Oligosaccharyltransferase: A Gatekeeper of Health and Tumor Progression


Abstract
1. Introduction
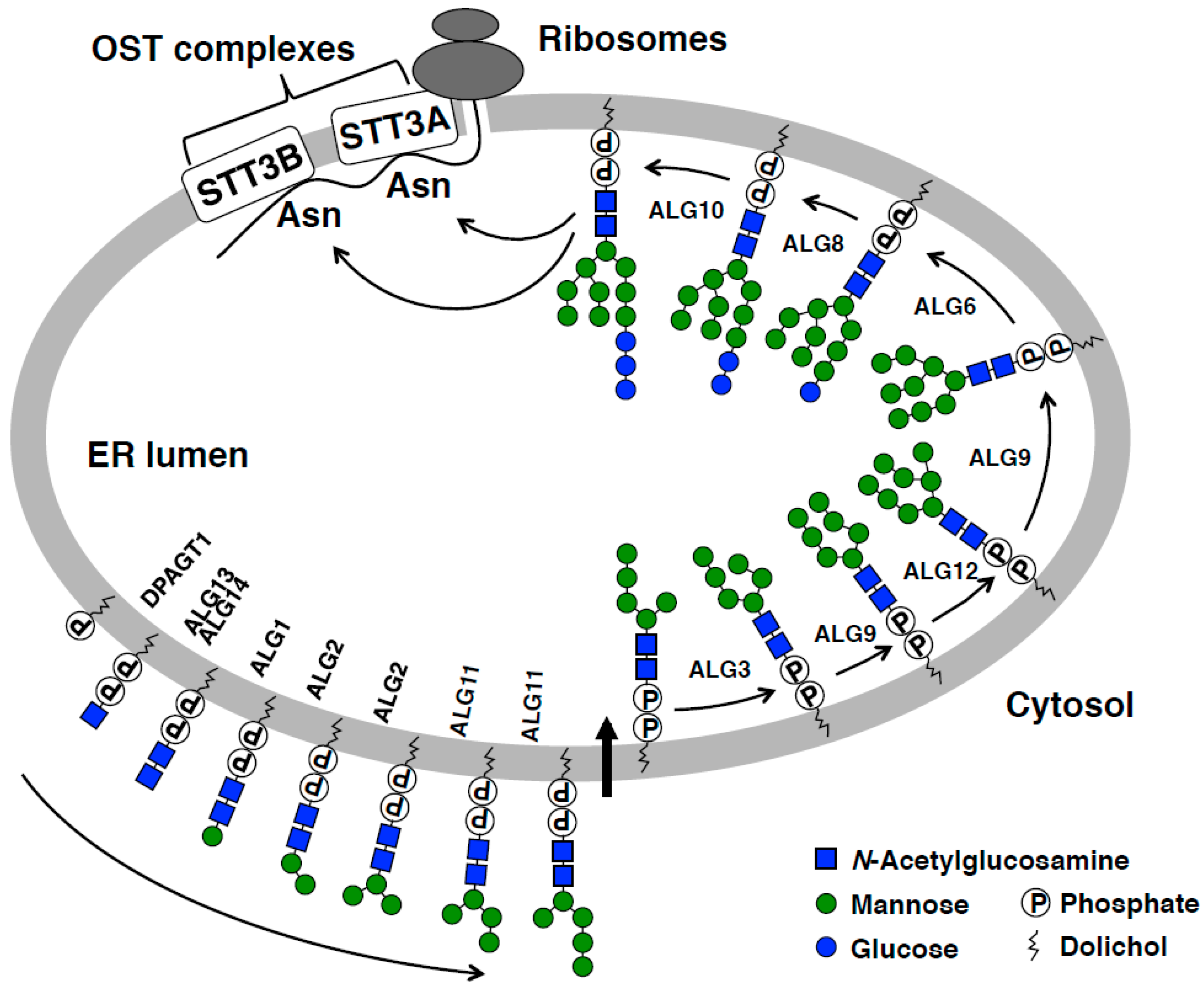
2. Overview of N-Glycosylation in the ER
3. OST and Its Action
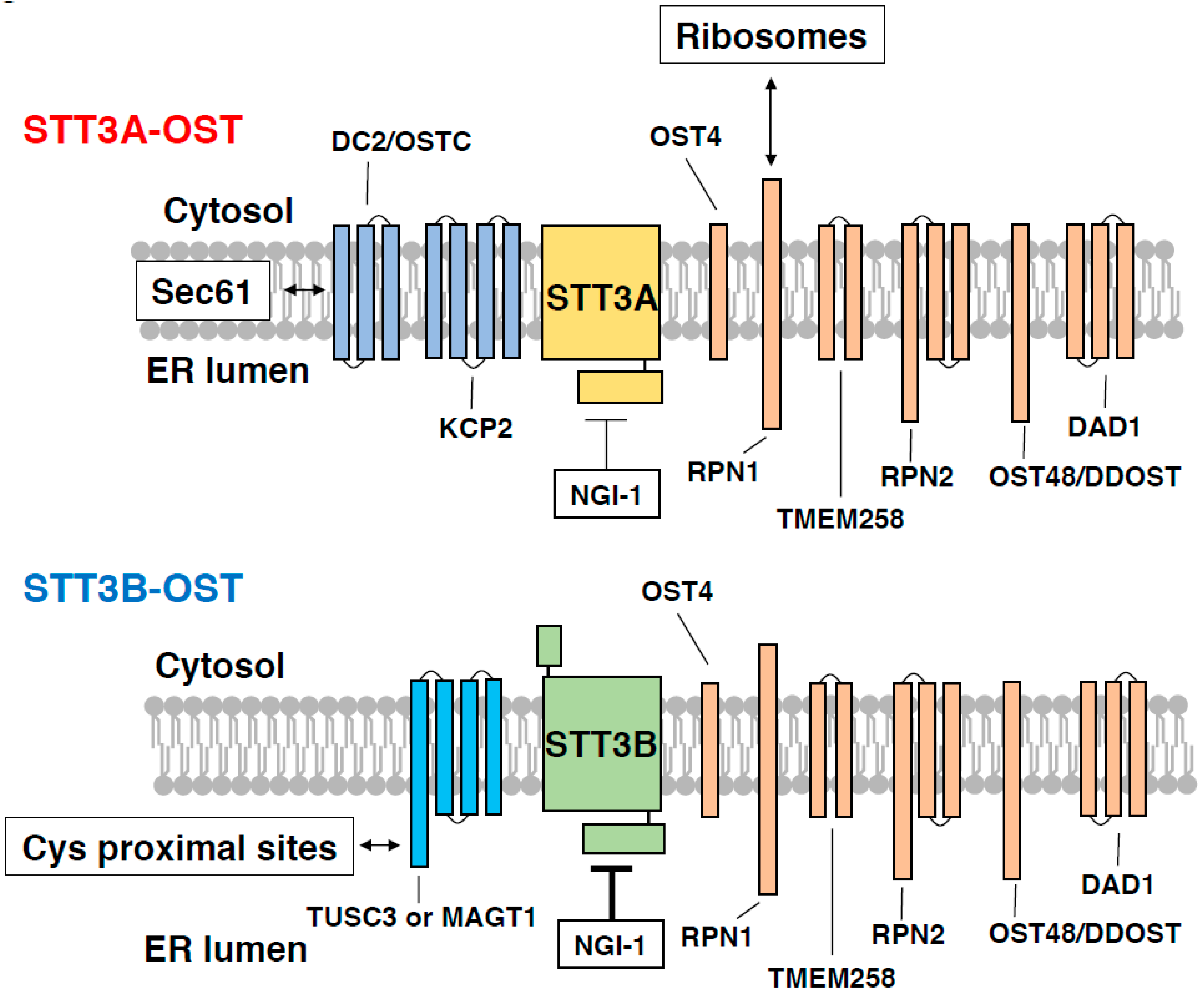
4. Roles of Accessory OST Subunits in N-Glycosylation and Health
4.1. Shared Subunits
4.2. Subunits Specific for STT3A-OST and STT3B-OST
5. Roles of OST in Tumor Progression
6. Association of RPN2 and TUSC3 with Tumor Progression
7. OST as a Potential Druggable Target for Cancer Treatment
8. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Apweiler, R.; Hermjakob, H.; Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the swiss-prot database. Biochim. Biophys. Acta 1999, 1473, 4–8. [Google Scholar] [CrossRef]
- Kelleher, D.J.; Gilmore, R. An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 2006, 16, 47R–62R. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Hirayama, H.; Suzuki, T. Generation and degradation of free asparagine-linked glycans. Cell Mol. Life Sci. 2015, 72, 2509–2533. [Google Scholar] [CrossRef] [PubMed]
- Stanley, P.; Taniguchi, N.; Aebi, M. N-glycans. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 99–111. [Google Scholar]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Taniguchi, N.; Kizuka, Y. Glycans and cancer: Role of n-glycans in cancer biomarker, progression and metastasis, and therapeutics. Adv. Cancer Res. 2015, 126, 11–51. [Google Scholar]
- Chiaradonna, F.; Ricciardiello, F.; Palorini, R. The nutrient-sensing hexosamine biosynthetic pathway as the hub of cancer metabolic rewiring. Cells 2018, 7, 53. [Google Scholar] [CrossRef]
- Hsu, J.M.; Li, C.W.; Lai, Y.J.; Hung, M.C. Posttranslational modifications of pd-l1 and their applications in cancer therapy. Cancer Res. 2018, 78, 6349–6353. [Google Scholar] [CrossRef]
- Marsico, G.; Russo, L.; Quondamatteo, F.; Pandit, A. Glycosylation and integrin regulation in cancer. Trends Cancer 2018, 4, 537–552. [Google Scholar] [CrossRef]
- Pang, X.; Li, H.; Guan, F.; Li, X. Multiple roles of glycans in hematological malignancies. Front. Oncol. 2018, 8, 364. [Google Scholar] [CrossRef]
- RodrIguez, E.; Schetters, S.T.T.; van Kooyk, Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat. Rev. Immunol. 2018, 18, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Chandler, K.B.; Costello, C.E.; Rahimi, N. Glycosylation in the tumor microenvironment: Implications for tumor angiogenesis and metastasis. Cells 2019, 8, 544. [Google Scholar] [CrossRef] [PubMed]
- Mereiter, S.; Balmana, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the era of cancer-targeted therapy: Where are we heading? Cancer Cell 2019, 36, 6–16. [Google Scholar] [CrossRef]
- Peixoto, A.; Relvas-Santos, M.; Azevedo, R.; Santos, L.L.; Ferreira, J.A. Protein glycosylation and tumor microenvironment alterations driving cancer hallmarks. Front. Oncol. 2019, 9, 380. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Shrimal, S.; Cherepanova, N.A.; Gilmore, R. Cotranslational and posttranslocational n-glycosylation of proteins in the endoplasmic reticulum. Semin. Cell Dev. Biol. 2015, 41, 71–78. [Google Scholar] [CrossRef]
