Abstract
Drought stress, especially during the seedling stage, seriously limits the growth of maize and reduces production in the northeast of China. To investigate the molecular mechanisms of drought response in maize seedlings, proteome changes were analyzed. Using an isotopic tagging relative quantitation (iTRAQ) based method, a total of 207 differentially accumulated protein species (DAPS) were identified under drought stress in maize seedlings. The DAPS were classified into ten essential groups and analyzed thoroughly, which involved in signaling, osmotic regulation, protein synthesis and turnover, reactive oxygen species (ROS) scavenging, membrane trafficking, transcription related, cell structure and cell cycle, fatty acid metabolism, carbohydrate and energy metabolism, as well as photosynthesis and photorespiration. The enhancements of ROS scavenging, osmotic regulation, protein turnover, membrane trafficking, and photosynthesis may play important roles in improving drought tolerance of maize seedlings. Besides, the inhibitions of some protein synthesis and slowdown of cell division could reduce the growth rate and avoid excessive water loss, which is possible to be the main reasons for enhancing drought avoidance of maize seedlings. The incongruence between protein and transcript levels was expectedly observed in the process of confirming iTRAQ data by quantitative real-time polymerase chain reaction (qRT-PCR) analysis, which further indicated that the multiplex post-transcriptional regulation and post-translational modification occurred in drought-stressed maize seedlings. Finally, a hypothetical strategy was proposed that maize seedlings coped with drought stress by improving drought tolerance (via. promoting osmotic adjustment and antioxidant capacity) and enhancing drought avoidance (via. reducing water loss). Our study provides valuable insight to mechanisms underlying drought response in maize seedlings.
1. Introduction
Drought is a major environmental factor affecting crop production. With global warming, the frequency of drought has also increased significantly. Maize (Zea mays L.), as an important crop, is often affected by drought or moisture deficit. Drought has seriously threatened maize production worldwide, especially under rain-fed conditions []. In China, more than 70% of maize growing areas are threatened by drought stress []. In the northeast of China, drought mostly occurs in spring and affects maize seedling growth and yield potential []. Therefore, investigating the mechanism of drought response at maize seedling stage is very helpful to breed drought-tolerant maize varieties.
Drought response of plants involves gene expression, hormone signaling, ionic equilibrium, metabolite changes, and other aspects [,,]. Drought stress always damages photosystem, reduces photosynthetic capacity, affects carbon fixation, and produces excessive reactive oxygen species (ROS). The content of osmotic regulators and the activity of relevant enzymes in ROS scavenging system often increase correspondingly, so as to maintain ROS homeostasis and reduce the damage of cell membrane system caused by ROS [,,,,]. Drought stress also causes seed germination delay, hinders plant growth, shortens flowering period, and leads to insufficient nutrient accumulation in seeds, which ultimately results in a reduction of maize production [,,]. Unfortunately, our understanding of drought response mechanisms is still unclear in maize seedlings.
In recent years, structural and functional genomics, transcriptomics, proteomics, and other omics methods have been wildly used to study molecular mechanisms of drought tolerance in plants. Such studies always accurately and efficiently detected the expression of all genes or the contents of proteins and metabolites in a specific tissue and organ of plants under drought conditions. Proteins are very important for plant stress responding because they are directly involved in plant cell composition and metabolism [,]. Therefore, proteomic study can provide new insights to dissect drought response mechanisms at the protein level. High-throughput plant proteomics has been developing rapidly. The isobaric tags for relative and absolute quantification (iTRAQ) analysis method is a second-generation proteomic technique that has been widely used in plant stress response studies []. However, just limited studies were recently reported on maize drought response by iTRAQ, in which drought-tolerant and drought-sensitive maize varieties were selected to compare protein profiles under the drought conditions [,,]. Usually, comparative proteomics analysis is an effective strategy to identify pivotal functional proteins and pathways, but it becomes very difficult in maize because of the great differences in genetic background among maize varieties. Consequently, investigating the proteomic changes of each important inbred line should be the first step to investigate the mechanisms of drought tolerance in maize.
Here, an iTRAQ-based quantitative strategy is employed to compare proteome profiles of an important maize inbred line, B73, under the well-watered and water-withheld conditions. The purpose of this study is to summarize the essential proteins and metabolic pathways involved in drought response and to speculate drought resistance strategies of maize seedlings.
2. Results
2.1. Phenotypic and Physiological Changes of Maize Seedlings in Response to Drought Stress
To validate a sampling time point and to investigate the response to drought stress in B73 seedlings, plants at 3-leaf stage were withheld water or not for 5 days in controlled conditions. Three days later, leaf relative water content (RWC) showed significant differences between control and drought treated seedlings, whilst no obviously phenotypic differences were observed, and soil moisture had decreased from ~52% to ~16% under the drought treatment (Table S1). Five days later, 3rd and 4th leaves of well-watered seedlings were obviously longer than that of water-withheld seedlings (Table S2). The activities of peroxidase (POD), superoxide dismutase (SOD), and glutathione S-transferase (GST) were also induced significantly increasing after 5-day water withholding treatment (Figure 1). These results indicated that ROS scavenging system had been activated to maintain ROS homeostasis. Therefore, comparing the proteome differences between the treated and untreated seedlings at this time point, we can explore the early response mechanism of drought tolerance in maize seedling stage. Subsequent results also showed that the abundance of POD (C4J6E4, A5H453, B4FN24, B4FLE3), SOD (P23346), and GST (B6SMJ6, A0A1D6PD99, A0A1D6JYM2, Q9FQA3, A0A1D6LSN2, A0A1D6L6U6) all significantly increased under the drought treatment, indicating that the accumulation of POD, SOD, and GST may be a major reason for the enhancement of ROS scavenging.
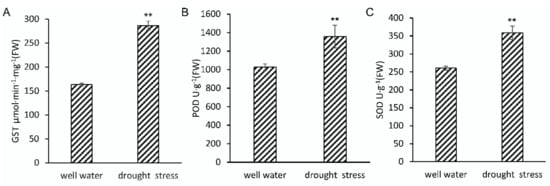
Figure 1.
Analysis of GST (A), POD (B), and SOD (C) activities in maize seedlings under drought stress. Data are the means ± SD calculated from four replicates. Statistical significance was determined by a two-sided t-test: * p < 0.05 and ** p < 0.01.
2.2. Identification of Differentially Accumulated Protein Species (DAPS) by iTRAQ
A total of 4504 proteins were identified by Paragon, and 3676 trusted proteins were screened based on the criteria described in Section 5. Compared with the well-watered group, the abundance of 2533 proteins in the drought treated group increased by 1.115 times on average, and the abundance of 1758 proteins decreased by an average of 0.911 times. According to the criteria that a protein species was considered differentially accumulated as it exhibited a fold change >1.3 and a p-value < 0.05 (t-test) with a false discovery rate (FDR) of <1%, 207 DAPS were identified, of which 111 were up-regulated with an average increase of 1.493 times, and 96 were down-regulated with an average decrease of 0.699 times (Table S3). In all DAPS, the most accumulated protein was maize dehydrin DHN1 (A3KLI1) under the drought stress, which was 4.308 times that of the well-watered group; the most significant decrease in protein abundance was the translation initiation factor TAB2 (B4FTR7) involved in the formation of photosystem I (PS I), which was only 0.449 times that of the well-watered group.
2.3. Bioinformatics Analysis of DAPS Identified by iTRAQ
DAPS were classified according to the GO functional categories of biological process, molecular function, and cell component. As shown in Table 1, DAPS induced by drought stress in maize seedlings involved in various functional groups, of which biological processes accounted for 15 GO terms (the most representative was “oxidation-reduction process”), molecular functions accounted for 15 GO terms (the most representative was “oxidoreductase activity”), and cellular components accounted for 27 GO terms (the most representative was “cytoplasm”).

Table 1.
Gene Ontology (GO) annotation of drought-responsive differentially accumulated protein species (DAPS) in maize seedlings.
According to GO analysis of biological processes, DAPS significantly enriched in the biological process of photosynthesis and photorespiration, energy metabolism, and carbon fixation. Some DAPS related to plant abiotic stress response, such as regulation of protein stability, oxidation-reduction, and water response, were also significantly changed. Although “oxidation-reduction process” (GO: 0055114, p = 8.93 × 10−3) was the most representative in all biological process, in which 19 DASP were enriched, the most significantly differential process was “photosynthesis” (GO: 0015979, p = 3.49 × 10−6). In the process of “response to water” (GO: 0009415), all enriched proteins were dehydrated proteins, and they all significantly accumulated under drought stress, especially DHN1 (A3KLI1), the most up-regulated in all DAPS. GO analysis of molecular functions showed DAPS were classified into 15 categories, in which “oxidoreductase activity” (GO: 0016491, p = 5.19 × 10−3) was the most representative. “Ribulose-bisphosphate carboxylase activity” was most significant differential function category, in which three DAPS (A0A096TUU6, P05348, and O24574) were down-accumulated. GO analysis of cell components showed that most of the drought-induced DAPS were located in chloroplasts, mitochondria, and related membrane systems. The results of GO analyses showed that photosynthesis was most affected by drought stress in maize seedlings. KEGG analysis also showed that photosynthesis (Pathway ID: zma00195, p = 4.06 × 10−8) was the most significant enriched pathway, in which eight DAPS, PsaE (B6TH55), PsaL (B6STG2), PsaA (P04966), PsaG (B6U534), PsbH (P24993), PsaG/K (B4G1K9), FNR (B4FI05), and Fd (B4FYW4), were enriched. Except for PsaA (P04966), other DAPS were up-regulated under the drought stress (Figure S1). All these results indicated that drought stress significantly affected photosynthesis in maize seedlings.
2.4. qRT PCR Verification
In order to clarify the correspondence between mRNA transcription and protein expression, as well as confirm the authenticity of the iTRAQ analysis, qRT-PCR analysis of 17 protein species were performed. We selected the genes from the 10 categories of DAPS based on the functional annotation (Table 2 and Table 3). Meanwhile, the selected genes should be highly differentiated in response to drought stress and reported to be potentially associated with drought tolerance. The results showed that 15 genes showed the same change tendency on the RNA level as the changes of the corresponding protein abundance, such as gibberellin receptor GID1, dehydrin DHN1, and superoxide dismutase. Besides, two genes of the 60S ribosomal protein and ABC transporter B family member 28 showed opposite trends to the abundance of their corresponding proteins (Table 2). The discrepancy between the transcription level and the abundance of the corresponding protein species probably resulted from various post-translational modification and post-translational modification under the drought stress, such as protein phosphorylation and glycosylation.
Table 2.
Quantitative real-time PCR (qRT-PCR) information of the selected genes encoding DAPS.
Table 3.
Classification of drought-responsive DAPS with detailed annotation.
3. Discussion
Plants have developed various strategies in response to drought stress, including drought escape via a short life cycle or developmental plasticity, drought avoidance via enhanced water uptake and reduced water loss, and drought tolerance via osmotic adjustment, antioxidant capacity, and desiccation tolerance []. Proteins are substance basis of life activities and directly involve in plant stress responses. Therefore, proteomics is a new suitable tool for the comprehensive identification of drought-responsive proteins in plants [,]. A large number of plant drought resistance related proteins have been identified, which have high potential for crop breeding [,,,,]. The integrative analysis of physiological, molecular, and proteomic data most probably provides new clues for further understanding the drought resistance in maize seedlings.
In order to explore the maize seedling responds to drought stress, a proteomics analysis was performed using iTRAQ technique. As a result, a total of 207 DAPS was identified. Due to the problems of repeated statistics and concept ambiguity in the results of GO and KEGG analysis, we performed a more detailed classification analysis of 142 DAPS with accurate functional annotations (Table 3). These DAPS were classified into 10 categories: signaling (7 DAPS), osmotic regulation (5 DAPS), protein synthesis and turnover (31 DAPS), ROS scavenging (14 DAPS), membrane trafficking (19 DAPS), transcription related (19 DAPS), cell structure and cell cycle (9 DAPS), fatty acid metabolism (3 DAPS), carbohydrate and energy metabolism (15 DAPS), and photosynthesis and photorespiration (20 DAPS).
ASR (ABA-, stress-, and ripening-induced) and CBL (Calcineurin B-like) are two important proteins involved in ABA signaling. Some studies have shown that ASR and CBL genes up-regulated rapidly when plants were under drought, cold, salt, and weak light stress [,]. In this study, the abundance of ASR2 (B4FKG5), ASR3 (A0A1D6EB22), and CBL(B4F9B4) increased significantly under the drought stress, which indicate that ABA signaling is involved in maize seedling drought response. In addition, we found that GID1(K7U051) protein abundance decreased significantly. The function of GID1 is to bind to GA in plants to induce the degradation of DELLAs that inhibit plant growth [,,,]. Slowing of leaf growth (Table S2) might be partly regulated through GA-GID1-DELLA mediated signaling. We presumed that GA signaling should be an important hormone signal pathway involved in the drought response of maize seedlings.
Thirty-one DAPS were involved in protein synthesis and turnover, which indicated that this process is very influential in response to drought stress in maize seedlings. The DAPS related to protein synthesis mainly included various ribosomal proteins, translation initiation factor, and peptide chain release factor. The abundance of these proteins all decreased under the drought stress (Table 3). This meant that protein synthesis was weakening in maize seedlings, which may cause slowing down of plant growth and decreasing of water consumption as found in Phaseolus vulgaris []. As described above, GID1 abundance decreasing may lead to growth inhibition mediated by GA-GID1-DELLA signaling. We inferred that there should be multiplex cross-talk between GA signaling and protein synthesis. Meanwhile, most up-regulated DAPS related to protein turnover were heat shock proteins and chaperones (Table 3), which were essential components for maintaining protein stability and repairing damaged proteins [,,]. The results indicated that the integrity of protein structure was necessary for maize seedlings to enhance drought tolerance.
Drought stress rapidly lowers the cell division rate in plant leaves []. The abundance of several proteins involved in cell structure and cell division were also decreased in this proteomic study, such as a cytoskeleton protein (tubulin: Q41785) and a cell proliferation related protein (nitrilase: B4FQE2). It has been demonstrated that inhibition of Nitrilase expression suppressed Arabidopsis growth and consequently avoided the effects of drought stress []. These results suggest that there may also be a mechanism similar to that in Arabidopsis in coping with drought stress by suppressing cell growth. Besides, the abundant of apoptosis related proteins, such as programmed cell death protein 5 (B4FN06) and CASP-like protein cysteinyl aspartate specific proteinase Caspase (A0A1D6QU75), were increased. Transcriptomic and proteomic studies have revealed that up-regulation of programmed apoptosis-related genes is important for maize drought tolerance [,,].
Transcription related proteins are crucial for plants to cope with drought stress. Nineteen DAPS involved in transcription regulation were identified in this study. Chromatin structure modification is a prerequisite to regulate gene transcription. Histones are the major proteins of chromatin and can regulate gene expression []. The abundance of histones, especially histone H1, was significantly changed under drought conditions in some plants [,,,,]. In addition, some studies have also shown that histone deacetylation promoted gene expression and further influenced the morphology, development, and stress tolerance of plants [,,,,]. In this study, we not only identified the abundance of histone H1 (A0A1D6NW49, B4FD93) increased significantly under the drought stress, but also identified the abundance of histone deacetylase (B4F939) decreased. These results indicate that the increase of histone content and the decrease of histone deacetylation in maize seedlings is a means of inhibiting the expression of some genes under drought stress. Unfortunately, the target genes regulated by this means are still unclear.
In this study, all DAPS in osmotic regulation were related to water regulation and up-regulated significantly, in which four DAPS were annotated to dehydrin (DHN) (Table 3). DNH is a class of hydrophilic proteins widely existing in plants. Under various abiotic stress conditions, DHN accumulates rapidly and plays an important role in stabilizing cell membranes and scavenging free radicals []. In addition, phosphorylated DHN binds calcium ions to perform the function of molecular chaperone under drought stress []. Therefore, the accumulation of DHN (B6SIK2, B4G1H1, A3KLI1, C4J477) suggests that DHN may play an important role in maintaining stable membrane structure, promoting protein synthesis and turnover, as well as ROS scavenging, while the detailed mechanism in drought response of maize seedlings needs to be further studied.
Abiotic stress can stimulate plants to produce excessive ROS and break ROS homeostasis. Therefore, ROS scavenging is very important to improve plant tolerance to abiotic stress. The known ROS scavenging pathways in plants include SOD, POD, and CAT pathways, the ascorbate-glutathione pathway, and the glutathione peroxidase/glutathione s-transferase (GPX/GST) pathway [,,,,]. In this study, all identified DAPS involved in ROS scavenging were accumulated. The accumulation of SOD, POD, and GST was consistent with activity increasing of them (Figure 1). According to the different ROS scavenging mechanisms, these DAPS can be classified into two groups. One was antioxidant enzymes, including SOD (P23346) and POD (C4J6E4, A5H453, B4FN24); the other was chemical antioxidant related proteins, including dehydro ascorbate reductase (DHAR: B4FT31), and glutathione s-transferase (GST: B6SMJ6, A0A1D6PD99, A0A1D6JYM2). These proteins should perform similar functions which have been reported in Arabidopsis, rice, wheat, and other plants [,,]. ROS homeostasis is also necessary for maize seedlings to enhance drought tolerance.
In this study, the DAPS classified in membrane trafficking were mostly located in mitochondrial, plasma, or vacuole membranes. Although many membrane proteins have been studied in soybean, wheat, barley, cucumber, and other plants [,,,], no clear regulatory mechanism was concluded that membrane proteins participated in drought responding process. Moreover, three DAPS related to the fatty acid metabolism were identified in this study. Esterase (B6TZ91) and GDSL esterase (A0A1D6HJU1) were up-regulated under drought stress, while fatty acid export 3 (B4G272) was down-regulated. Based on limited reports, differential expression of fatty acid metabolism-related proteins probably helped to maintain cell membrane integrity and stability under drought stress []. The DAPS related cell membrane could be used as reference data for investigating changes of membrane structure and function during drought stress in maize.
Inhibition of photosynthesis is another major influence of drought stress on plants. Recovering photosynthesis is a strategy for plants to cope with drought stress. In this study, the most of drought-increased DAPS involved in photoreactions, which suggested photosynthesis in maize seedlings was maintained under the water-deficient condition. For example, the expression of cytochrome b related proteins were also significantly up-regulated in response to drought in Arabidopsis [] and apples []. Unexpectedly, TAB2 (B4FTR7), a regulatory protein related to PS I assembly, was significantly down-regulated under drought stress and then probably inhibited photosynthesis, which contradicted with the abundance increasing of other photosynthesis related DAPS identified in this study. Some studies verified that TAB2 was closely related to the transcriptional regulation of glycolytic enzymes [,]. These results indicated that TAB2 might be involved in complex interactions between photosynthesis and glucose metabolism under drought stress.
4. Materials and Methods
4.1. Plant Materials and Drought Stress Treatments
Maize inbred line B73 was used in this experiment because of its abundant database resources and important breeding value. Seeds were surface sterilized with 70% alcohol for 1 min and then 3% NaClO for 10 min, thoroughly rinsed with distilled water and germinated on filter paper wetted with distilled water in plates at 26 °C for 3 days. Germinated seeds were transplanted into pots containing 200 g fully dried soil, and then the water content of soil was maintained at 50%. The seedlings were grown under controlled conditions (light/dark cycles: 14h/10h; light intensity: 70 mmol/m2s; temperature: 28/22 °C; relative humidity: 60% ± 5%) to 3-leaf stage. Then, a half of seedlings were exposed to drought stress that water was withheld from the seedlings for 5 days, while the rest seedlings were still grown under the well-watered conditions. Leaf samples were collected after 0, 1, 3, and 5 days of treatment (for measuring leaf length and leaf relative water content (RWC)). On the fifth day after water withdrawing treatment, all green tissues from every 5 individual plants were mixed as one biological replicate to be stored in liquid nitrogen. Four biological replicates were respectively collected from the treatment and control groups for protein extraction and subsequent analysis
4.2. iTRAQ Analysis
iTRAQ analysis was carried out by Shanghai Luming Biotechnology Co., LTD. The standard iTRAQ analysis was performed with minor modifications as previously described [,] briefly, including protein preparation, iTRAQ labeling and SCX fractionation, LC-ESI-MS/MS analysis, protein identification and data analysis, and bioinformatics analysis.
Data was processed with Protein Pilot Software v. 5.0 (AB SCIEX, USA) against the Uniprot database (available online: https://www.uniprot.org; accessed on 12 January 2018; uniprot_Zea mays_132339_20180112.FASTA; 76,417 sequences) using the Paragon algorithm []. The experimental data from tandem mass spectrometry (MS) was used to match the theory data to obtain result of protein identification. Protein identification was performed with the search option: emphasis on biological modifications.
To reduce the probability of false peptide identification, only peptides with significance scores (≥20) at the 95% confidence interval by a Paragon probability analysis greater than “identity” were counted as identified. Each confident protein identification involves at least one unique peptide. For protein quantitation, it was required that a protein contains at least two unique peptides. The quantitative protein ratios were weighted and normalized by the median ratio in Paragon. We only used ratios with p-values < 0.05 (t-test), and only fold changes of >1.3 were considered as significant on the basis of the related iTRAQ studies [,,].
Functional annotations of differentially accumulated protein species were performed using Gene Ontology (GO) (http://www.geneontology.org). The Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg) was used to predict the main metabolic pathways and biochemical signals transduction pathways that involved the DAPS. A p-value <0.05 (Fisher’s exact test) was used as the threshold to determine the significant enrichments of GO and KEGG pathways.
4.3. qRT PCR Verification
Total RNA was isolated using RNAiso Plus reagent (TaKaRa) from no less than 3 seedlings. To remove genomic DNA contamination, total RNA was treated with the TURBO DNA-free™ Kit (Ambion). The concentration of total RNA was determined using a Nanodrop2000c (Thermo Scientific, USA). One microgram of total RNA was used to synthesize cDNA with the TransScript All-in-One First-Strand cDNA Synthesis SuperMix for qPCR kit (Transgen Biotech). To validate the differentially accumulated protein obtained from iTRAQ, 17 genes (Table 2) were subjected to quantitative real-time PCR. All reactions were performed in triplicate, including the non-template controls. Data were quantified using the comparative CT method (2−ΔΔCT method) [].
4.4. Antioxidants Assays
The activities of POD, SOD, and GST in shoots were respectively assayed using detection kits (POD-1-Y, SOD-1-Y, and GST-2-W) from Suzhou Comin Biotechnology Co. Ltd., following the manufacturer’s instructions. Statistical data were obtained from four independent experiments. All values were the means of four assays carried out for each value. Data analysis was performed using the SPSS statistical software package (version 19.0; SPSS Institute Ltd., Armonk, NY, USA), and the significance of differences were tested by t-test with a p values < 0.05 set as statistically significant.
5. Conclusions
Proteomics is a powerful tool to analyze the mechanisms of drought response and tolerance in maize which hardly revealed by transcriptomic or genomic technologies. However, the studies on maize proteomes related with drought response are still very limited. In this study, we found that more than 200 DAPS were drought-responsive in maize seedlings, which were involved in drought signal transduction, ROS scavenging, osmotic regulation, specific gene expression regulation, protein synthesis and turnover, cell structure modulation, as well as other metabolisms. A hypothetical strategy was proposed that maize seedlings coped with drought stress by improving drought tolerance (via promoting osmotic adjustment and antioxidant capacity, maintaining membrane integrity and stability, as well as recovering photosynthesis) and enhancing drought avoidance (via inhibiting cell division and protein synthesis to reduce water loss) (Figure 2). All these findings enrich the proteome data of maize. The DAPS will provide candidate genes/proteins for genetic improvement in maize drought tolerance. In the future, the integration of genomics, proteomics, transcriptomics, and metabolomics will help us to understand the drought response mechanism of maize seedlings.
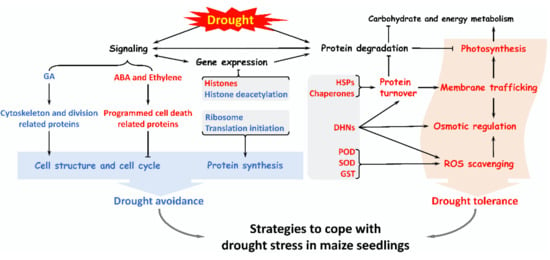
Figure 2.
A summary of various pathways in maize seedlings in response to drought stress. Drought stress activates several signaling to regulates some gene expression, and enhances ROS scavenging, osmotic regulation, protein turnover, membrane trafficking, and photosynthesis, which improves drought tolerance of maize seedlings. Besides, drought stress inhibits some protein synthesis as well as the cytoskeleton and cell division to avoid excessive water loss. Importantly, maize seedlings enable a complex set of strategies to cope with drought stress.
Supplementary Materials
The following are available online at https://www.mdpi.com/1422-0067/20/23/5956/s1.
Author Contributions
Z.J., F.J. and Y.L. conceived and designed the experiments; Z.J. and F.J. performed the experiments; Z.J., X.S. and Y.L. analyzed the data; F.J. provided reagents/materials/analysis tools; X.S. and Y.L. wrote the paper.
Funding
This work was funded by the Science Development Planning of Jilin Province, grant number 20170101014JC, and the Agricultural Science and Technology Innovation Project, grant number CXGC2018ZY025 and CXGC2017TD004.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rao, G.J.N.; Reddy, J.N.; Variar, M.; Mahender, A. Molecular Breeding to Improve Plant Resistance to Abiotic Stresses. In Advances in Plant Breeding Strategies: Agronomic, Abiotic and Biotic Stress Traits; Springer: Cincinnati, OH, USA, 2016; pp. 283–326. [Google Scholar]
- Min, H.; Chen, C.; Wei, S.; Shang, X.; Sun, M.; Xia, R.; Liu, X.; Hao, D.; Chen, H.; Xie, Q. Identification of Drought Tolerant Mechanisms in Maize Seedlings Based on Transcriptome Analysis of Recombination Inbred Lines. Front Plant Sci. 2016, 7, 1080. [Google Scholar] [CrossRef]
- Zenda, T.; Liu, S.; Wang, X.; Jin, H.; Liu, G.; Duan, H. Comparative Proteomic and Physiological Analyses of Two Divergent Maize Inbred Lines Provide More Insights into Drought-Stress Tolerance Mechanisms. Int. J. Mol. Sci. 2018, 19, 3225. [Google Scholar] [CrossRef]
- Budak, H.; Hussain, B.; Khan, Z.; Ozturk, N.Z.; Ullah, N. From Genetics to Functional Genomics: Improvement in Drought Signaling and Tolerance in Wheat. Front. Plant Sci. 2015, 6, 1012. [Google Scholar] [CrossRef]
- Wang, X.; Cai, X.; Xu, C.; Wang, Q.; Dai, S. Drought-Responsive Mechanisms in Plant Leaves Revealed by Proteomics. Int. J. Mol. Sci. 2016, 17, 1706. [Google Scholar] [CrossRef]
- Laloum, T.; Martin, G.; Duque, P. Alternative Splicing Control of Abiotic Stress Responses. Trends Plant Sci. 2018, 23, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liu, Y.; Rao, J.; Wang, G.; Li, H.; Ge, F.; Chen, C. Overexpression of the glutathione S-transferase gene from Pyrus pyrifolia fruit improves tolerance to abiotic stress in transgenic tobacco plants. Mol. Biol. 2013, 47, 591–601. [Google Scholar] [CrossRef]
- Ji, W.; Zhu, Y.; Li, Y.; Yang, L.; Zhao, X.; Cai, H.; Bai, X. Over-expression of a glutathione S-transferase gene, GsGST, from wild soybean (Glycine soja) enhances drought and salt tolerance in transgenic tobacco. Biotechnol. Lett. 2010, 32, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- McKersie, B.D.; Bowley, S.R.; Harjanto, E.; Leprince, O. Water-Deficit Tolerance and Field Performance of Transgenic Alfalfa Overexpressing Superoxide Dismutase. Plant Physiol. 1996, 111, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.B.; Kim, Y.H.; Lee, H.S.; Kim, K.Y.; Deng, X.P.; Kwak, S.S. Analysis of antioxidant enzyme activity during germination of alfalfa under salt and drought stresses. Plant Physiol. Biochem. 2009, 47, 570–577. [Google Scholar] [CrossRef]
- Miller, G.; Suzuki, N.; Ciftci-Yilmaz, S.; Mittler, R. Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant Cell Environ. 2010, 33, 453–467. [Google Scholar] [CrossRef]
- Benesova, M.; Hola, D.; Fischer, L.; Jedelsky, P.L.; Hnilicka, F.; Wilhelmova, N.; Rothova, O.; Kocova, M.; Prochazkova, D.; Honnerova, J.; et al. The physiology and proteomics of drought tolerance in maize: Early stomatal closure as a cause of lower tolerance to short-term dehydration? PLoS ONE 2012, 7, e38017. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Li, M.; Guan, Q.; Liu, F.; Zhang, S.; Chen, W.; Yin, L.; Qin, Y.; Ma, F. Physiological and proteome analysis suggest critical roles for the photosynthetic system for high water-use efficiency under drought stress in Malus. Plant Sci. 2015, 236, 44–60. [Google Scholar] [CrossRef] [PubMed]
- Abreu, I.A.; Farinha, A.P.; Negrao, S.; Goncalves, N.; Fonseca, C.; Rodrigues, M.; Batista, R.; Saibo, N.J.; Oliveira, M.M. Coping with abiotic stress: Proteome changes for crop improvement. J. Proteom. 2013, 93, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Hiraga, S.; Yanagawa, Y. Proteomics techniques for the development of flood tolerant crops. J. Proteome Res. 2012, 11, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Ning, F.; Zhang, Q.; Wu, X.; Wang, W. Enhancing Omics Research of Crop Responses to Drought under Field Conditions. Front Plant Sci. 2017, 8, 174. [Google Scholar] [CrossRef]
- Wu, X.; Wang, W. Increasing Confidence of Proteomics Data Regarding the Identification of Stress-Responsive Proteins in Crop Plants. Front Plant Sci. 2016, 7, 702. [Google Scholar] [CrossRef]
- Wang, X.; Zenda, T.; Liu, S.; Liu, G.; Jin, H.; Dai, L.; Dong, A.; Yang, Y.; Duan, H. Comparative Proteomics and Physiological Analyses Reveal Important Maize Filling-Kernel Drought-Responsive Genes and Metabolic Pathways. Int. J. Mol. Sci. 2019, 20, 3743. [Google Scholar] [CrossRef]
- Zeng, W.; Peng, Y.; Zhao, X.; Wu, B.; Chen, F.; Ren, B.; Zhuang, Z.; Gao, Q.; Ding, Y. Comparative Proteomics Analysis of the Seedling Root Response of Drought-sensitive and Drought-tolerant Maize Varieties to Drought Stress. Int. J. Mol. Sci. 2019, 20, 2793. [Google Scholar] [CrossRef]
- Zhang, Q. Strategies for developing Green Super Rice. Proc. Natl. Acad. Sci. USA 2007, 104, 16402–16409. [Google Scholar] [CrossRef]
- Baerenfaller, K.; Massonnet, C.; Walsh, S.; Baginsky, S.; Buhlmann, P.; Hennig, L.; Hirsch-Hoffmann, M.; Howell, K.A.; Kahlau, S.; Radziejwoski, A.; et al. Systems-based analysis of Arabidopsis leaf growth reveals adaptation to water deficit. Mol. Syst. Biol. 2012, 8, 606. [Google Scholar] [CrossRef]
- Mohammadi, P.P.; Moieni, A.; Hiraga, S.; Komatsu, S. Organ-specific proteomic analysis of drought-stressed soybean seedlings. J. Proteom. 2012, 75, 1906–1923. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, L.; Valot, B.; Tardieu, F.; Zivy, M. Phosphoproteome dynamics upon changes in plant water status reveal early events associated with rapid growth adjustment in maize leaves. Mol. Cell Proteom. 2012, 11, 957–972. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Han, G.; He, H.; Li, J. Differential regulation of proteins and phosphoproteins in rice under drought stress. Biochem. Biophys. Res. Commun. 2009, 379, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Zheng, H.; Yang, Z.; Guo, J.; Liu, T.; Sun, L.; Xiao, Y.; Yang, J.; Yang, Q.; Guo, L. Physiological and proteomic analysis of maize seedling response to water deficiency stress. J. Plant Physiol. 2018, 228, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Krannich, C.T.; Maletzki, L.; Kurowsky, C.; Horn, R. Network Candidate Genes in Breeding for Drought Tolerant Crops. Int. J. Mol. Sci. 2015, 16, 16378–16400. [Google Scholar] [CrossRef]
- Cakir, B.; Agasse, A.; Gaillard, C.; Saumonneau, A.; Delrot, S.; Atanassova, R. A grape ASR protein involved in sugar and abscisic acid signaling. Plant Cell 2003, 15, 2165–2180. [Google Scholar] [CrossRef]
- Zhang, F.; Li, L.; Jiao, Z.; Chen, Y.; Liu, H.; Chen, X.; Fu, J.; Wang, G.; Zheng, J. Characterization of the calcineurin B-Like (CBL) gene family in maize and functional analysis of ZmCBL9 under abscisic acid and abiotic stress treatments. Plant Sci. 2016, 253, 118–129. [Google Scholar] [CrossRef]
- Colebrook, E.H.; Thomas, S.G.; Phillips, A.L.; Hedden, P. The role of gibberellin signalling in plant responses to abiotic stress. J. Exp. Biol. 2014, 217, 67–75. [Google Scholar] [CrossRef]
- Hedden, P.; Thomas, S.G. Gibberellin biosynthesis and its regulation. Biochem. J. 2012, 444, 11–25. [Google Scholar] [CrossRef]
- Sun, T.P. Gibberellin-GID1-DELLA: A pivotal regulatory module for plant growth and development. Plant Physiol. 2010, 154, 567–570. [Google Scholar] [CrossRef]
- Van De Velde, K.; Ruelens, P.; Geuten, K.; Rohde, A.; Van Der Straeten, D. Exploiting DELLA Signaling in Cereals. Trends Plant Sci. 2017, 22, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Zadraznik, T.; Hollung, K.; Egge-Jacobsen, W.; Meglic, V.; Sustar-Vozlic, J. Differential proteomic analysis of drought stress response in leaves of common bean (Phaseolus vulgaris L.). J. Proteom. 2013, 78, 254–272. [Google Scholar] [CrossRef] [PubMed]
- Sabehat, A.; Lurie, S.; Weiss, D. Expression of small heat-shock proteins at low temperatures. A possible role in protecting against chilling injuries. Plant Physiol. 1998, 117, 651–658. [Google Scholar] [CrossRef]
- Swindell, W.R.; Huebner, M.; Weber, A.P. Transcriptional profiling of Arabidopsis heat shock proteins and transcription factors reveals extensive overlap between heat and non-heat stress response pathways. BMC Genom. 2007, 8, 125. [Google Scholar] [CrossRef]
- Timperio, A.M.; Egidi, M.G.; Zolla, L. Proteomics applied on plant abiotic stresses: Role of heat shock proteins (HSP). J. Proteom. 2008, 71, 391–411. [Google Scholar] [CrossRef]
- Tardieu, F.; Granier, C.; Muller, B. Water deficit and growth. Co-ordinating processes without an orchestrator? Curr. Opin. Plant Biol. 2011, 14, 283–289. [Google Scholar] [CrossRef]
- Doskocilova, A.; Kohoutova, L.; Volc, J.; Kourova, H.; Benada, O.; Chumova, J.; Plihal, O.; Petrovska, B.; Halada, P.; Bogre, L.; et al. NITRILASE1 regulates the exit from proliferation, genome stability and plant development. New Phytol. 2013, 198, 685–698. [Google Scholar] [CrossRef]
- Ghabooli, M.; Khatabi, B.; Ahmadi, F.S.; Sepehri, M.; Mirzaei, M.; Amirkhani, A.; Jorrin-Novo, J.V.; Salekdeh, G.H. Proteomics study reveals the molecular mechanisms underlying water stress tolerance induced by Piriformospora indica in barley. J. Proteom. 2013, 94, 289–301. [Google Scholar] [CrossRef]
- Koh, J.; Chen, G.; Yoo, M.J.; Zhu, N.; Dufresne, D.; Erickson, J.E.; Shao, H.; Chen, S. Comparative Proteomic Analysis of Brassica napus in Response to Drought Stress. J. Proteome Res. 2015, 14, 3068–3081. [Google Scholar] [CrossRef]
- Trivedi, I.; Rai, K.M.; Singh, S.K.; Kumar, V.; Singh, M.; Ranjan, A.; Lodhi, N.; Sawant, S.V. Analysis of histones and histone variants in plants. Methods Mol. Biol. 2012, 833, 225–236. [Google Scholar]
- Brossa, R.; Pinto-Marijuan, M.; Francisco, R.; Lopez-Carbonell, M.; Chaves, M.M.; Alegre, L. Redox proteomics and physiological responses in Cistus albidus shrubs subjected to long-term summer drought followed by recovery. Planta 2015, 241, 803–822. [Google Scholar] [CrossRef]
- Trivedi, I.; Ranjan, A.; Sharma, Y.K.; Sawant, S. The histone H1 variant accumulates in response to water stress in the drought tolerant genotype of Gossypium herbaceum L. Protein J. 2012, 31, 477–486. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Du, Y.; Zhao, Z.; Zhu, X.; Jiang, X.; Shu, Z.; Yin, Y.; Li, X. Overexpression of Camellia sinensis H1 histone gene confers abiotic stress tolerance in transgenic tobacco. Plant Cell Rep. 2014, 33, 1829–1841. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.T.; Luo, M.; Wang, Y.Y.; Wu, K. Involvement of Arabidopsis histone deacetylase HDA6 in ABA and salt stress response. J. Exp. Bot. 2010, 61, 3345–3353. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.T.; Wu, K. Role of histone deacetylases HDA6 and HDA19 in ABA and abiotic stress response. Plant Signal Behav. 2010, 5, 1318–1320. [Google Scholar] [CrossRef] [PubMed]
- Rossi, V.; Locatelli, S.; Varotto, S.; Donn, G.; Pirona, R.; Henderson, D.A.; Hartings, H.; Motto, M. Maize histone deacetylase hda101 is involved in plant development, gene transcription, and sequence-specific modulation of histone modification of genes and repeats. Plant Cell 2007, 19, 1145–1162. [Google Scholar] [CrossRef]
- Yang, H.; Liu, X.; Xin, M.; Du, J.; Hu, Z.; Peng, H.; Rossi, V.; Sun, Q.; Ni, Z.; Yao, Y. Genome-Wide Mapping of Targets of Maize Histone Deacetylase HDA101 Reveals Its Function and Regulatory Mechanism during Seed Development. Plant Cell 2016, 28, 629–645. [Google Scholar] [CrossRef]
- Zheng, Y.; Ding, Y.; Sun, X.; Xie, S.; Wang, D.; Liu, X.; Su, L.; Wei, W.; Pan, L.; Zhou, D.X. Histone deacetylase HDA9 negatively regulates salt and drought stress responsiveness in Arabidopsis. J. Exp. Bot. 2016, 67, 1703–1713. [Google Scholar] [CrossRef]
- Allagulova Ch, R.; Gimalov, F.R.; Shakirova, F.M.; Vakhitov, V.A. The plant dehydrins: Structure and putative functions. Biochemistry 2003, 68, 945–951. [Google Scholar]
- Kim, Y.S.; Kim, I.S.; Bae, M.J.; Choe, Y.H.; Kim, Y.H.; Park, H.M.; Kang, H.G.; Yoon, H.S. Homologous expression of cytosolic dehydroascorbate reductase increases grain yield and biomass under paddy field conditions in transgenic rice (Oryza sativa L. japonica). Planta 2013, 237, 1613–1625. [Google Scholar] [CrossRef]
- Xu, J.; Xing, X.J.; Tian, Y.S.; Peng, R.H.; Xue, Y.; Zhao, W.; Yao, Q.H. Transgenic Arabidopsis Plants Expressing Tomato Glutathione S-Transferase Showed Enhanced Resistance to Salt and Drought Stress. PLoS ONE 2015, 10, e0136960. [Google Scholar] [CrossRef] [PubMed]
- Ford, K.L.; Cassin, A.; Bacic, A. Quantitative proteomic analysis of wheat cultivars with differing drought stress tolerance. Front Plant Sci. 2011, 2, 44. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, A.; Tanaka, Y. Effects of ABA, auxin, and gibberellin on the expression of genes for vacuolar H+ -inorganic pyrophosphatase, H+ -ATPase subunit A, and Na+/H+ antiporter in barley. Plant Physiol. Biochem. 2006, 44, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Kabala, K.; Janicka-Russak, M.; Reda, M.; Migocka, M. Transcriptional regulation of the V-ATPase subunit c and V-PPase isoforms in Cucumis sativus under heavy metal stress. Physiol. Plant 2014, 150, 32–45. [Google Scholar] [CrossRef]
- Gigon, A.; Matos, A.R.; Laffray, D.; Zuily-Fodil, Y.; Pham-Thi, A.T. Effect of drought stress on lipid metabolism in the leaves of Arabidopsis thaliana (ecotype Columbia). Ann. Bot. 2004, 94, 345–351. [Google Scholar] [CrossRef]
- Xu, Y.H.; Liu, R.; Yan, L.; Liu, Z.Q.; Jiang, S.C.; Shen, Y.Y.; Wang, X.F.; Zhang, D.P. Light-harvesting chlorophyll a/b-binding proteins are required for stomatal response to abscisic acid in Arabidopsis. J. Exp. Bot. 2012, 63, 1095–1106. [Google Scholar] [CrossRef]
- Gargouri, M.; Bates, P.D.; Park, J.J.; Kirchhoff, H.; Gang, D.R. Functional photosystem I maintains proper energy balance during nitrogen depletion in Chlamydomonas reinhardtii, promoting triacylglycerol accumulation. Biotechnol. Biofuels 2017, 10, 89. [Google Scholar] [CrossRef][Green Version]
- Gargouri, M.; Park, J.J.; Holguin, F.O.; Kim, M.J.; Wang, H.; Deshpande, R.R.; Shachar-Hill, Y.; Hicks, L.M.; Gang, D.R. Identification of regulatory network hubs that control lipid metabolism in Chlamydomonas reinhardtii. J. Exp. Bot. 2015, 66, 4551–4566. [Google Scholar] [CrossRef]
- Xie, H.; Yang, D.H.; Yao, H.; Bai, G.; Zhang, Y.H.; Xiao, B.G. iTRAQ-based quantitative proteomic analysis reveals proteomic changes in leaves of cultivated tobacco (Nicotiana tabacum) in response to drought stress. Biochem. Biophys. Res. Commun. 2016, 469, 768–775. [Google Scholar] [CrossRef]
- Liu, B.; Shan, X.; Wu, Y.; Su, S.; Li, S.; Liu, H.; Han, J.; Yuan, Y. iTRAQ-Based Quantitative Proteomic Analysis of Embryogenic and Non-embryogenic Calli Derived from a Maize (Zea mays L.) Inbred Line Y423. Int. J. Mol. Sci. 2018, 19, 4004. [Google Scholar] [CrossRef]
- Shilov, I.V.; Seymour, S.L.; Patel, A.A.; Loboda, A.; Tang, W.H.; Keating, S.P.; Hunter, C.L.; Nuwaysir, L.M.; Schaeffer, D.A. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell Proteom. 2007, 6, 1638–1655. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shan, X.; Wu, Y.; Su, S.; Li, S.; Liu, H.; Han, J.; Xue, C.; Yuan, Y. iTRAQ-based quantitative proteomic analysis reveals new metabolic pathways responding to chilling stress in maize seedlings. J. Proteom. 2016, 146, 14–24. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Xu, X.Y.; Gong, Q.Q.; Xie, C.; Fan, W.; Yang, J.L.; Lin, Q.S.; Zheng, S.J. Root proteome of rice studied by iTRAQ provides integrated insight into aluminum stress tolerance mechanisms in plants. J. Proteom. 2014, 98, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Cong, R.; Li, S.; Li, R.; Qin, Z.; