p53 Isoforms in Cellular Senescence- and Ageing-Associated Biological and Physiological Functions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
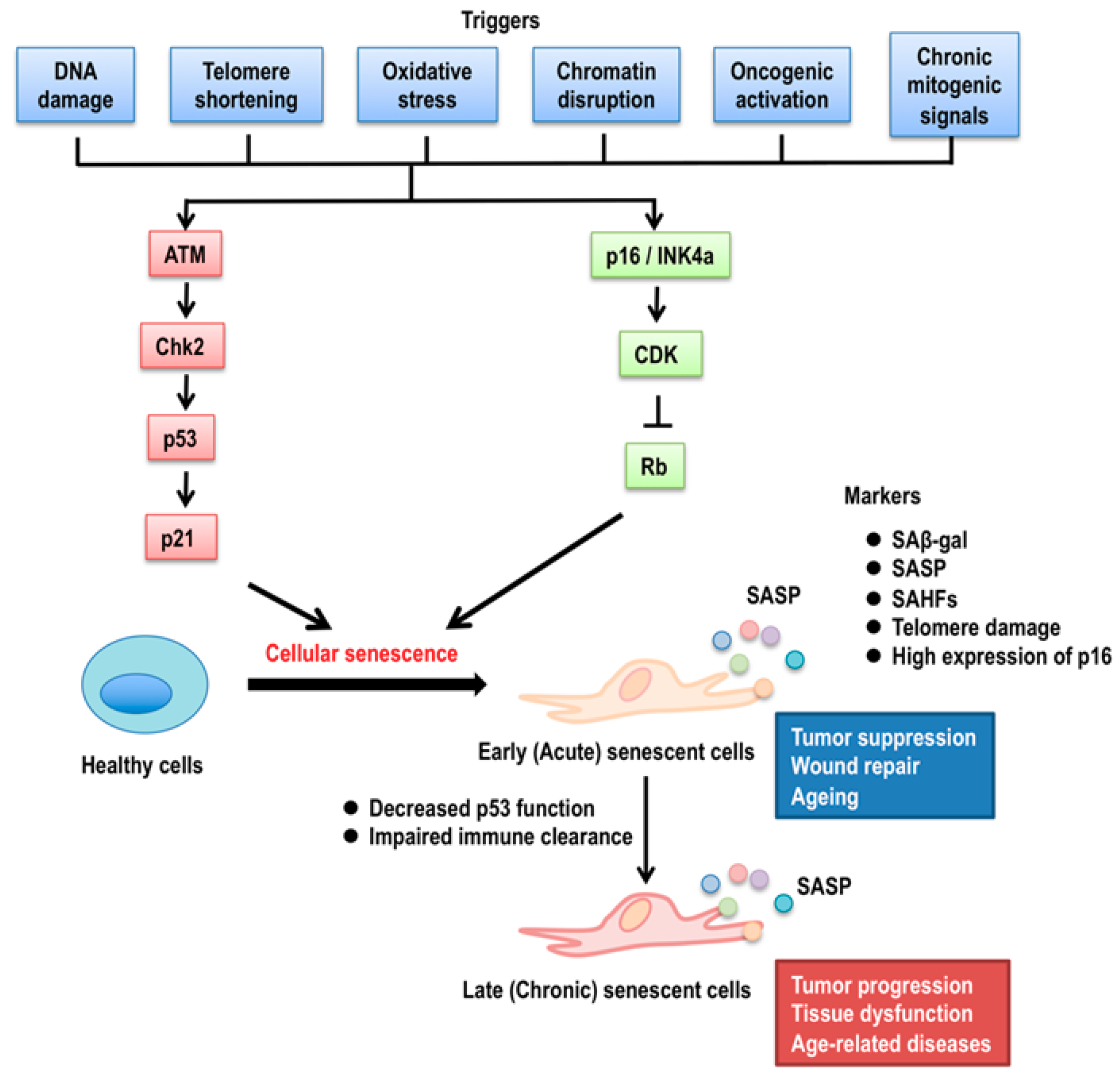
2. p53 Isoforms in Cellular Senescence
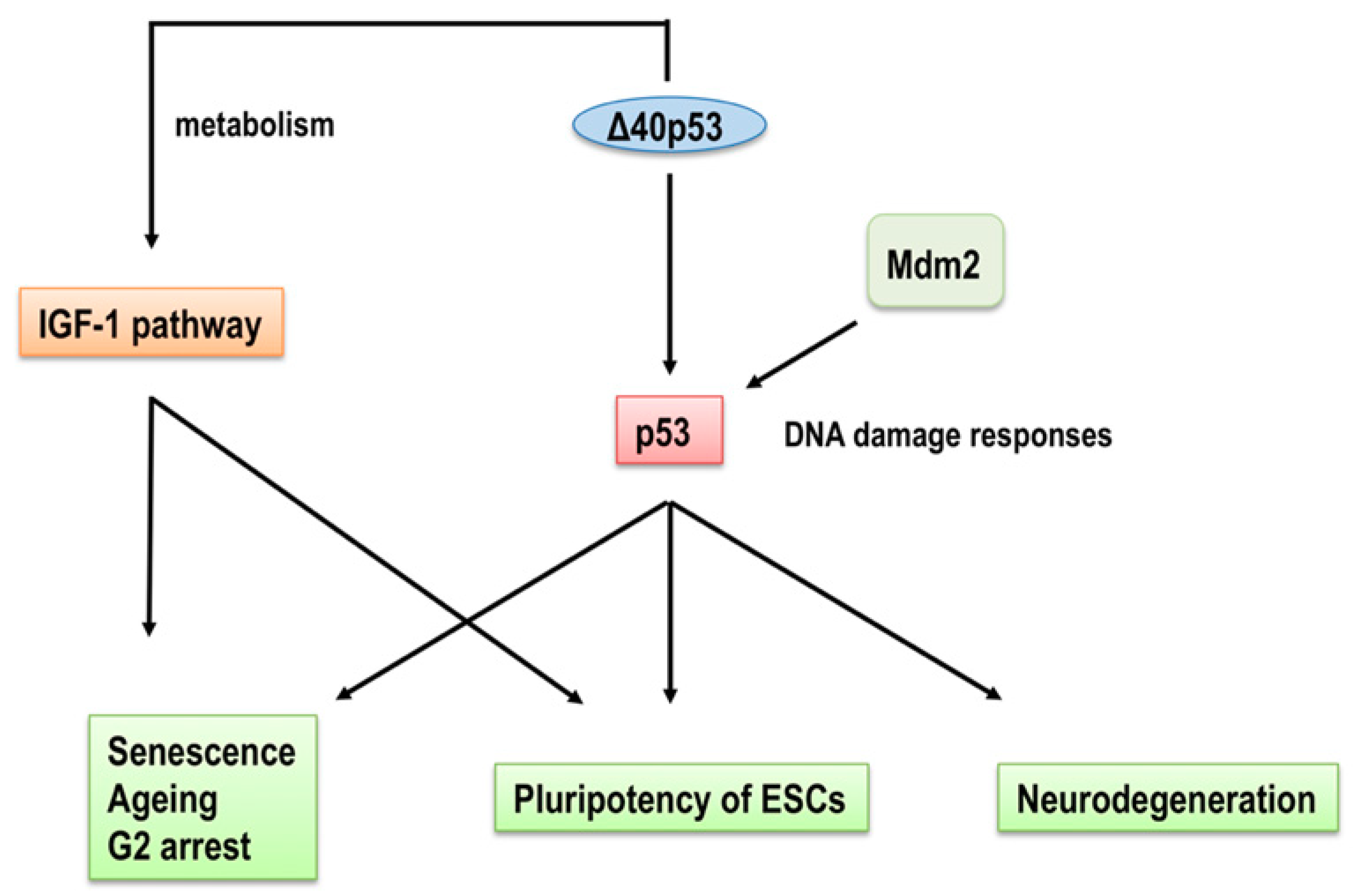
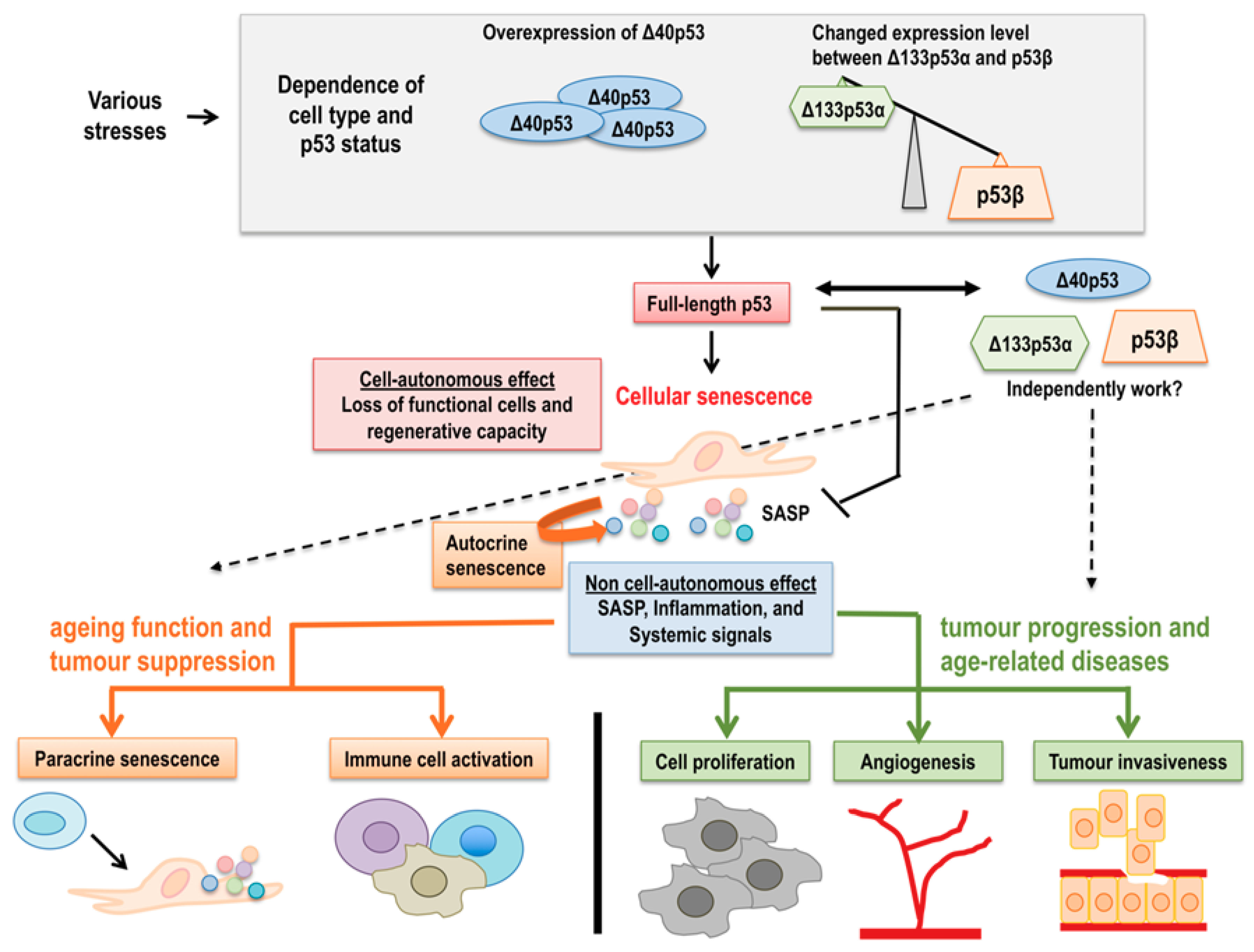
3. p53 Isoforms in Ageing and Age-Related Functional Decline
4. p53 Isoforms in Cell Reprogramming to Pluripotent Cells
5. p53 Isoforms in Cancer
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The establishment of a line (WISH) of human amnion cells in continuous cultivation. Exp. Cell Res. 1961, 23, 14–20. [Google Scholar] [CrossRef]
- Harley, C.B.; Vaziri, H.; Counter, C.M.; Allsopp, R.C. The telomere hypothesis of cellular aging. Exp. Gerontol. 1992, 27, 375–382. [Google Scholar] [CrossRef]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Zhu, J.; Woods, D.; McMahon, M.; Bishop, J.M. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998, 12, 2997–3007. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S. Replicative senescence: The human fibroblast comes of age. Science 1990, 249, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Narita, M.; Krizhanovsky, V.; Nunez, S.; Chicas, A.; Hearn, S.A.; Myers, M.P.; Lowe, S.W. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 2006, 126, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Kauser, K.; Campisi, J.; Beausejour, C.M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 2006, 281, 29568–29574. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, K.; Jorres, A.; Witowski, J. Senescence induces a proangiogenic switch in human peritoneal mesothelial cells. Rejuvenation Res. 2008, 11, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Krtolica, A.; Beausejour, C.M.; Parrinello, S.; Hodgson, J.G.; Chin, K.; Desprez, P.Y.; Campisi, J. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS ONE 2010, 5, e9188. [Google Scholar] [CrossRef]
- Millis, A.J.; Hoyle, M.; McCue, H.M.; Martini, H. Differential expression of metalloproteinase and tissue inhibitor of metalloproteinase genes in aged human fibroblasts. Exp. Cell Res. 1992, 201, 373–379. [Google Scholar] [CrossRef]
- Kang, M.K.; Kameta, A.; Shin, K.H.; Baluda, M.A.; Kim, H.R.; Park, N.H. Senescence-associated genes in normal human oral keratinocytes. Exp. Cell Res. 2003, 287, 272–281. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Canamero, M.; Maraver, A.; Gomez-Lopez, G.; Contreras, J.; Murillo-Cuesta, S.; Rodriguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef] [PubMed]
- Pribluda, A.; Elyada, E.; Wiener, Z.; Hamza, H.; Goldstein, R.E.; Biton, M.; Burstain, I.; Morgenstern, Y.; Brachya, G.; Billauer, H.; et al. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell 2013, 24, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular senescence in aging primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sanoff, H.K.; Cho, H.; Burd, C.E.; Torrice, C.; Ibrahim, J.G.; Thomas, N.E.; Sharpless, N.E. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell 2009, 8, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Helman, A.; Klochendler, A.; Azazmeh, N.; Gabai, Y.; Horwitz, E.; Anzi, S.; Swisa, A.; Condiotti, R.; Granit, R.Z.; Nevo, Y.; et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med. 2016, 22, 412–420. [Google Scholar] [CrossRef]
- Wang, C.; Jurk, D.; Maddick, M.; Nelson, G.; Martin-Ruiz, C.; von Zglinicki, T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009, 8, 311–323. [Google Scholar] [CrossRef]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef]
