TCGA-TCIA Impact on Radiogenomics Cancer Research: A Systematic Review



Abstract
1. Introduction
2. Results
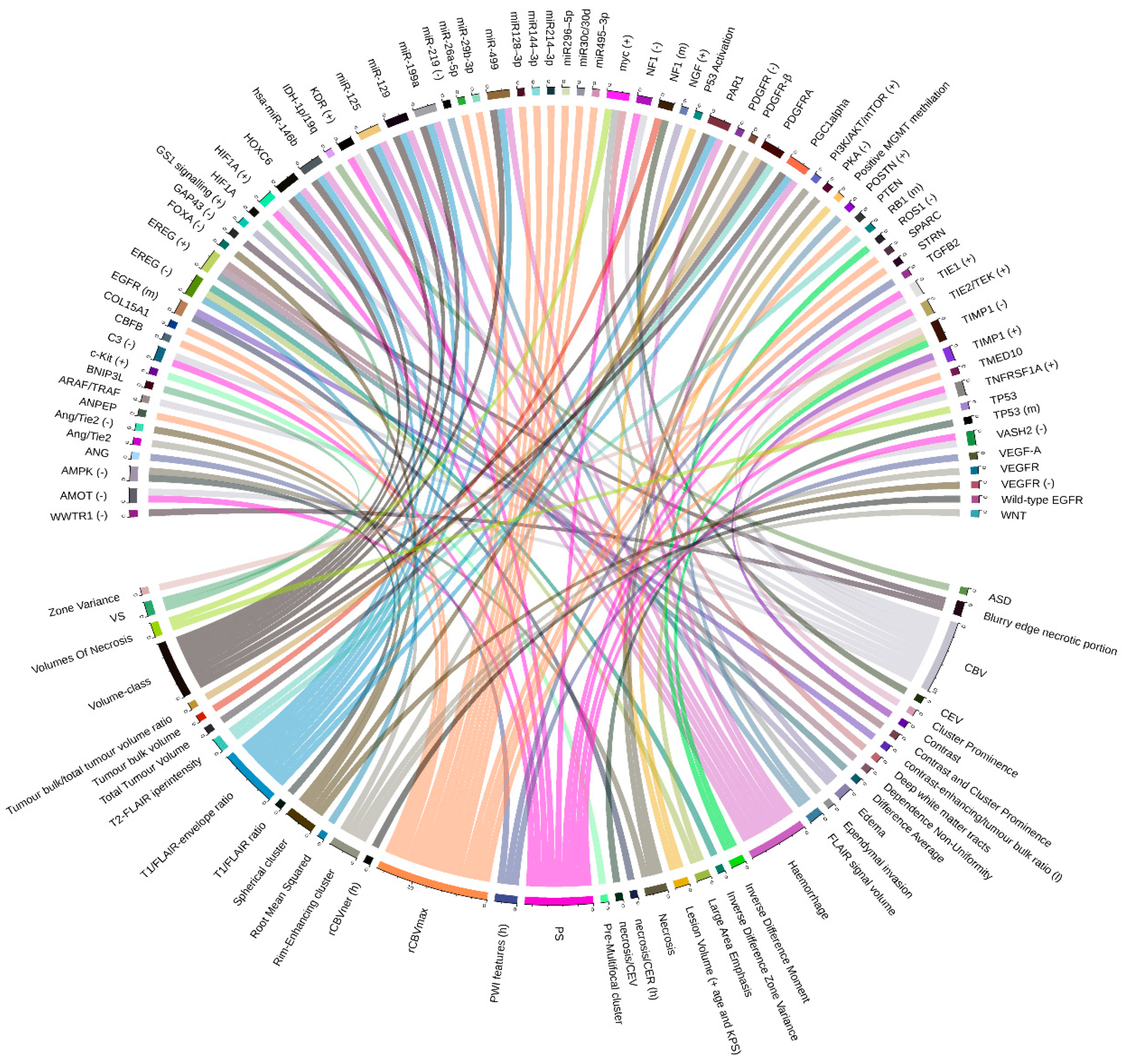
2.1. Glioblastoma Multiforme (GBM) and Low-Grade Glioma (LGG)
2.1.1. Cancer Sub-Groups Classification
2.1.2. Relations between Imaging Features and Gene Expression Data
2.1.3. Relations between Imaging Features and Mutation Data
2.1.4. Relations between Imaging Features and Proteomic Data
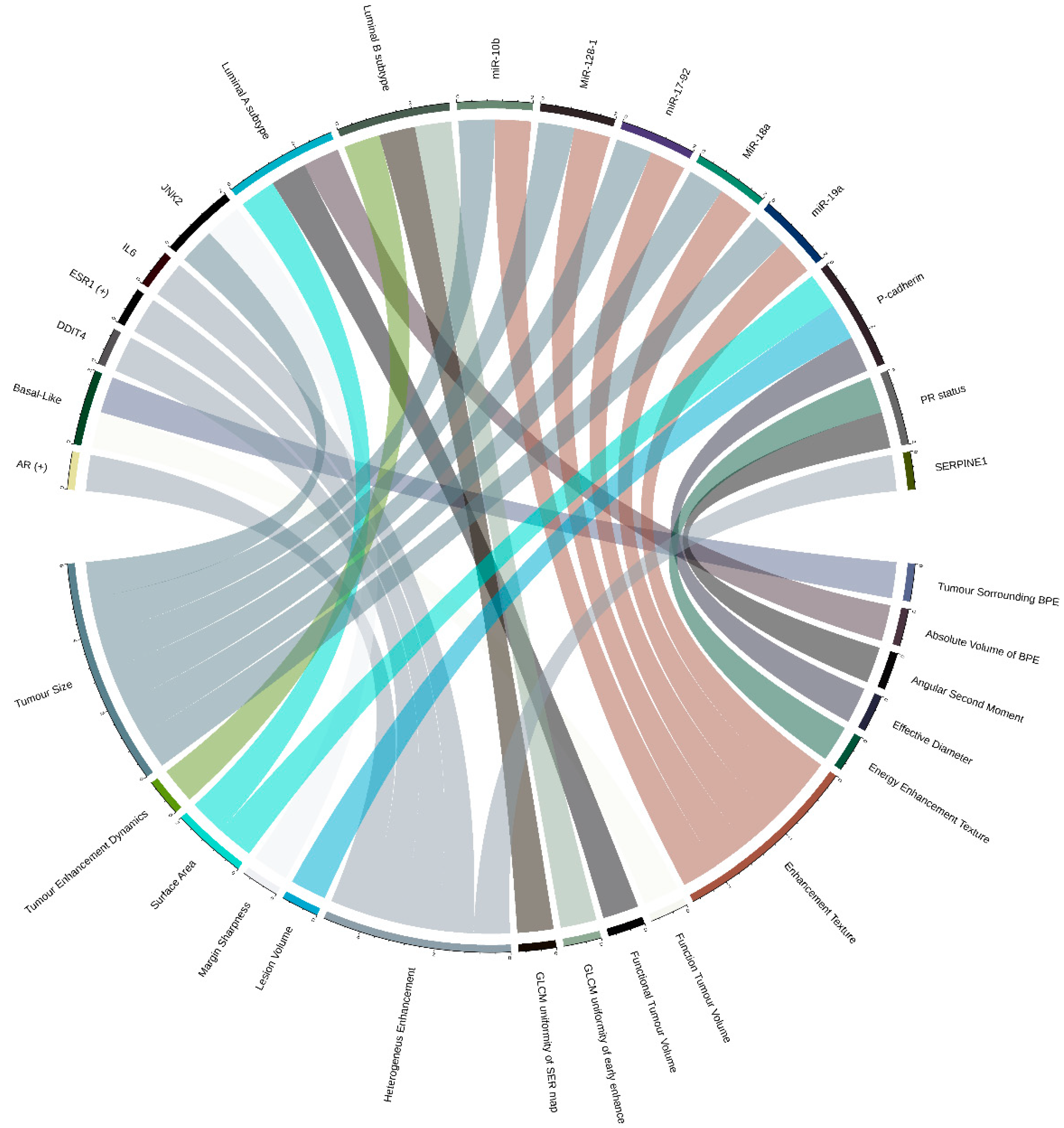
2.2. Breast Cancer (BRCA)
2.2.1. Relations between Imaging Features and Cancer Subtypes
2.2.2. Relations between Imaging Features and Gene Expression Data
2.2.3. Relations between Imaging Features and Multiple Molecular ‘Omic Features
2.3. Clear Cell Renal Cell Carcinoma (KIRC)
2.4. Other Tumors
3. Discussion
- lack of image-sample registration (gene expression profiles cannot be matched to a specific location on MRI),
- manually annotated ROIs that could introduce potential interobserver variability,
- unknown location of biopsy (given tumor heterogeneity, tissue origin is needed to obtain more accurate samples),
- data acquired with varying protocols from different MRI systems. Image data can be extremely heterogeneous due to different scanner modalities, manufacturers, and acquisition protocols,
- in most cases, data were not acquired as part of a research protocol or clinical trial.