- Aebi, M. N-linked protein glycosylation in the er. Biochim. Biophys. Acta 2013, 1833, 2430–2437. [Google Scholar] [CrossRef]
- Harada, Y. Biosynthesis and degradation of dolichol-linked oligosaccharides. Trends Glycosci. Glycotechnol. 2016, 28, E91–E96. [Google Scholar] [CrossRef]
- Dell, A.; Galadari, A.; Sastre, F.; Hitchen, P. Similarities and differences in the glycosylation mechanisms in prokaryotes and eukaryotes. Int. J. Microbiol. 2010, 2010, 148178. [Google Scholar] [CrossRef]
- Larkin, A.; Imperiali, B. The expanding horizons of asparagine-linked glycosylation. Biochemistry 2011, 50, 4411–4426. [Google Scholar] [CrossRef]
- Magidovich, H.; Eichler, J. Glycosyltransferases and oligosaccharyltransferases in archaea: Putative components of the n-glycosylation pathway in the third domain of life. FEMS Microbiol. Lett. 2009, 300, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Canada, C.; Kelleher, D.J.; Gilmore, R. Cotranslational and posttranslational n-glycosylation of polypeptides by distinct mammalian ost isoforms. Cell 2009, 136, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, D.J.; Karaoglu, D.; Mandon, E.C.; Gilmore, R. Oligosaccharyltransferase isoforms that contain different catalytic stt3 subunits have distinct enzymatic properties. Mol. Cell 2003, 12, 101–111. [Google Scholar] [CrossRef]
- Shibatani, T.; David, L.L.; McCormack, A.L.; Frueh, K.; Skach, W.R. Proteomic analysis of mammalian oligosaccharyltransferase reveals multiple subcomplexes that contain sec61, trap, and two potential new subunits. Biochemistry 2005, 44, 5982–5992. [Google Scholar] [CrossRef]
- Graham, D.B.; Lefkovith, A.; Deelen, P.; de Klein, N.; Varma, M.; Boroughs, A.; Desch, A.N.; Ng, A.C.Y.; Guzman, G.; Schenone, M.; et al. Tmem258 is a component of the oligosaccharyltransferase complex controlling er stress and intestinal inflammation. Cell Rep. 2016, 17, 2955–2965. [Google Scholar] [CrossRef]
- Dumax-Vorzet, A.; Roboti, P.; High, S. Ost4 is a subunit of the mammalian oligosaccharyltransferase required for efficient n-glycosylation. J. Cell Sci. 2013, 126, 2595–2606. [Google Scholar] [CrossRef]
- Shrimal, S.; Cherepanova, N.A.; Gilmore, R. Dc2 and kcp2 mediate the interaction between the oligosaccharyltransferase and the er translocon. J. Cell Biol. 2017, 216, 3625–3638. [Google Scholar] [CrossRef]
- Cherepanova, N.A.; Gilmore, R. Mammalian cells lacking either the cotranslational or posttranslocational oligosaccharyltransferase complex display substrate-dependent defects in asparagine linked glycosylation. Sci. Rep. 2016, 6, 20946. [Google Scholar] [CrossRef]
- Bas, T.; Gao, G.Y.; Lvov, A.; Chandrasekhar, K.D.; Gilmore, R.; Kobertz, W.R. Post-translational n-glycosylation of type i transmembrane kcne1 peptides: Implications for membrane protein biogenesis and disease. J. Biol. Chem. 2011, 286, 28150–28159. [Google Scholar] [CrossRef]
