duplication 294–658 kb upstream of SOX9 Pt 2: 141-kb duplication 31–572 kb upstream of SOX9 Pt 3: Reciprocal translocation t(11;17)(p13;q24.3). | |
| [97] | Ambiguous | Ovotestes/Testes | Pt 1: 143-kb duplication 516–659 kb upstream of SOX9 Pt 2: 444-kb duplication 259–703 kb upstream of SOX9 Pt 3: At least 480-kb duplication 264–744 kb upstream of SOX9 | |
| [98] | Ambiguous | One scrotal testis Histol. NA | Translocation t(12;17)(q14.3;q24.3) involving regulatory elements of pseudogene LOC204010 or of gene Deynar 776–811 kb upstream of SOX9 | |
| [99] | Ambiguous | Pt 1. Scrotal, Histol. NA Pt 2: Scrotal testis and abdominal ovary Pt 3: Scrotal ovotestis and abdominal streak | Pt 1: 605–694-kb duplication 353 kb upstream of SOX9 Pt 2: 148-kb duplication 447–595 kb upstream of SOX9 Pt3. 762–780-kb duplication 508 kb upstream of SOX9 | |
| [100] | NA | Pt 1: Bilateral testes Pt 2: Ovotestes | Duplicated minimum critical region of 5.2 kb located ~600 kb upstream of SOX9, common to Pt 1 and Pt 2 | |
| [101] | Male | Pt 1 and Pt 2: Testicular dysgenesis Pt 3: Histol. NA | Duplicated minimum critical region of ~41 kb located ~600 kb upstream of SOX9, common to all 3 patients | |
| [102] | Ambiguous | Ovary and ovotestis | Duplication from −581 kb upstream of to 4.4 kb coding sequence of SOX9 | |
| [103,104] | Male | Left Scrotal Ovotestis Right ectopic gonad (Histol. NA) | Duplicated minimum critical region of 24 kb located ~600 kb upstream of SOX9 | |
| [105] | Hypospadias | Scrotal Histol. NA | 74-kb duplication 510–584 kb upstream of SOX9 |
| Genetics | Ref. | Genitalia | Gonads | Molecular Findings |
|---|---|---|---|---|
| Duplication of SOX3 gene | [109] | Pt 1 and Pt 2: males Pt 3: scrotal, retractile testes | Histol. NA | Pt 1: Microduplication of ~123 kb involving the entire SOX3 gene Pt 3: ~6-Mb duplication involving the entire SOX3 gene |
| [96] | Male | Histol. NA | 5.6-Mb duplication involving the entire SOX3 gene | |
| [110] | Ambiguous | Abdominal ovotestes | ~500-kb duplication involving the entire SOX3 gene | |
| [111] | Ambiguous | Scrotal Histol. NA | 494-kb duplication involving the entire SOX3 gene | |
| Rearrangement of SOX3 regulatory regions | [96,109] | Male | Scrotal Dysgenetic testes | 343-kb microdeletion immediately upstream of SOX3 |
| [112] | Ambiguous | Testes | Duplicated minimum critical region of ~41 kb located ~566 kb upstream of SOX3, common to all 4 patients | |
| [113] | Ambiguous | Scrotal One testis, one ovary | ~774-kb insertion of chromosome 1 ~80 kb downstream of SOX3 gene | |
| SOX10 duplication | [114] | Ambiguous | One testis, one ovary | Duplication involving the entire SOX10 gene |
| [115] | Male, micropenis | Cryptorchid testes | Trisomy of chromosome 22, where SOX10 maps | |
| [116] | Male | Scrotal Histol. NA | Partial duplication of chromosome 22q including the entire SOX10 gene | |
| [117] | Male | Scrotal Histol. NA | 22q11.2q13 duplication, including the entire SOX10 gene | |