{kind=link}
{kind=link}
| Paper | Imaging Features | Molecular Omic Features | TCGA/TCIA Number of Patients | Internal Cohort/ n° Patients | Statistical Analysis |
|---|---|---|---|---|---|
| Glioblastoma Multiforme (GBM) and Low Grade Glioma (LGG) | |||||
| [10] | FLAIR signal volume | POSTN (+), miR-219 (–) | 78 (39 of which as validation set) | No | Comparative marker selection (CMS) |
| [12] | Lesion Volume (+ age and KPS) | P53 activation, MGMT methylation | 78 (+ 64 from TCGA and Rembrandt) | No | Cox proportional hazards likelihood ratio |
| [18] | rCBVner (h) | Wild-type EGFR | 45 | No | Analysis of variance |
| [19] | PS, CBV | TNFRSF1A, HIF1A, KDR, TIE1, TIE2/TEK (+), VASH2, C3, AMOT, and NF1 (–) | 18 | No | Pearson correlation coefficient |
| [20] | rCBVmax | miR-29b-3p, miR495-3p, miR30c/30d, miR-26a-5p, miR296-5p, miR128-3p, miR144-3p and miR214-3p, PTEN, COL15A1, SPARC, ANPEP, CBFB, STRN, TMED10 | 50 | No | Two-sided t-test |
| [21] | CBV | EGF pathway (ARAF/TRAF) | 484 (validation set) | Yes/ 21 (discovery cohort) | Cox-regression tests |
| VS | HIF1A, BNIP3L | ||||
| [13] | Blurry edge necrotic portion | GAP43 (–), WWTR1 (–) | 426 | No | Amaretto modules |
| [15] | Deep white matter tracts, ependymal invasion | myc (+) | 92 | No | Comparative marker selection (CMS) |
| [23] | Volumes of necrosis | myc (+ in female), TP53 (– in male) | 99 | Yes/ 369 (validation set) | Comparative marker selection (CMS) |
| [24] | Volume-class, hemorrhage, T1/FLAIR-envelope ratio | PGC1alpha, PAR1, HOXC6, miR-199a, miR-125 and miR-129, miR-499, miR-146b | 92 (48 of which as validation set) | No | Comparative marker selection (CMS) |
| [31] | Necrosis/contrast enhancing ratio (h), Contrast-enhancing/tumor bulk ratio (l) | EGFR (m) | 76 | No | Two-sided student’s t test |
| T2-FLAIR hyperintensity | RB1 (m) | ||||
| Necrosis/contrast enhancing volume | TP53 (m) | ||||
| Contrast enhancing volume, Tumor bulk volume | NF1 (m) | ||||
| T2-FLAIR hyperintensity, Total tumor volume, Tumor bulk/total tumor volume ratio | PDGFRA (m) | ||||
| [26] | Pre-multifocal cluster | c-Kit (+) | 144 (validation set) | Yes/121 (discovery cohort) | SAM (FDR < 15% for imaging features and FDR < 5% for signaling pathways) |
| Spherical cluster | VEGFR (–), PDGFR (–), FOXA (–), Ang/Tie2 (–) | ||||
| Rim-enhancing cluster | WNT, PDGFR-β, VEGFR, Ang/Tie2 | ||||
| [27] | PsP | IRF9 (+), XRCC1 (+) | 21 (validation set) | Yes/17 (discovery cohort) | Multi-task longitudinal sparse regression |
| [28] | PWI features (h) | ANG, VEGF-A, TGFB2 | 48 | Yes/79 (validation set) | Random forest model |
| [16] | ASD | IDH-1p/19q (m) | 110 | ||
| [33] | T1/FLAIR ratio | AMPK (–) | 57 | No | Agglomerative unsupervised hierarchical clustering (FDR < 0.25) |
| Necrosis | PI3K/AKT/mTOR (+), AMPK (–), PKA (–) | ||||
| Edema | NGF (+), GS1 signalling (+) | ||||
| [30] | Dependence non-uniformity, Difference average, Contrast and cluster prominence | EREG (+) | 46 | No | Pearson correlation analysis |
| Inverse difference zone variance, Large area emphasis, Root mean squared | EREG (–) | ||||
| Inverse difference moment | ROS1 (–) | ||||
| Contrast, Cluster prominence | TIMP1 (+) | ||||
| Inverse difference moment, Zone variance, Large area emphasis | TIMP1 (–) | ||||
| Breast Cancer (BRCA) | |||||
| [37] | Tumor enhancement dynamics | Luminal B subtype | 48 | No | Multivariate logistic regression models (associations FDR < 0.0022) |
| [38] | Surface area, Functional tumor volume, Absolute volume of BPE | Luminal A subtype | 126 (validation set) | Yes/ 84 (discovery cohort) | Multivariate logistic regression models (associations FDR < 0.25) |
| GLCM uniformity of SER map, GLCM uniformity of early enhancement map | Luminal B subtype | ||||
| Function tumor volume, Tumor surrounding BPE | Basal-Like | ||||
| [49] | Angular second moment, Energy enhancement texture | PR status | 91 | No | Logistic regression and t-test (associations FDR < 0.1) |
| [50] | Tumor size, Enhancement texture | MiR-128-1, MiR-18a, miR-19a, miR-17-92, miR-10b | 91 | No | Regression analysis and clustering analysis (FDR ≤ 0.05) |
| Effective diameter, Surface area, Lesion volume | P-cadherin | ||||
| Tumor size, Margin sharpness | JNK2 | ||||
| [45] | Heterogeneus enhancement | AR (+), ESR1 (+) | 70 | No | Unsupervised hierarchical cluster and t-test (no multiple hypothesis testing) |
| [48] | Heterogeneus enhancement | IL6, SERPINE1, DDIT4 | 126 | No/ 879 (Independent cohort) + 159 (Independent cohort, GEO data set) | Univariate analysis (associations FDR < 0.1) |
| Clear Cell Renal Cell Carcinoma (ccRCC) | |||||
| [52] | Ill-defined tumor margins | BAP1 | 103 | No | Pearson’s χ2 test and the Mann–Whitney U test (no significant associations after adjusting for multiple hypothesis testing) |
| Exophytic growth | MUC4 | ||||
| [55] | Well-defined margin | PBRM1 (m) (+) | 177 | No | Multivariate logistic regression analysis |
| Well-defined margin, Renal vein invasion, Urinary collecting system invasion | CDKN2A (m), PTEN (m) | ||||
| Others | |||||