- Shrimal, S.; Gilmore, R. Glycosylation of closely spaced acceptor sites in human glycoproteins. J. Cell Sci. 2013, 126, 5513–5523. [Google Scholar] [CrossRef]
- Shrimal, S.; Trueman, S.F.; Gilmore, R. Extreme c-terminal sites are posttranslocationally glycosylated by the stt3b isoform of the ost. J. Cell Biol. 2013, 201, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Cherepanova, N.A.; Shrimal, S.; Gilmore, R. Oxidoreductase activity is necessary for n-glycosylation of cysteine-proximal acceptor sites in glycoproteins. J. Cell Biol. 2014, 206, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Napiorkowska, M.; Boilevin, J.; Sovdat, T.; Darbre, T.; Reymond, J.L.; Aebi, M.; Locher, K.P. Molecular basis of lipid-linked oligosaccharide recognition and processing by bacterial oligosaccharyltransferase. Nat. Struct. Mol. Biol. 2017, 24, 1100–1106. [Google Scholar] [CrossRef]
- Braunger, K.; Pfeffer, S.; Shrimal, S.; Gilmore, R.; Berninghausen, O.; Mandon, E.C.; Becker, T.; Forster, F.; Beckmann, R. Structural basis for coupling protein transport and n-glycosylation at the mammalian endoplasmic reticulum. Science 2018, 360, 215–219. [Google Scholar] [CrossRef]
- Nilsson, I.M.; von Heijne, G. Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J. Biol. Chem. 1993, 268, 5798–5801. [Google Scholar]
- Harada, Y.; Li, H.; Li, H.; Lennarz, W.J. Oligosaccharyltransferase directly binds to ribosome at a location near the translocon-binding site. Proc. Natl. Acad. Sci. USA 2009, 106, 6945–6949. [Google Scholar] [CrossRef]
- Sato, T.; Sako, Y.; Sho, M.; Momohara, M.; Suico, M.A.; Shuto, T.; Nishitoh, H.; Okiyoneda, T.; Kokame, K.; Kaneko, M.; et al. Stt3b-dependent posttranslational n-glycosylation as a surveillance system for secretory protein. Mol. Cell 2012, 47, 99–110. [Google Scholar] [CrossRef]
- Wei, W.; Zheng, C.; Zhu, M.; Zhu, X.; Yang, R.; Misra, S.; Zhang, B. Missense mutations near the n-glycosylation site of the a2 domain lead to various intracellular trafficking defects in coagulation factor viii. Sci. Rep. 2017, 7, 45033. [Google Scholar] [CrossRef]
- Wei, W.; Misra, S.; Cannon, M.V.; Yang, R.; Zhu, X.; Gilmore, R.; Zhu, M.; Zhang, B. Molecular mechanisms of missense mutations that generate ectopic n-glycosylation sites in coagulation factor viii. Biochem. J. 2018, 475, 873–886. [Google Scholar] [CrossRef]
- Fan, Y.; Hu, Y.; Yan, C.; Goldman, R.; Pan, Y.; Mazumder, R.; Dingerdissen, H.M. Loss and gain of n-linked glycosylation sequons due to single-nucleotide variation in cancer. Sci. Rep. 2018, 8, 4322. [Google Scholar] [CrossRef]
- Shrimal, S.; Ng, B.G.; Losfeld, M.E.; Gilmore, R.; Freeze, H.H. Mutations in stt3a and stt3b cause two congenital disorders of glycosylation. Hum. Mol. Genet. 2013, 22, 4638–4645. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sambrooks, C.; Shrimal, S.; Khodier, C.; Flaherty, D.P.; Rinis, N.; Charest, J.C.; Gao, N.; Zhao, P.; Wells, L.; Lewis, T.A.; et al. Oligosaccharyltransferase inhibition induces senescence in rtk-driven tumor cells. Nat. Chem. Biol. 2016, 12, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Kreibich, G.; Czako-Graham, M.; Grebenau, R.; Mok, W.; Rodriguez-Boulan, E.; Sabatini, D.D. Characterization of the ribosomal binding site in rat liver rough microsomes: Ribophorins i and ii, two integral membrane proteins related to ribosome binding. J. Supramol. Struct. 1978, 8, 279–302. [Google Scholar] [CrossRef] [PubMed]