| FGF9 duplication | [118] | Ambiguous | Histol. NA | Duplication of 13q12.11, where FGF9 maps |
| Genetics | Ref. | Genitalia | Gonads | Molecular Findings |
|---|---|---|---|---|
| WNT4 mutations | [131] | Pt 1: Ambiguous Pt 2: Male + SERKAL syndrome | Pt 1: Dysgenetic testes Pt 2: Ovotestes | WNT4 gene: c.C341T (p.A114V) |
| RSPO1 mutations | [134,135] | Ambiguous | Histol. NA | Family 1: RSPO1: c.108_109insG (frameshift mutation in exon 5) Family 2: Homozygous deletion of 2752 bp (exon 4 and adjacent introns) |
| [136] | Ambiguous | Ovotestes + Seminoma | RSPO1: homozygous splice-donor-site mutation c.28611G > A | |
| [137] | Ambiguous | Histol. NA | RSPO1: c.43_43delA (frameshift mutation in exon 4) | |
| [138] | Ambiguous | Pt 1: Dysgenetic testes Pt 2: Not reported | RSPO1: homozygous mutation c.332G > A (p.Cys111Tyr) |
| Genetics | Ref. | Genitalia | Gonads | Molecular Findings |
|---|---|---|---|---|
| NR5A1 mutations | [140] | Ambiguous (n:3) | Pt 1: Dysgenetic testis and fibrous streak Pt 2: Bilateral ovotestes Pt 3: Bilateral dysgenetic testes | NR5A1: c.274C > T (p.Arg92Trp) |
| [141] | Family 1 (n:.2): Ambiguous. Family 2 (n:1): Micropenis, bilateral cryptorchidism Family 3 (n:1): Male | Family 1: Ovotestes. Family 2: Histol. NA Family 3 (n:1) Testes | NR5A1: c.274C > T (p.Arg92Trp) | |
| [139] | Ambiguous | Scrotal Histol. NA | NR5A1: c.274C > T (p.Arg92Trp) | |
| [143,146] | Ambiguous | Pt 1: Dysgenetic testis + ovotestis Pt 2: Testes | NR5A1: c.274C > T (p.Arg92Trp) | |
| [145] | Ambiguous | Pt 1: Testes Pt 2: Ovotestes, intratubular Pt 3: Histol. NA Pt 4: Ovotestis + ovary | Pt 1 to 3: NR5A1 c.274C > T p.(Arg92Trp), Pt 4: NR5A1 c.779C > T (p.Ala260Val). | |
| [147] | Ambiguous | Ovotestes | NR5A1 c.275G > A (p.Arg92Gln) | |
| WT1 mutation | [148] | Ambiguous | Dysgenetic testes | WT1: c.1453_1456del; (p.Arg485Glyfs*14) |
| NR2F2 mutation | [149] | Pt 1: Male genitalia, non-palpable gonads Pt 2 and Pt 3: Ambiguous genitalia | Pt 1 and Pt 2: Histol. NA Pt 3: Ovotestes | Pt 1: NR2F2: c.103_109delGGCGCCC (p.Gly35Argfs*75) Pt 2 and Pt 3: NR2F2: c.97_103delCCGCCCG (p.Pro33Alafs*77) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grinspon, R.P.; Rey, R.A. Molecular Characterization of XX Maleness. Int. J. Mol. Sci. 2019, 20, 6089. https://doi.org/10.3390/ijms20236089
Grinspon RP, Rey RA. Molecular Characterization of XX Maleness. International Journal of Molecular Sciences. 2019; 20(23):6089. https://doi.org/10.3390/ijms20236089
Chicago/Turabian StyleGrinspon, Romina P., and Rodolfo A. Rey. 2019. "Molecular Characterization of XX Maleness" International Journal of Molecular Sciences 20, no. 23: 6089. https://doi.org/10.3390/ijms20236089
APA StyleGrinspon, R. P., & Rey, R. A. (2019). Molecular Characterization of XX Maleness. International Journal of Molecular Sciences, 20(23), 6089. https://doi.org/10.3390/ijms20236089