| [57] | Presence of peritoneal disease in the pouch of Douglas, Higher number peritoneal disease sites | The mesenchymal subtype of high-grade serous ovarian cancer | 92 | No | Multivariate logistic regression analysis (associations FDR < 0.1) |
| [59] | Smaller tumor diameter and acute tumor transition angle | Contrast-induced nephropathy (CIN) status | 40 | Not specified/18 validation cohort | Multivariate logistic regression analysis (no multiple hypothesis testing) |
4. Methods
4.1. Search Strategy and Articles Selection
4.2. Graphic Representation of Feature Associations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NIMH | National Institute of Mental Health |
| NDA | National Institute of Mental Health Data Archive |
| TCGA | The Cancer Genome Atlas |
| TCIA | The Cancer Imaging Archiv |
| GBM | Glioblastoma multiforme |
| LGG | Low-grade glioma |
| MRI | Magnetic resonance imaging |
| mRNA | Messenger ribonucleic acid |
| microRNA | Micro ribonucleic acid |
| IPA | Ingenuity pathway analysis |
| POSTN | Periostin |
| KPS | Karnofsky performance status |
| VAK | Volume, Age, and KPS |
| MGMT | O6-methylguanine-DNA-methyltransferase |
| EGFR | Epidermal growth factor receptor |
| rCBVner | Relative cerebral blood volume of non-enhancing region |
| OS | Overall survival |
| PS | Permeability surface |
| CBV | Cerebral blood volume |
| VEGF | Vascular endothelial growth factor |
| HIF1A | Hypoxia-inducible factor 1-alpha |
| VASH2 | Vasohibin-2 |
| C3 | Complement component 3 |
| AMOT | Angiomotin |
| NF1 | Neurofibromin 1 |
| WHO | World health organization |
| rCBV | Relative cerebral blood volume |
| PTEN | Phosphatase and tensin homolog |
| COL15A1 | Collagen alpha-1(XV) chain |
| SPARC | Secreted protein acidic and rich in cysteine |
| ANPEP | Alanyl aminopeptidase |
| CBFB | Core-binding factor subunit beta |
| STRN | Striatin |
| TMED10 | Transmembrane P24 trafficking protein 10 |
| EGF | Epithelial growth factor |
| ARAF | A-Raf proto-oncogene |
| TRAF | TNF receptor-associated factor 1 |
| ROI | Region of interest |
| GAP43 | Growth associated protein 43 |
| WWTR1 | WW domain-containing transcription regulator 1 |
| TP53 | Tumor protein P53 |
| PGC1alpha | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PAR1 | Prader Willi/Angelman region RNA 1 |
| HOXC6 | Homeobox C6 |
| RB1 | Retinoblastoma protein |
| PDGFRA | Platelet-derived growth factor receptor A |
| AUC | Area under the curve |
| HRAS | Harvey rat sarcoma viral oncogene homolog |
| PARADIGM | Pathway recognition algorithm using data integration on genomic models |
| VEGFR | Vascular endothelial growth factor receptor |
| Ang | Angiopoietin |
| Tie2 | Tyrosine-protein kinase receptor Tie-2 |
| WNT | Wnt family member 1 |
| IRF9 | Interferon regulatory factor 9 |
| XRCC1 | X-ray cross-complementing |
| PsP | Pseudoprogression |
| TTP | True tumor progression |
| GSEA | Gene set enrichment analysis |
| PWI | Perfusion weighted imaging |
| ANG | Angiogenin |
| TGFB2 | Transforming growth factor-beta 2 |
| BEVR | Bounding ellipsoid volume ratio |
| BEDR1 | Bounding ellipsoid diameter ratio 1 |
| BEDR2 | Bounding ellipsoid diameter ratio 2 |
| MF | Margin fluctuation |
| ASD | Angular standard deviation |
| AMKP | AMP-activated protein kinase |
| PI3k | Phosphoinositide 3-kinase |
| AKT | Serine/threonine kinase 1 |
| mTOR | Mechanistic target of rapamycin kinase |
| NGF | Nerve growth factor; Phosphatidylinositol-4,5-bisphosphate 3-kinase |
| AKT | RAC-alpha serine/threonine-protein kinase |
| IL-8 | Interleukin 8 |
| CGGA | Chinese glioma genome atlas |
| TIMP1 | TIMP metallopeptidase inhibitor 1 |
| ROS1 | ROS proto-oncogene 1 |
| EREG | Epiregulin |
| DCE | Dynamic contrast-enhanced |
| PET | Positron emission tomography |
| BPE | Breast parenchymal enhancement |
| GLCM | Gray-level-co-occurrence matrix |
| PR | Progesterone receptor |
| CDK4 | Cyclin-dependent kinase 4 |
| BCL2 | B-cell lymphoma 2 |
| ER | Estrogen receptor |
| BI-RADS | Breast imaging reporting and data system |
| AR | Androgen receptor |
| ESR1 | Estrogen receptor 1 |
| IL6- | Interleukin 6 |
| DDIT4 | DNA damage-inducible transcript 4 |
| ccRCC | Clear cell renal cell carcinoma |
| pRCC | Papillary renal cell carcinoma |
| RCC | Renal cell carcinoma |
| VHL | Von Hippel–Lindau |
| CT | Computed tomography |
| BAP1 | BRCA1 associated protein 1 |
| MUC4 | Mucin 4, Cell surface associated |
| ANN | Artificial neural network |
| RF | Random forest |
| PBRM | Polybromo-1 |
| CLOVAR | Classification of ovarian cancer |
| CIN | Contrast-induced nephropathy |
| CER | Contrast-enhancing ratio |
| CEV | Contrast-enhancing volume |
References
- Hariri, A.R.; Weinberger, D.R. Imaging genomics. Br. Med. Bull. 2003, 65, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Baltrušaitis, T.; Ahuja, C.; Morency, L.P. Multimodal Machine Learning: A Survey and Taxonomy. IEEE Trans. Pattern. Anal. Mach. Intell. 2018, 41. [Google Scholar] [CrossRef]
- Bodalal, Z.; Trebeschi, S.; Nguyen-Kim, T.D.L.; Schats, W.; Beets-Tan, R. Radiogenomics: Bridging imaging and genomics. Abdom. Radiol. 2019, 44, 1960–1984. [Google Scholar] [CrossRef] [PubMed]
- NIMH Data Archive. Available online: https://nda.nih.gov/ (accessed on 29 November 2019).