- Honma, K.; Iwao-Koizumi, K.; Takeshita, F.; Yamamoto, Y.; Yoshida, T.; Nishio, K.; Nagahara, S.; Kato, K.; Ochiya, T. Rpn2 gene confers docetaxel resistance in breast cancer. Nat. Med. 2008, 14, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, N.; Hagiwara, K.; Kosaka, N.; Honma, K.; Nakagama, H.; Ochiya, T. Rpn2-mediated glycosylation of tetraspanin cd63 regulates breast cancer cell malignancy. Mol. Cancer 2014, 13, 134. [Google Scholar] [CrossRef]
- Nakashima, T.; Sekiguchi, T.; Kuraoka, A.; Fukushima, K.; Shibata, Y.; Komiyama, S.; Nishimoto, T. Molecular cloning of a human cdna encoding a novel protein, dad1, whose defect causes apoptotic cell death in hamster bhk21 cells. Mol. Cell Biol. 1993, 13, 6367–6374. [Google Scholar] [CrossRef]
- Sanjay, A.; Fu, J.; Kreibich, G. Dad1 is required for the function and the structural integrity of the oligosaccharyltransferase complex. J. Biol. Chem. 1998, 273, 26094–26099. [Google Scholar] [CrossRef]
- Roboti, P.; High, S. The oligosaccharyltransferase subunits ost48, dad1 and kcp2 function as ubiquitous and selective modulators of mammalian n-glycosylation. J. Cell Sci. 2012, 125, 3474–3484. [Google Scholar] [CrossRef]
- Jones, M.A.; Ng, B.G.; Bhide, S.; Chin, E.; Rhodenizer, D.; He, P.; Losfeld, M.E.; He, M.; Raymond, K.; Berry, G.; et al. Ddost mutations identified by whole-exome sequencing are implicated in congenital disorders of glycosylation. Am. J. Hum. Genet. 2012, 90, 363–368. [Google Scholar] [CrossRef]
- Garshasbi, M.; Hadavi, V.; Habibi, H.; Kahrizi, K.; Kariminejad, R.; Behjati, F.; Tzschach, A.; Najmabadi, H.; Ropers, H.H.; Kuss, A.W. A defect in the tusc3 gene is associated with autosomal recessive mental retardation. Am. J. Hum. Genet. 2008, 82, 1158–1164. [Google Scholar] [CrossRef]
- Zhou, H.; Clapham, D.E. Mammalian magt1 and tusc3 are required for cellular magnesium uptake and vertebrate embryonic development. Proc. Natl. Acad. Sci. USA 2009, 106, 15750–15755. [Google Scholar] [CrossRef]
- Vasickova, K.; Horak, P.; Vanhara, P. Tusc3: Functional duality of a cancer gene. Cell Mol. Life Sci. 2018, 75, 849–857. [Google Scholar] [CrossRef]
- MacGrogan, D.; Levy, A.; Bova, G.S.; Isaacs, W.B.; Bookstein, R. Structure and methylation-associated silencing of a gene within a homozygously deleted region of human chromosome band 8p22. Genomics 1996, 35, 55–65. [Google Scholar] [CrossRef]
- Bova, G.S.; MacGrogan, D.; Levy, A.; Pin, S.S.; Bookstein, R.; Isaacs, W.B. Physical mapping of chromosome 8p22 markers and their homozygous deletion in a metastatic prostate cancer. Genomics 1996, 35, 46–54. [Google Scholar] [CrossRef]