- Jung, J.H.; Bae, S.; Lee, J.Y.; Woo, S.R.; Cha, H.J.; Yoon, Y.; Suh, K.S.; Lee, S.J.; Park, I.C.; Jin, Y.W.; et al. E3 ubiquitin ligase Hades negatively regulates the exonuclear function of p53. Cell Death Differ. 2011, 18, 1865–1875. [Google Scholar] [CrossRef]
- Gao, W.; Shen, Z.; Shang, L.; Wang, X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. 2011, 18, 1598–1607. [Google Scholar] [CrossRef]
- Gogna, R.; Madan, E.; Kuppusamy, P.; Pati, U. Re-oxygenation causes hypoxic tumor regression through restoration of p53 wild-type conformation and post-translational modifications. Cell Death Dis. 2012, 3, e286. [Google Scholar] [CrossRef]
- Tapia, N.; Scholer, H.R. p53 connects tumorigenesis and reprogramming to pluripotency. J. Exp. Med. 2010, 207, 2045–2048. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Kanagawa, O.; Nakagawa, M.; Okita, K.; Yamanaka, S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 2009, 460, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Lowe, S.W. Stem cells: The promises and perils of p53. Nature 2009, 460, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Marion, R.M.; Strati, K.; Li, H.; Murga, M.; Blanco, R.; Ortega, S.; Fernandez-Capetillo, O.; Serrano, M.; Blasco, M.A. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 2009, 460, 1149–1153. [Google Scholar] [CrossRef]
- Menendez, S.; Camus, S.; Izpisua Belmonte, J.C. p53: Guardian of reprogramming. Cell Cycle 2010, 9, 3887–3891. [Google Scholar] [CrossRef]
- Sarig, R.; Rivlin, N.; Brosh, R.; Bornstein, C.; Kamer, I.; Ezra, O.; Molchadsky, A.; Goldfinger, N.; Brenner, O.; Rotter, V. Mutant p53 facilitates somatic cell reprogramming and augments the malignant potential of reprogrammed cells. J. Exp. Med. 2010, 207, 2127–2140. [Google Scholar] [CrossRef]
- Zhao, T.; Xu, Y. p53 and stem cells: New developments and new concerns. Trends Cell Biol. 2010, 20, 170–175. [Google Scholar] [CrossRef]
- Li, M.; He, Y.; Dubois, W.; Wu, X.; Shi, J.; Huang, J. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol. Cell 2012, 46, 30–42. [Google Scholar] [CrossRef]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef]
- Yu, X.; Narayanan, S.; Vazquez, A.; Carpizo, D.R. Small molecule compounds targeting the p53 pathway: Are we finally making progress? Apoptosis 2014, 19, 1055–1068. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Gatta, R.; Dolfini, D.; Mantovani, R. NF-Y joins E2Fs, p53 and other stress transcription factors at the apoptosis table. Cell Death Dis. 2011, 2, e162. [Google Scholar] [CrossRef] [PubMed]
- Osawa, T.; Davies, D.; Hartley, J.A. Mechanism of cell death resulting from DNA interstrand cross-linking in mammalian cells. Cell Death Dis. 2011, 2, e187. [Google Scholar] [CrossRef] [PubMed]
- Medema, R.H.; Macurek, L. Checkpoint control and cancer. Oncogene 2012, 31, 2601–2613. [Google Scholar] [CrossRef] [PubMed]
- Sermeus, A.; Michiels, C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011, 2, e164. [Google Scholar] [CrossRef]
- Qin, J.Z.; Chaturvedi, V.; Denning, M.F.; Bacon, P.; Panella, J.; Choubey, D.; Nickoloff, B.J. Regulation of apoptosis by p53 in UV-irradiated human epidermis, psoriatic plaques and senescent keratinocytes. Oncogene 2002, 21, 2991–3002. [Google Scholar] [CrossRef]
- Lewis, D.A.; Yi, Q.; Travers, J.B.; Spandau, D.F. UVB-induced senescence in human keratinocytes requires a functional insulin-like growth factor-1 receptor and p53. Mol. Biol. Cell 2008, 19, 1346–1353. [Google Scholar] [CrossRef]
- Tavana, O.; Benjamin, C.L.; Puebla-Osorio, N.; Sang, M.; Ullrich, S.E.; Ananthaswamy, H.N.; Zhu, C. Absence of p53-dependent apoptosis leads to UV radiation hypersensitivity, enhanced immunosuppression and cellular senescence. Cell Cycle 2010, 9, 3328–3336. [Google Scholar] [CrossRef]