- The Cancer Genome Atlas Program. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 29 November 2019).
- Cancer Imaging Archive. Available online: https://www.cancerimagingarchive.net/ (accessed on 29 November 2019).
- Huret, J.L.; Ahmad, M.; Arsaban, M.; Bernheim, A.; Cigna, J.; Desangles, F.; Guignard, J.C.; Jacquemot-Perbal, M.C.; Labarussias, M.; Leberre, V.; et al. Atlas of genetics and cytogenetics in oncology and haematology in 2013. Nucleic Acids Res. 2013, 41, D920–D924. [Google Scholar] [CrossRef]
- Davis, M.E. Glioblastoma: Overview of disease and treatment. Clin. J. Oncol. Nurs. 2016, 20, S2. [Google Scholar] [CrossRef]
- Kazerooni, A.F.; Bakas, S.; Saligheh, H.; Davatzikos, R.C. Imaging signatures of glioblastoma molecular characteristics: A radiogenomics review. J. Magn. Reson. Imaging 2019. [CrossRef]
- Zinn, P.O.; Mahajan, B.; Sathyan, P.; Singh, S.K.; Majumder, S.; Jolesz, F.A.; Colen, R.R. Radiogenomic mapping of edema/cellular invasion MRI-phenotypes in glioblastoma multiforme. PLoS ONE 2011, 6, e25451. [Google Scholar] [CrossRef]
- Gusev, Y.; Bhuvaneshwar, K.; Song, L.; Zenklusen, J.C.; Fine, H.; Madhavan, S. The REMBRANDT study, a large collection of genomic data from brain cancer patients. Sci. Data 2018, 5, 180158. [Google Scholar] [CrossRef]
- Zinn, P.O.; Sathyan, P.; Mahajan, B.; Bruyere, J.; Hegi, M.; Majumder, S.; Colen, R.R. A novel volume-age-KPS (VAK) glioblastoma classification identifies a prognostic cognate microRNA-gene signature. PLoS ONE 2012, 7, e41522. [Google Scholar] [CrossRef]
- Gevaert, O.; Mitchell, L.A.; Achrol, A.S.; Xu, J.; Echegaray, S.; Steinberg, G.K.; Cheshier, S.H.; Napel, S.; Zaharchuk, G.; Plevritis, S.K. Glioblastoma multiforme: Exploratory radiogenomic analysis by using quantitative image features. Radiology 2014, 273, 168–174. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Colen, R.R.; Vangel, M.; Wang, J.; Gutman, D.A.; Hwang, S.N.; Wintermark, M.; Jain, R.; Jilwan-Nicolas, M.; Chen, J.Y.; Raghavan, P.; et al. Imaging genomic mapping of an invasive MRI phenotype predicts patient outcome and metabolic dysfunction: A TCGA glioma phenotype research group project. BMC Med. Genom. 2014, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Mazurowski, M.A.; Clark, K.; Czarnek, N.M.; Shamsesfandabadi, P.; Peters, K.B.; Saha, A. Radiogenomics of lower-grade glioma: Algorithmically-assessed tumor shape is associated with tumor genomic subtypes and patient outcomes in a multi-institutional study with The Cancer Genome Atlas data. J. Neurooncol. 2017, 133, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; Morozova, O.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef]
- Jain, R.; Poisson, L.M.; Gutman, D.; Scarpace, L.; Hwang, S.N.; Holder, C.A.; Wintermark, M.; Rao, A.; Colen, R.R.; Kirby, J.; et al. Outcome prediction in patients with glioblastoma by using imaging, clinical, and genomic biomarkers: Focus on the nonenhancing component of the tumor. Radiology 2014, 272, 484–493. [Google Scholar] [CrossRef]
- Jain, R.; Poisson, L.; Narang, J.; Scarpace, L.; Rosenblum, M.L.; Rempel, S.; Mikkelsen, T. Correlation of perfusion parameters with genes related to angiogenesis regulation in glioblastoma: A feasibility study. AJNR Am. J. Neuroradiol. 2012, 33, 1343–1348. [Google Scholar] [CrossRef]
- Rao, A.; Manyam, G.; Rao, G.; Jain, R. Integrative Analysis of mRNA, microRNA, and Protein Correlates of Relative Cerebral Blood Volume Values in GBM Reveals the Role for Modulators of Angiogenesis and Tumor Proliferation. Cancer Inform. 2016, 15, 29–33. [Google Scholar] [CrossRef]
- Heiland, D.H.; Demerath, T.; Kellner, E.; Kiselev, V.G.; Pfeifer, D.; Schnell, O.; Staszewski, O.; Urbach, H.; Weyerbrock, A.; Mader, I. Molecular differences between cerebral blood volume and vessel size in glioblastoma multiforme. Oncotarget 2017, 8, 11083–11093. [Google Scholar] [CrossRef]
- Gutman, D.A.; Cooper, L.A.; Hwang, S.N.; Holder, C.A.; Gao, J.; Aurora, T.D.; Dunn, W.D., Jr.; Scarpace, L.; Mikkelsen, T.; Jain, R.; et al. MR imaging predictors of molecular profile and survival: Multi-institutional study of the TCGA glioblastoma data set. Radiology 2013, 267, 560–569. [Google Scholar] [CrossRef]
- Colen, R.R.; Wang, J.; Singh, S.K.; Gutman, D.A.; Zinn, P.O. Glioblastoma: Imaging Genomic Mapping Reveals Sex-specific Oncogenic Associations of Cell Death. Radiology 2014, 275, 215–227. [Google Scholar] [CrossRef]
- Rao, A.; Rao, G.; Gutman, D.A.; Flanders, A.E.; Hwang, S.N.; Rubin, D.L.; Colen, R.R.; Zinn, P.O.; Jain, R.; Wintermark, M.; et al. A combinatorial radiographic phenotype may stratify patient survival and be associated with invasion and proliferation characteristics in glioblastoma. J. Neurosurg. 2016, 124, 1008–1017. [Google Scholar] [CrossRef]
- Vaske, C.J.; Benz, S.C.; Sanborn, J.Z.; Earl, D.; Szeto, C.; Zhu, J.; Haussler, D.; Stuart, J.M. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 2010, 26, i237–i245. [Google Scholar] [CrossRef] [PubMed]
- Itakura, H.; Achrol, A.S.; Mitchell, L.A.; Loya, J.J.; Liu, T.; Westbroek, E.M.; Feroze, A.H.; Rodriguez, S.; Echegaray, S.; Azad, T.D.; et al. Magnetic resonance image features identify glioblastoma phenotypic subtypes with distinct molecular pathway activities. Sci. Transl. Med. 2015, 7, 303ra138. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Tan, H.; Zhang, J.; Liu, K.; Yang, T.; Wang, M.; Debinskie, W.; Zhao, W.; Chan, M.D.; Zhou, X. Identification of biomarkers for pseudo and true progression of GBM based on radiogenomics study. Oncotarget 2016, 7, 55377–55394. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.T.; Achrol, A.S.; Mitchell, L.A.; Rodriguez, S.A.; Feroze, A.; Iv, M.; Kim, C.; Chaudhary, N.; Gevaert, O.; Stuart, J.M.; et al. Magnetic resonance perfusion image features uncover an angiogenic subgroup of glioblastoma patients with poor survival and better response to antiangiogenic treatment. Neuro. Oncol. 2017, 19, 997–1007. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.; Qian, Z.; Sun, Z.; Xu, K.; Wang, K.; Liu, S.; Fan, X.; Li, S.; Zhang, Z.; et al. A radiomic signature as a non-invasive predictor of progression-free survival in patients with lower-grade gliomas. Neuroimage Clin. 2018, 20, 1070–1077. [Google Scholar] [CrossRef]
- Liao, X.; Cai, B.; Tian, B.; Luo, Y.; Song, W.; Li, Y. Machine-learning based radiogenomics analysis of MRI features and metagenes in glioblastoma multiforme patients with different survival time. J. Cell Mol. Med. 2019, 23, 4375–4385. [Google Scholar] [CrossRef]
- Gutman, D.A.; Dunn, W.D., Jr.; Grossmann, P.; Cooper, L.A.; Holder, C.A.; Ligon, K.L.; Alexander, B.M.; Aerts, H.J. Somatic mutations associated with MRI-derived volumetric features in glioblastoma. Neuroradiology 2015, 57, 1227–1237. [Google Scholar] [CrossRef]
- Nicolasjilwan, M.; Hu, Y.; Yan, C.; Meerzaman, D.; Holder, C.A.; Gutman, D.; Jain, R.; Colen, R.; Rubin, D.L.; Zinn, P.O.; et al. Addition of MR imaging features and genetic biomarkers strengthens glioblastoma survival prediction in TCGA patients. J. Neuroradiol. 2015, 42, 212–221. [Google Scholar] [CrossRef]
- Lehrer, M.; Bhadra, A.; Ravikumar, V.; Chen, J.Y.; Wintermark, M.; Hwang, S.N.; Holder, C.A.; Huang, E.P.; Fevrier-Sullivan, B.; Freymann, J.B.; et al. Multiple-response regression analysis links magnetic resonance imaging features to de-regulated protein expression and pathway activity in lower grade glioma. Oncoscience 2017, 4, 57–66. [Google Scholar] [CrossRef][Green Version]
- Pinker, K.; Chin, J.; Melsaether, A.N.; Morris, E.A.; Moy, L. Precision Medicine and Radiogenomics in Breast Cancer: New Approaches toward Diagnosis and Treatment. Radiology 2018, 287, 732–747. [Google Scholar] [CrossRef] [PubMed]
- Incoronato, M.; Grimaldi, A.M.; Mirabelli, P.; Cavaliere, C.; Parente, C.A.; Franzese, M.; Staibano, S.; Ilardi, G.; Russo, D.; Soricelli, A.; et al. Circulating miRNAs in Untreated Breast Cancer: An Exploratory Multimodality Morpho-Functional Study. Cancers 2019, 11, 876. [Google Scholar] [CrossRef] [PubMed]
- Schiano, C.; Franzese, M.; Pane, K.; Garbino, N.; Soricelli, A.; Salvatore, M.; de Nigris, F.; Napoli, C. Hybrid 18F-FDG-PET/MRI Measurement of Standardized Uptake Value Coupled with Yin Yang 1 Signature in Metastatic Breast Cancer. A Preliminary Study. Cancers 2019, 26, 11. [Google Scholar] [CrossRef] [PubMed]
- Mazurowski, M.A.; Zhang, J.; Grimm, L.J.; Yoon, S.C.; Silber, J.I. Radiogenomic analysis of breast cancer: Luminal B molecular subtype is associated with enhancement dynamics at MR imaging. Radiology 2014, 273, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, X.; Wang, J.; Cui, Y.; Kato, F.; Shirato, H.; Ikeda, D.M.; Li., R. Identifying relations between imaging phenotypes and molecular subtypes of breast cancer: Model discovery and external validation. J. Magn. Reson. Imaging 2017, 46, 1017–1027. [Google Scholar] [CrossRef]
- Li, H.; Zhu, Y.; Burnside, E.S.; Drukker, K.; Hoadley, K.A.; Fan, C.; Conzen, S.D.; Zuley, M.; Net, J.M.; Sutton, E.; et al. Quantitative MRI radiomics in the prediction of molecular classifications of breast cancer subtypes in the TCGA/TCIA data set. NPJ Breast Cancer 2016, 2, 16012. [Google Scholar] [CrossRef]
- Li, H.; Zhu, Y.; Burnside, E.S.; Hoadley, K.A.; Fan, C.; Conzen, S.D.; Whitman, G.J.; Sutton, E.J.; Net, J.M.; Ganott, M.; et al. MR Imaging Radiomics Signatures for Predicting the Risk of Breast Cancer Recurrence as Given by Research Versions of MammaPrint, Oncotype DX, and PAM50 Gene Assays. Radiology 2016, 281, 382–391. [Google Scholar] [CrossRef]
- van‘t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Wu, J.; Cui, Y.; Sun, X.; Cao, G.; Li, B.; Ikeda, D.M.; Kurian, A.W.; Li, R. Unsupervised Clustering of Quantitative Image Phenotypes Reveals Breast Cancer Subtypes with Distinct Prognoses and Molecular Pathways. Clin. Cancer Res. 2017, 23, 3334–3342. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.R.; Ku, Y.J.; Cho, S.G.; Kim, S.J.; Min, B.S. Associations between gene expression profiles of invasive breast cancer and Breast Imaging Reporting and Data System MRI lexicon. Ann. Surg. Treat. Res. 2017, 93, 18–26. [Google Scholar] [CrossRef] [PubMed]
- D’Orsi, C.J. ACR BI-RADS Atlas: Breast Imaging Reporting and Data System, 5th ed.; American College of Radiology: Reston, VA, USA, 2013. [Google Scholar]
- Fan, M.; Xia, P.; Liu, B.; Zhang, L.; Wang, Y.; Gao, X.; Li, L. Tumour heterogeneity revealed by unsupervised decomposition of dynamic contrast-enhanced magnetic resonance imaging is assReferences. Breast Cancer Res. 2019, 21, 112. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, B.; Sun, X.; Cao, G.; Rubin, D.L.; Napel, S.; Ikeda, D.M.; Kurian, A.W.; Li, R. Heterogeneous Enhancement Patterns of Tumor-adjacent Parenchyma at MR Imaging Are Associated with Dysregulated Signaling Pathways and Poor Survival in Breast Cancer. Radiology 2017, 285, 401–413. [Google Scholar] [CrossRef]
- Guo, W.; Li, H.; Zhu, Y.; Lan, L.; Yang, S.; Drukker, K.; Morris, E.; Burnside, E.; Whitman, G.; Giger, M.L.; et al. Prediction of clinical phenotypes in invasive breast carcinomas from the integration of radiomics and genomics data. J. Med. Imaging 2015, 2, 041007. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, H.; Guo, W.; Drukker, K.; Lan, L.; Giger, M.L.; Ji, Y. Deciphering Genomic Underpinnings of Quantitative MRI-based Radiomic Phenotypes of Invasive Breast Carcinoma. Sci. Rep. 2015, 5, 17787. [Google Scholar] [CrossRef]
- Alessandrino, F.; Shinagare, A.B.; Bossé, D.; Choueiri, T.K.; Krajewski, K.M. Radiogenomics in renal cell carcinoma. Abdom. Radiol. 2019, 44, 1990–1998. [Google Scholar] [CrossRef]
- Shinagare, A.B.; Vikram, R.; Jaffe, C.; Akin, O.; Kirby, J.; Huang, E.; Freymann, J.; Sainani, N.I.; Sadow, C.A.; Bathala, T.K.; et al. Radiogenomics of clear cell renal cell carcinoma: Preliminary findings of The Cancer Genome Atlas-Renal Cell Carcinoma (TCGA-RCC) Imaging Research Group. Abdom. Imaging 2015, 40, 1684–1692. [Google Scholar] [CrossRef]
- Ghosh, P.; Tamboli, P.; Vikram, R.; Rao, A. Imaging-genomic pipeline for identifying gene mutations using three-dimensional intra-tumor heterogeneity features. J. Med. Imaging 2015, 2, 041009. [Google Scholar] [CrossRef]
- Kocak, B.; Durmaz, E.S.; Ates, E.; Ulusan, M.B. Radiogenomics in Clear Cell Renal Cell Carcinoma: Machine Learning-Based High-Dimensional Quantitative CT Texture Analysis in Predicting PBRM1 Mutation Status. Am. J. Roentgenol. 2019, 212, W55–W63. [Google Scholar] [CrossRef]
- Bowen, L.; Xiaojing, L. Radiogenomics of Clear Cell Renal Cell Carcinoma: Associations Between mRNA-Based Subtyping and CT Imaging Features. Acad. Radiol. 2019, 26, e32–e37. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Vargas, H.A.; Huang, E.P.; Lakhman, Y.; Ippolito, J.E.; Bhosale, P.; Mellnick, V.; Shinagare, A.B.; Anello, M.; Kirby, J.; Fevrier-Sullivan, B.; et al. Radiogenomics of High-Grade Serous Ovarian Cancer: Multireader Multi-Institutional Study from the Cancer Genome Atlas Ovarian Cancer Imaging Research Group. Radiology 2017, 285, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Tamayo, P.; Yang, J.Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Investig. 2013, 123, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.C.; Yeh, T.S.; Wu, R.C.; Tsai, C.K.; Yang., L.Y.; Lin, G.; Kuo, M.D. Acute Tumor Transition Angle on Computed Tomography Predicts Chromosomal Instability Status of Primary Gastric Cancer: Radiogenomics Analysis from TCGA and Independent Validation. Cancers 2019, 11, 641. [Google Scholar] [CrossRef]
- Incoronato, M.; Aiello, M.; Infante, T.; Cavaliere, C.; Grimaldi, A.M.; Mirabelli, P.; Monti, S.; Salvatore, M. Radiogenomic Analysis of Oncological Data: A Technical Survey. Int. J. Mol. Sci. 2017, 18, 805. [Google Scholar] [CrossRef]
- Jansen, R.W.; van Amstel, P.; Martens, R.M.; Kooi, I.E.; Wesseling, P.; de Langen, A.J.; Menke-Van der Houven van Oordt, C.W.; Jansen, B.H.E.; Moll, A.C.; Dorsman., J.C.; et al. Non-invasive tumor genotyping using radiogenomic biomarkers, a systematic review and oncology-wide pathway analysis. Oncotarget 2018, 9, 20134–20155. [Google Scholar] [CrossRef]
- Zanfardino, M.; Franzese, M.; Pane, K.; Cavaliere, C.; Monti, S.; Esposito, G.; Salvatore, M.; Aiello, M. Bringing radiomics into a multi-omics framework for a comprehensive genotype-phenotype characterization of oncological diseases. J. Transl. Med. 2019, 17, 337. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]


| Tumor Type | TCGA/TCIA Project | Sum of Study |
|---|---|---|
| Glioblastoma and low grade glioma | GBM and LGG | 21 |
| Breast cancer | BRCA | 10 |
| Clear cell renal carcinoma | KIRC | 2 |
| Other | OV and STAD | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanfardino, M.; Pane, K.; Mirabelli, P.; Salvatore, M.; Franzese, M. TCGA-TCIA Impact on Radiogenomics Cancer Research: A Systematic Review. Int. J. Mol. Sci. 2019, 20, 6033. https://doi.org/10.3390/ijms20236033
Zanfardino M, Pane K, Mirabelli P, Salvatore M, Franzese M. TCGA-TCIA Impact on Radiogenomics Cancer Research: A Systematic Review. International Journal of Molecular Sciences. 2019; 20(23):6033. https://doi.org/10.3390/ijms20236033
Chicago/Turabian StyleZanfardino, Mario, Katia Pane, Peppino Mirabelli, Marco Salvatore, and Monica Franzese. 2019. "TCGA-TCIA Impact on Radiogenomics Cancer Research: A Systematic Review" International Journal of Molecular Sciences 20, no. 23: 6033. https://doi.org/10.3390/ijms20236033
APA StyleZanfardino, M., Pane, K., Mirabelli, P., Salvatore, M., & Franzese, M. (2019). TCGA-TCIA Impact on Radiogenomics Cancer Research: A Systematic Review. International Journal of Molecular Sciences, 20(23), 6033. https://doi.org/10.3390/ijms20236033