- Pils, D.; Horak, P.; Vanhara, P.; Anees, M.; Petz, M.; Alfanz, A.; Gugerell, A.; Wittinger, M.; Gleiss, A.; Auner, V.; et al. Methylation status of tusc3 is a prognostic factor in ovarian cancer. Cancer 2013, 119, 946–954. [Google Scholar] [CrossRef]
- Duppel, U.; Woenckhaus, M.; Schulz, C.; Merk, J.; Dietmaier, W. Quantitative detection of tusc3 promoter methylation—A potential biomarker for prognosis in lung cancer. Oncol. Lett. 2016, 12, 3004–3012. [Google Scholar] [CrossRef][Green Version]
- Taniue, K.; Hayashi, T.; Kamoshida, Y.; Kurimoto, A.; Takeda, Y.; Negishi, L.; Iwasaki, K.; Kawamura, Y.; Goshima, N.; Akiyama, T. Uhrf1-kat7-mediated regulation of tusc3 expression via histone methylation/acetylation is critical for the proliferation of colon cancer cells. Oncogene 2019. [Google Scholar] [CrossRef]
- Horak, P.; Tomasich, E.; Vanhara, P.; Kratochvilova, K.; Anees, M.; Marhold, M.; Lemberger, C.E.; Gerschpacher, M.; Horvat, R.; Sibilia, M.; et al. Tusc3 loss alters the er stress response and accelerates prostate cancer growth in vivo. Sci. Rep. 2014, 4, 3739. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Kim, T.; Park, D.; Nuovo, G.J.; Rhee, S.; Joshi, P.; Lee, B.K.; Jeong, J.; Suh, S.S.; Grotzke, J.E.; et al. Mirna-mediated tusc3 deficiency enhances upr and erad to promote metastatic potential of nsclc. Nat. Commun. 2018, 9, 5110. [Google Scholar] [CrossRef]
- Feng, S.; Zhai, J.; Lu, D.; Lin, J.; Dong, X.; Liu, X.; Wu, H.; Roden, A.C.; Brandi, G.; Tavolari, S.; et al. Tusc3 accelerates cancer growth and induces epithelial-mesenchymal transition by upregulating claudin-1 in non-small-cell lung cancer cells. Exp. Cell Res. 2018, 373, 44–56. [Google Scholar] [CrossRef]
- Kratochvilova, K.; Horak, P.; Esner, M.; Soucek, K.; Pils, D.; Anees, M.; Tomasich, E.; Drafi, F.; Jurtikova, V.; Hampl, A.; et al. Tumor suppressor candidate 3 (tusc3) prevents the epithelial-to-mesenchymal transition and inhibits tumor growth by modulating the endoplasmic reticulum stress response in ovarian cancer cells. Int. J. Cancer 2015, 137, 1330–1340. [Google Scholar] [CrossRef]
- Gu, Y.; Wang, Q.; Guo, K.; Qin, W.; Liao, W.; Wang, S.; Ding, Y.; Lin, J. Tusc3 promotes colorectal cancer progression and epithelial-mesenchymal transition (emt) through wnt/beta-catenin and mapk signalling. J. Pathol. 2016, 239, 60–71. [Google Scholar] [CrossRef]
- Gu, Y.; Pei, X.; Ren, Y.; Cai, K.; Guo, K.; Chen, J.; Qin, W.; Lin, M.; Wang, Q.; Tang, N.; et al. Oncogenic function of tusc3 in non-small cell lung cancer is associated with hedgehog signalling pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1749–1760. [Google Scholar] [CrossRef]
- Li, F.Y.; Chaigne-Delalande, B.; Kanellopoulou, C.; Davis, J.C.; Matthews, H.F.; Douek, D.C.; Cohen, J.I.; Uzel, G.; Su, H.C.; Lenardo, M.J. Second messenger role for Mg2+ revealed by human t-cell immunodeficiency. Nature 2011, 475, 471–476. [Google Scholar] [CrossRef]
- Matsuda-Lennikov, M.; Biancalana, M.; Zou, J.; Ravell, J.C.; Zheng, L.; Kanellopoulou, C.; Jiang, P.; Notarangelo, G.; Jing, H.; Masutani, E.; et al. Magnesium transporter 1 (magt1) deficiency causes selective defects in n-linked glycosylation and expression of immune-response genes. J. Biol. Chem. 2019, 294, 13638–13656. [Google Scholar] [CrossRef]
- Kelleher, D.J.; Gilmore, R. Dad1, the defender against apoptotic cell death, is a subunit of the mammalian oligosaccharyltransferase. Proc. Natl. Acad. Sci. USA 1997, 94, 4994–4999. [Google Scholar] [CrossRef]
- Fu, J.; Kreibich, G. Retention of subunits of the oligosaccharyltransferase complex in the endoplasmic reticulum. J. Biol. Chem. 2000, 275, 3984–3990. [Google Scholar] [CrossRef]
- Mahoney, K.M.; Freeman, G.J.; McDermott, D.F. The next immune-checkpoint inhibitors: Pd-1/pd-l1 blockade in melanoma. Clin. Ther. 2015, 37, 764–782. [Google Scholar] [CrossRef]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses t-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Pang, X.X.; Bai, Q.; Wu, F.; Chen, G.J.; Zhang, A.H.; Tang, C.S. Urotensin ii induces er stress and emt and increase extracellular matrix production in renal tubular epithelial cell in early diabetic mice. Kidney Blood Press Res. 2016, 41, 434–449. [Google Scholar] [CrossRef]
- Zhang, J.; Jamaluddin, M.; Zhang, Y.; Widen, S.G.; Sun, H.; Brasier, A.R.; Zhao, Y. Type ii epithelial-mesenchymal transition upregulates protein n-glycosylation to maintain proteostasis and extracellular matrix production. J. Proteome Res. 2019, 18, 3447–3460. [Google Scholar] [CrossRef]
- Hsu, J.M.; Xia, W.; Hsu, Y.H.; Chan, L.C.; Yu, W.H.; Cha, J.H.; Chen, C.T.; Liao, H.W.; Kuo, C.W.; Khoo, K.H.; et al. Stt3-dependent pd-l1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Tan, Z.; Chen, S.; Guan, F. Role of glycans in cancer cells undergoing epithelial-mesenchymal transition. Front. Oncol. 2016, 6, 33. [Google Scholar] [CrossRef]
- Lucena, M.C.; Carvalho-Cruz, P.; Donadio, J.L.; Oliveira, I.A.; de Queiroz, R.M.; Marinho-Carvalho, M.M.; Sola-Penna, M.; de Paula, I.F.; Gondim, K.C.; McComb, M.E.; et al. Epithelial mesenchymal transition induces aberrant glycosylation through hexosamine biosynthetic pathway activation. J. Biol. Chem. 2016, 291, 12917–12929. [Google Scholar] [CrossRef]
- Li, C.W.; Lim, S.O.; Chung, E.M.; Kim, Y.S.; Park, A.H.; Yao, J.; Cha, J.H.; Xia, W.; Chan, L.C.; Kim, T.; et al. Eradication of triple-negative breast cancer cells by targeting glycosylated pd-l1. Cancer Cell 2018, 33, 187–201.e110. [Google Scholar] [CrossRef]
- Ono, M.; Tsuda, H.; Kobayashi, T.; Takeshita, F.; Takahashi, R.U.; Tamura, K.; Akashi-Tanaka, S.; Moriya, T.; Yamasaki, T.; Kinoshita, T.; et al. The expression and clinical significance of ribophorin ii (rpn2) in human breast cancer. Pathol. Int. 2015, 65, 301–308. [Google Scholar] [CrossRef]
- Fujita, Y.; Yagishita, S.; Takeshita, F.; Yamamoto, Y.; Kuwano, K.; Ochiya, T. Prognostic and therapeutic impact of rpn2-mediated tumor malignancy in non-small-cell lung cancer. Oncotarget 2015, 6, 3335–3345. [Google Scholar] [CrossRef]
- Bi, C.; Jiang, B. Downregulation of rpn2 induces apoptosis and inhibits migration and invasion in colon carcinoma. Oncol. Rep. 2018, 40, 283–293. [Google Scholar] [CrossRef]
- Fujimoto, D.; Goi, T.; Koneri, K.; Hirono, Y. Rpn2 is effective biomarker to predict the outcome of combined chemotherapy docetaxel and cisplatin for advanced gastric cancer. Oncotarget 2018, 9, 15208–15218. [Google Scholar] [CrossRef]
- Li, Y.; Huang, C.; Bai, Q.; Yu, J. Ribophorin ii promotes cell proliferation, migration, and invasion in esophageal cancer cells in vitro and in vivo. Biosci. Rep. 2019, 39, BSR20182448. [Google Scholar] [CrossRef]
- Li, H.; Al-Japairai, K.; Tao, Y.; Xiang, Z. Rpn2 promotes colorectal cancer cell proliferation through modulating the glycosylation status of egfr. Oncotarget 2017, 8, 72633–72651. [Google Scholar] [CrossRef]
- Zhou, T.; Wu, L.; Wang, Q.; Jiang, Z.; Li, Y.; Ma, N.; Chen, W.; Hou, Z.; Gan, W.; Chen, S. Microrna-128 targeting rpn2 inhibits cell proliferation and migration through the akt-p53-cyclin pathway in colorectal cancer cells. Oncol. Lett. 2018, 16, 6940–6949. [Google Scholar] [CrossRef]
- Tan, C.L.; Plotkin, J.L.; Veno, M.T.; von Schimmelmann, M.; Feinberg, P.; Mann, S.; Handler, A.; Kjems, J.; Surmeier, D.J.; O’Carroll, D.; et al. Microrna-128 governs neuronal excitability and motor behavior in mice. Science 2013, 342, 1254–1258. [Google Scholar] [CrossRef]
- Huang, W.; Feng, Y.; Liang, J.; Yu, H.; Wang, C.; Wang, B.; Wang, M.; Jiang, L.; Meng, W.; Cai, W.; et al. Loss of microrna-128 promotes cardiomyocyte proliferation and heart regeneration. Nat. Commun. 2018, 9, 700. [Google Scholar] [CrossRef]
- Rinis, N.; Golden, J.E.; Marceau, C.D.; Carette, J.E.; Van Zandt, M.C.; Gilmore, R.; Contessa, J.N. Editing n-glycan site occupancy with small-molecule oligosaccharyltransferase inhibitors. Cell Chem. Biol. 2018, 25, 1231–1241. [Google Scholar] [CrossRef]
- Tsuda, T.; Ikeda, Y.; Taniguchi, N. The asn-420-linked sugar chain in human epidermal growth factor receptor suppresses ligand-independent spontaneous oligomerization. Possible role of a specific sugar chain in controllable receptor activation. J. Biol. Chem. 2000, 275, 21988–21994. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J.; et al. Irreversible inhibitors of the egf receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the egfr kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired egfr c797s mutation mediates resistance to azd9291 in non-small cell lung cancer harboring egfr t790m. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. Met amplification leads to gefitinib resistance in lung cancer by activating erbb3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of met amplification in egfr mutant nsclc. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. Met amplification occurs with or without t790m mutations in egfr mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef]
- Lopez Sambrooks, C.; Baro, M.; Quijano, A.; Narayan, A.; Cui, W.; Greninger, P.; Egan, R.; Patel, A.; Benes, C.H.; Saltzman, W.M.; et al. Oligosaccharyltransferase inhibition overcomes therapeutic resistance to egfr tyrosine kinase inhibitors. Cancer Res. 2018, 78, 5094–5106. [Google Scholar] [CrossRef]
- Tax, G.; Lia, A.; Santino, A.; Roversi, P. Modulation of erqc and erad: A broad-spectrum spanner in the works of cancer cells? J. Oncol. 2019, 2019, 8384913. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| OST Subunits | Type of OST | Functions in OST Complexes | Phenotypes Caused by Mutations | Phenotypes Caused by Downregulation | References |
|---|---|---|---|---|---|
| STT3A | STT3A-OST | Catalytic subunit, co-translational glycosylation | STT3A-CDG | Impaired co-translational glycosylation | [42] |
| STT3B | STT3B-OST | Catalytic subunit, post-translational glycosylation | STT3B-CDG | Impaired post-translational glycosylation | [42] |
| RPN1 | Shared | Binding to ribosome | Not known | Reduced expression of STT3A and STT3B | [23,35] |
| RPN2 | Shared | Binding to ribosome? | Not known | Hypoglycosylation of P-glycoprotein and CD63 | [44,45,46] |
| TMEM258 | Shared | Association with RPN1 | Not known | Increased intestinal inflammation in dextran sodium sulfate-treated haploinsufficient mice | [26] |
| DAD1 | Shared | Stabilization of RPN1, RPN2 and OST48 | Increased susceptibility to apoptotic cell death at non-permissive temperature | Reduced expression of STT3A, STT3B, OST48/DDOST and KCP2 | [47,48,49] |
| OST48/DDOST | Shared | Stabilization of STT3A-OST and STT3B-OST | DDOST-CDG | Reduced expression of STT3A, STT3B, DAD1 and KCP2 | [49,50] |
| OST4 | Shared | Stabilization of STT3A-OST, but not STT3B-OST | Not known | Hypoglycosylation of prosaposin and reduced expression of STT3A and KCP2 | [27] |
| DC2/OSTC and KCP2 | STT3A-OST | Association with the Sec61 protein-conducting channel, co-translational glycosylation | Not known | Impaired co-translational glycosylation | [28,35] |
| TUSC3 | STT3B-OST | Thioredoxin, glycosylation at Cys-proximal sites | Autosomal recessive mental retardation | Impaired Mg2+ uptake and tumor progression | [33,51,52,53,54,55,56,57,58,59,60,61,62,63,64] |
| MAGT1 | STT3B-OST | Thioredoxin, glycosylation at Cys-proximal sites | X-linked immunodeficiency | Impaired Mg2+ uptake | [33,52,65,66] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harada, Y.; Ohkawa, Y.; Kizuka, Y.; Taniguchi, N. Oligosaccharyltransferase: A Gatekeeper of Health and Tumor Progression. Int. J. Mol. Sci. 2019, 20, 6074. https://doi.org/10.3390/ijms20236074
Harada Y, Ohkawa Y, Kizuka Y, Taniguchi N. Oligosaccharyltransferase: A Gatekeeper of Health and Tumor Progression. International Journal of Molecular Sciences. 2019; 20(23):6074. https://doi.org/10.3390/ijms20236074
Chicago/Turabian StyleHarada, Yoichiro, Yuki Ohkawa, Yasuhiko Kizuka, and Naoyuki Taniguchi. 2019. "Oligosaccharyltransferase: A Gatekeeper of Health and Tumor Progression" International Journal of Molecular Sciences 20, no. 23: 6074. https://doi.org/10.3390/ijms20236074
APA StyleHarada, Y., Ohkawa, Y., Kizuka, Y., & Taniguchi, N. (2019). Oligosaccharyltransferase: A Gatekeeper of Health and Tumor Progression. International Journal of Molecular Sciences, 20(23), 6074. https://doi.org/10.3390/ijms20236074