- Al-Ejeh, F.; Kumar, R.; Wiegmans, A.; Lakhani, S.R.; Brown, M.P.; Khanna, K.K. Harnessing the complexity of DNA-damage response pathways to improve cancer treatment outcomes. Oncogene 2010, 29, 6085–6098. [Google Scholar] [CrossRef]
- Wong, K.K.; Maser, R.S.; Bachoo, R.M.; Menon, J.; Carrasco, D.R.; Gu, Y.; Alt, F.W.; DePinho, R.A. Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature 2003, 421, 643–648. [Google Scholar] [CrossRef]
- Dulic, V.; Drullinger, L.F.; Lees, E.; Reed, S.I.; Stein, G.H. Altered regulation of G1 cyclins in senescent human diploid fibroblasts: Accumulation of inactive cyclin E-Cdk2 and cyclin D1-Cdk2 complexes. Proc. Natl. Acad. Sci. USA 1993, 90, 11034–11038. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Wei, W.; Dutriaux, A.; Jobling, W.A.; Sedivy, J.M. Real-time imaging of transcriptional activation in live cells reveals rapid up-regulation of the cyclin-dependent kinase inhibitor gene CDKN1A in replicative cellular senescence. Aging Cell 2003, 2, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Lanigan, F.; Geraghty, J.G.; Bracken, A.P. Transcriptional regulation of cellular senescence. Oncogene 2011, 30, 2901–2911. [Google Scholar] [CrossRef]
- Wang, X.D.; Lapi, E.; Sullivan, A.; Ratnayaka, I.; Goldin, R.; Hay, R.; Lu, X. SUMO-modified nuclear cyclin D1 bypasses Ras-induced senescence. Cell Death Differ. 2011, 18, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Chandrasekaran, C.; Gordon, J.I.; Beach, D.; Jacks, T.; Hannon, G.J. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 1995, 377, 552–557. [Google Scholar] [CrossRef]
- Sherr, C.J. The Pezcoller lecture: Cancer cell cycles revisited. Cancer Res. 2000, 60, 3689–3695. [Google Scholar]
- Brown, J.P.; Wei, W.; Sedivy, J.M. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science 1997, 277, 831–834. [Google Scholar] [CrossRef]
- McConnell, B.B.; Starborg, M.; Brookes, S.; Peters, G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr. Biol. 1998, 8, 351–354. [Google Scholar] [CrossRef]
- Chang, B.D.; Watanabe, K.; Broude, E.V.; Fang, J.; Poole, J.C.; Kalinichenko, T.V.; Roninson, I.B. Effects of p21Waf1/Cip1/Sdi1 on cellular gene expression: Implications for carcinogenesis, senescence, and age-related diseases. Proc. Natl. Acad. Sci. USA 2000, 97, 4291–4296. [Google Scholar] [CrossRef]
- Fang, L.; Igarashi, M.; Leung, J.; Sugrue, M.M.; Lee, S.W.; Aaronson, S.A. p21Waf1/Cip1/Sdi1 induces permanent growth arrest with markers of replicative senescence in human tumor cells lacking functional p53. Oncogene 1999, 18, 2789–2797. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, W.; Gao, Y.F.; Su, X.Q.; Zhai, Z.H. Senescence-like changes induced by expression of p21(waf1/Cip1) in NIH3T3 cell line. Cell Res. 2002, 12, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Matlashewski, G.; Lamb, P.; Pim, D.; Peacock, J.; Crawford, L.; Benchimol, S. Isolation and characterization of a human p53 cDNA clone: Expression of the human p53 gene. EMBO J. 1984, 3, 3257–3262. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Harris, N.; Goldfinger, N.; Rotter, V. Isolation of a full-length mouse cDNA clone coding for an immunologically distinct p53 molecule. Mol. Cell Biol. 1985, 5, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Laver-Rudich, Z.; Rotter, V. In vitro expression of human p53 cDNA clones and characterization of the cloned human p53 gene. Mol. Cell Biol. 1985, 5, 1887–1893. [Google Scholar] [CrossRef]
- Wolf, D.; Rotter, V. Major deletions in the gene encoding the p53 tumor antigen cause lack of p53 expression in HL-60 cells. Proc. Natl. Acad. Sci. USA 1985, 82, 790–794. [Google Scholar] [CrossRef]
- Courtois, S.; Caron de Fromentel, C.; Hainaut, P. p53 protein variants: Structural and functional similarities with p63 and p73 isoforms. Oncogene 2004, 23, 631–638. [Google Scholar] [CrossRef][Green Version]
- Marcel, V.; Hainaut, P. p53 isoforms—A conspiracy to kidnap p53 tumor suppressor activity? Cell. Mol. Life Sci. 2009, 66, 391–406. [Google Scholar] [CrossRef]
- Hollstein, M.; Hainaut, P. Massively regulated genes: The example of TP53. J. Pathol. 2010, 220, 164–173. [Google Scholar] [CrossRef]
- Marine, J.C.; Lozano, G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 2010, 17, 93–102. [Google Scholar] [CrossRef]
- Bourdon, J.C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef]
- Marcel, V.; Vijayakumar, V.; Fernandez-Cuesta, L.; Hafsi, H.; Sagne, C.; Hautefeuille, A.; Olivier, M.; Hainaut, P. p53 regulates the transcription of its Delta133p53 isoform through specific response elements contained within the TP53 P2 internal promoter. Oncogene 2010, 29, 2691–2700. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Stephen, C.W.; Luciani, M.G.; Fahraeus, R. p53 Stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat. Cell Biol. 2002, 4, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Courtois, S.; Verhaegh, G.; North, S.; Luciani, M.G.; Lassus, P.; Hibner, U.; Oren, M.; Hainaut, P. DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene 2002, 21, 6722–6728. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Stewart, D.; Matlashewski, G. Regulation of human p53 activity and cell localization by alternative splicing. Mol. Cell Biol. 2004, 24, 7987–7997. [Google Scholar] [CrossRef] [PubMed]
- Candeias, M.M.; Powell, D.J.; Roubalova, E.; Apcher, S.; Bourougaa, K.; Vojtesek, B.; Bruzzoni-Giovanelli, H.; Fahraeus, R. Expression of p53 and p53/47 are controlled by alternative mechanisms of messenger RNA translation initiation. Oncogene 2006, 25, 6936–6947. [Google Scholar] [CrossRef] [PubMed]
- Candeias, M.M.; Malbert-Colas, L.; Powell, D.J.; Daskalogianni, C.; Maslon, M.M.; Naski, N.; Bourougaa, K.; Calvo, F.; Fahraeus, R. P53 mRNA controls p53 activity by managing Mdm2 functions. Nat. Cell Biol. 2008, 10, 1098–1105. [Google Scholar] [CrossRef]
- Bourougaa, K.; Naski, N.; Boularan, C.; Mlynarczyk, C.; Candeias, M.M.; Marullo, S.; Fahraeus, R. Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol. Cell 2010, 38, 78–88. [Google Scholar] [CrossRef]
- Olivares-Illana, V.; Fahraeus, R. p53 isoforms gain functions. Oncogene 2010, 29, 5113–5119. [Google Scholar] [CrossRef]
- Maier, B.; Gluba, W.; Bernier, B.; Turner, T.; Mohammad, K.; Guise, T.; Sutherland, A.; Thorner, M.; Scrable, H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004, 18, 306–319. [Google Scholar] [CrossRef]
- Gambino, V.; De Michele, G.; Venezia, O.; Migliaccio, P.; Dall’Olio, V.; Bernard, L.; Minardi, S.P.; Della Fazia, M.A.; Bartoli, D.; Servillo, G.; et al. Oxidative stress activates a specific p53 transcriptional response that regulates cellular senescence and aging. Aging Cell 2013, 12, 435–445. [Google Scholar] [CrossRef]
- Medrano, S.; Burns-Cusato, M.; Atienza, M.B.; Rahimi, D.; Scrable, H. Regenerative capacity of neural precursors in the adult mammalian brain is under the control of p53. Neurobiol. Aging 2009, 30, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Ungewitter, E.; Scrable, H. Delta40p53 controls the switch from pluripotency to differentiation by regulating IGF signaling in ESCs. Genes Dev. 2010, 24, 2408–2419. [Google Scholar] [CrossRef] [PubMed]
- Ota, A.; Nakao, H.; Sawada, Y.; Karnan, S.; Wahiduzzaman, M.; Inoue, T.; Kobayashi, Y.; Yamamoto, T.; Ishii, N.; Ohashi, T.; et al. Delta40p53alpha suppresses tumor cell proliferation and induces cellular senescence in hepatocellular carcinoma cells. J. Cell Sci. 2017, 130, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Kumamoto, K.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, S.R.; et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009, 11, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Friedmann, P.D.; Zhang, Z.; Hendrickson, J.; Stein, M.D.; Gerstein, D.R. Effect of primary medical care on addiction and medical severity in substance abuse treatment programs. J. Gen. Intern. Med. 2003, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, I.; Fujita, K.; Jenkins, L.M.; Hiyoshi, Y.; Mondal, A.M.; Vojtesek, B.; Lane, D.P.; Appella, E.; Harris, C.C. Autophagic degradation of the inhibitory p53 isoform Delta133p53alpha as a regulatory mechanism for p53-mediated senescence. Nat. Commun. 2014, 5, 4706. [Google Scholar] [CrossRef] [PubMed]
- Turnquist, C.; Horikawa, I.; Foran, E.; Major, E.O.; Vojtesek, B.; Lane, D.P.; Lu, X.; Harris, B.T.; Harris, C.C. p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death Differ. 2016, 23, 1515–1528. [Google Scholar] [CrossRef]
- Gong, L.; Gong, H.; Pan, X.; Chang, C.; Ou, Z.; Ye, S.; Yin, L.; Yang, L.; Tao, T.; Zhang, Z.; et al. p53 isoform Delta113p53/Delta133p53 promotes DNA double-strand break repair to protect cell from death and senescence in response to DNA damage. Cell Res. 2015, 25, 351–369. [Google Scholar] [CrossRef]
- Marcel, V.; Petit, I.; Murray-Zmijewski, F.; Goullet de Rugy, T.; Fernandes, K.; Meuray, V.; Diot, A.; Lane, D.P.; Aberdam, D.; Bourdon, J.C. Diverse p63 and p73 isoforms regulate Delta133p53 expression through modulation of the internal TP53 promoter activity. Cell Death Differ. 2012, 19, 816–826. [Google Scholar] [CrossRef]
- Bernard, H.; Garmy-Susini, B.; Ainaoui, N.; Van Den Berghe, L.; Peurichard, A.; Javerzat, S.; Bikfalvi, A.; Lane, D.P.; Bourdon, J.C.; Prats, A.C. The p53 isoform, Delta133p53alpha, stimulates angiogenesis and tumour progression. Oncogene 2013, 32, 2150–2160. [Google Scholar] [CrossRef]
- Tang, Y.; Horikawa, I.; Ajiro, M.; Robles, A.I.; Fujita, K.; Mondal, A.M.; Stauffer, J.K.; Zheng, Z.M.; Harris, C.C. Downregulation of splicing factor SRSF3 induces p53beta, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene 2013, 32, 2792–2798. [Google Scholar] [CrossRef] [PubMed]
- Graupner, V.; Schulze-Osthoff, K.; Essmann, F.; Janicke, R.U. Functional characterization of p53beta and p53gamma, two isoforms of the tumor suppressor p53. Cell Cycle 2009, 8, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Tatar, M.; Bartke, A.; Antebi, A. The endocrine regulation of aging by insulin-like signals. Science 2003, 299, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef]
- Aguilaniu, H.; Durieux, J.; Dillin, A. Metabolism, ubiquinone synthesis, and longevity. Genes Dev. 2005, 19, 2399–2406. [Google Scholar] [CrossRef]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef]
- Pehar, M.; O’Riordan, K.J.; Burns-Cusato, M.; Andrzejewski, M.E.; del Alcazar, C.G.; Burger, C.; Scrable, H.; Puglielli, L. Altered longevity-assurance activity of p53:p44 in the mouse causes memory loss, neurodegeneration and premature death. Aging Cell 2010, 9, 174–190. [Google Scholar] [CrossRef]
- Effros, R.B.; Boucher, N.; Porter, V.; Zhu, X.; Spaulding, C.; Walford, R.L.; Kronenberg, M.; Cohen, D.; Schachter, F. Decline in CD28+ T cells in centenarians and in long-term T cell cultures: A possible cause for both in vivo and in vitro immunosenescence. Exp. Gerontol. 1994, 29, 601–609. [Google Scholar] [CrossRef]
- Monteiro, J.; Batliwalla, F.; Ostrer, H.; Gregersen, P.K. Shortened telomeres in clonally expanded CD28-CD8+ T cells imply a replicative history that is distinct from their CD28+CD8+ counterparts. J. Immunol. 1996, 156, 3587–3590. [Google Scholar]
- Brenchley, J.M.; Karandikar, N.J.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Crotty, L.E.; Casazza, J.P.; Kuruppu, J.; Migueles, S.A.; Connors, M.; et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood 2003, 101, 2711–2720. [Google Scholar] [CrossRef]
- Effros, R.B.; Dagarag, M.; Spaulding, C.; Man, J. The role of CD8+ T-cell replicative senescence in human aging. Immunol. Rev. 2005, 205, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Parish, S.T.; Wu, J.E.; Effros, R.B. Sustained CD28 expression delays multiple features of replicative senescence in human CD8 T lymphocytes. J. Clin. Immunol. 2010, 30, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.M.; Horikawa, I.; Pine, S.R.; Fujita, K.; Morgan, K.M.; Vera, E.; Mazur, S.J.; Appella, E.; Vojtesek, B.; Blasco, M.A.; et al. p53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J. Clin. Invest. 2013, 123, 5247–5257. [Google Scholar] [CrossRef] [PubMed]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Das, M.M.; Svendsen, C.N. Astrocytes show reduced support of motor neurons with aging that is accelerated in a rodent model of ALS. Neurobiol. Aging 2015, 36, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A. The mystery and magic of glia: A perspective on their roles in health and disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef]
- Liu, B.; Wang, J.; Chan, K.M.; Tjia, W.M.; Deng, W.; Guan, X.; Huang, J.D.; Li, K.M.; Chau, P.Y.; Chen, D.J.; et al. Genomic instability in laminopathy-based premature aging. Nat. Med. 2005, 11, 780–785. [Google Scholar] [CrossRef]
- Varela, I.; Cadinanos, J.; Pendas, A.M.; Gutierrez-Fernandez, A.; Folgueras, A.R.; Sanchez, L.M.; Zhou, Z.; Rodriguez, F.J.; Stewart, C.L.; Vega, J.A.; et al. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 2005, 437, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rusinol, A.; Sinensky, M.; Wang, Y.; Zou, Y. DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A. J. Cell Sci. 2006, 119, 4644–4649. [Google Scholar] [CrossRef] [PubMed]
- Musich, P.R.; Zou, Y. Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A. Aging 2009, 1, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Musich, P.R.; Zou, Y. DNA-damage accumulation and replicative arrest in Hutchinson-Gilford progeria syndrome. Biochem. Soc. Trans. 2011, 39, 1764–1769. [Google Scholar] [CrossRef]
- von Muhlinen, N.; Horikawa, I.; Alam, F.; Isogaya, K.; Lissa, D.; Vojtesek, B.; Lane, D.P.; Harris, C.C. p53 isoforms regulate premature aging in human cells. Oncogene 2018, 37, 2379–2393. [Google Scholar] [CrossRef]
- Molchadsky, A.; Rivlin, N.; Brosh, R.; Rotter, V.; Sarig, R. p53 is balancing development, differentiation and de-differentiation to assure cancer prevention. Carcinogenesis 2010, 31, 1501–1508. [Google Scholar] [CrossRef]
- Banito, A.; Rashid, S.T.; Acosta, J.C.; Li, S.; Pereira, C.F.; Geti, I.; Pinho, S.; Silva, J.C.; Azuara, V.; Walsh, M.; et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009, 23, 2134–2139. [Google Scholar] [CrossRef]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marion, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Munoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354. [Google Scholar] [CrossRef]
- Qin, H.; Yu, T.; Qing, T.; Liu, Y.; Zhao, Y.; Cai, J.; Li, J.; Song, Z.; Qu, X.; Zhou, P.; et al. Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J. Biol. Chem. 2007, 282, 5842–5852. [Google Scholar] [CrossRef]
- Dannenmann, B.; Lehle, S.; Hildebrand, D.G.; Kubler, A.; Grondona, P.; Schmid, V.; Holzer, K.; Froschl, M.; Essmann, F.; Rothfuss, O.; et al. High glutathione and glutathione peroxidase-2 levels mediate cell-type-specific DNA damage protection in human induced pluripotent stem cells. Stem Cell Rep. 2015, 4, 886–898. [Google Scholar] [CrossRef]
- Horikawa, I.; Park, K.Y.; Isogaya, K.; Hiyoshi, Y.; Li, H.; Anami, K.; Robles, A.I.; Mondal, A.M.; Fujita, K.; Serrano, M.; et al. Delta133p53 represses p53-inducible senescence genes and enhances the generation of human induced pluripotent stem cells. Cell Death Differ. 2017, 24, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Pan, X.; Chen, H.; Rao, L.; Zeng, Y.; Hang, H.; Peng, J.; Xiao, L.; Chen, J. p53 isoform Delta133p53 promotes efficiency of induced pluripotent stem cells and ensures genomic integrity during reprogramming. Sci. Rep. 2016, 6, 37281. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Bardeesy, N.; Lee, K.H.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; DePinho, R.A. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar] [CrossRef]
- Garcia-Cao, I.; Garcia-Cao, M.; Martin-Caballero, J.; Criado, L.M.; Klatt, P.; Flores, J.M.; Weill, J.C.; Blasco, M.A.; Serrano, M. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002, 21, 6225–6235. [Google Scholar] [CrossRef]
- Matheu, A.; Pantoja, C.; Efeyan, A.; Criado, L.M.; Martin-Caballero, J.; Flores, J.M.; Klatt, P.; Serrano, M. Increased gene dosage of Ink4a/Arf results in cancer resistance and normal aging. Genes Dev. 2004, 18, 2736–2746. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Hornsby, P.J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007, 67, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, B.; Multani, A.S.; Patrawala, L.; Chen, X.; Calhoun-Davis, T.; Zhou, J.; Schroeder, L.; Schneider-Broussard, R.; Shen, J.; Pathak, S.; et al. Evidence that senescent human prostate epithelial cells enhance tumorigenicity: Cell fusion as a potential mechanism and inhibition by p16INK4a and hTERT. Int. J. Cancer 2008, 122, 1483–1495. [Google Scholar] [CrossRef]
- Bartholomew, J.N.; Volonte, D.; Galbiati, F. Caveolin-1 regulates the antagonistic pleiotropic properties of cellular senescence through a novel Mdm2/p53-mediated pathway. Cancer Res. 2009, 69, 2878–2886. [Google Scholar] [CrossRef]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Friedman, M.H. Adaptive response of vascular endothelial cells to an acute increase in shear stress magnitude. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H983–H991. [Google Scholar] [CrossRef] [PubMed]
- Anensen, N.; Haaland, I.; D’Santos, C.; Van Belle, W.; Gjertsen, B.T. Proteomics of p53 in diagnostics and therapy of acute myeloid leukemia. Curr. Pharm. Biotechnol. 2006, 7, 199–207. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Boldrup, L.; Bourdon, J.C.; Coates, P.J.; Sjostrom, B.; Nylander, K. Expression of p53 isoforms in squamous cell carcinoma of the head and neck. Eur. J. Cancer 2007, 43, 617–623. [Google Scholar] [CrossRef]
- Avery-Kiejda, K.A.; Zhang, X.D.; Adams, L.J.; Scott, R.J.; Vojtesek, B.; Lane, D.P.; Hersey, P. Small molecular weight variants of p53 are expressed in human melanoma cells and are induced by the DNA-damaging agent cisplatin. Clin. Cancer Res. 2008, 14, 1659–1668. [Google Scholar] [CrossRef]
- Marabese, M.; Marchini, S.; Marrazzo, E.; Mariani, P.; Cattaneo, D.; Fossati, R.; Compagnoni, A.; Signorelli, M.; Moll, U.M.; Codegoni, A.M.; et al. Expression levels of p53 and p73 isoforms in stage I and stage III ovarian cancer. Eur. J. Cancer 2008, 44, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Huo, S.W.; Lu, J.J.; Liu, Z.; Fang, X.L.; Jin, X.B.; Yuan, M.Z. Expression of p53 isoforms in renal cell carcinoma. Chin. Med. J. 2009, 122, 921–926. [Google Scholar]
- Hofstetter, G.; Berger, A.; Fiegl, H.; Slade, N.; Zoric, A.; Holzer, B.; Schuster, E.; Mobus, V.J.; Reimer, D.; Daxenbichler, G.; et al. Alternative splicing of p53 and p73: The novel p53 splice variant p53delta is an independent prognostic marker in ovarian cancer. Oncogene 2010, 29, 1997–2004. [Google Scholar] [CrossRef]
- Bourdon, J.C.; Khoury, M.P.; Diot, A.; Baker, L.; Fernandes, K.; Aoubala, M.; Quinlan, P.; Purdie, C.A.; Jordan, L.B.; Prats, A.C.; et al. p53 mutant breast cancer patients expressing p53gamma have as good a prognosis as wild-type p53 breast cancer patients. Breast Cancer Res. 2011, 13, R7. [Google Scholar] [CrossRef]
- Nutthasirikul, N.; Limpaiboon, T.; Leelayuwat, C.; Patrakitkomjorn, S.; Jearanaikoon, P. Ratio disruption of the 133p53 and TAp53 isoform equilibrium correlates with poor clinical outcome in intrahepatic cholangiocarcinoma. Int. J. Oncol. 2013, 42, 1181–1188. [Google Scholar] [CrossRef]
- Takahashi, R.; Markovic, S.N.; Scrable, H.J. Dominant effects of Delta40p53 on p53 function and melanoma cell fate. J. Invest. Dermatol. 2014, 134, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Avery-Kiejda, K.A.; Morten, B.; Wong-Brown, M.W.; Mathe, A.; Scott, R.J. The relative mRNA expression of p53 isoforms in breast cancer is associated with clinical features and outcome. Carcinogenesis 2014, 35, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, G.; Berger, A.; Berger, R.; Zoric, A.; Braicu, E.I.; Reimer, D.; Fiegl, H.; Marth, C.; Zeimet, A.G.; Ulmer, H.; et al. The N-terminally truncated p53 isoform Delta40p53 influences prognosis in mucinous ovarian cancer. Int. J. Gynecol. Cancer 2012, 22, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, G.; Berger, A.; Schuster, E.; Wolf, A.; Hager, G.; Vergote, I.; Cadron, I.; Sehouli, J.; Braicu, E.I.; Mahner, S.; et al. Delta133p53 is an independent prognostic marker in p53 mutant advanced serous ovarian cancer. Br. J. Cancer 2011, 105, 1593–1599. [Google Scholar] [CrossRef]
- Chambers, S.K.; Martinez, J.D. The significance of p53 isoform expression in serous ovarian cancer. Future Oncol. 2012, 8, 683–686. [Google Scholar] [CrossRef]
- Mu, X.C.; Higgins, P.J. Differential growth state-dependent regulation of plasminogen activator inhibitor type-1 expression in senescent IMR-90 human diploid fibroblasts. J. Cell Physiol. 1995, 165, 647–657. [Google Scholar] [CrossRef]
- Stein, G.H.; Dulic, V. Origins of G1 arrest in senescent human fibroblasts. Bioessays 1995, 17, 537–543. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef] [PubMed]
- Tonnessen-Murray, C.A.; Lozano, G.; Jackson, J.G. The Regulation of Cellular Functions by the p53 Protein: Cellular Senescence. Cold Spring Harb. Perspect. Med. 2017, 7, a026112. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujita, K. p53 Isoforms in Cellular Senescence- and Ageing-Associated Biological and Physiological Functions. Int. J. Mol. Sci. 2019, 20, 6023. https://doi.org/10.3390/ijms20236023
Fujita K. p53 Isoforms in Cellular Senescence- and Ageing-Associated Biological and Physiological Functions. International Journal of Molecular Sciences. 2019; 20(23):6023. https://doi.org/10.3390/ijms20236023
Chicago/Turabian StyleFujita, Kaori. 2019. "p53 Isoforms in Cellular Senescence- and Ageing-Associated Biological and Physiological Functions" International Journal of Molecular Sciences 20, no. 23: 6023. https://doi.org/10.3390/ijms20236023
APA StyleFujita, K. (2019). p53 Isoforms in Cellular Senescence- and Ageing-Associated Biological and Physiological Functions. International Journal of Molecular Sciences, 20(23), 6023. https://doi.org/10.3390/ijms20236023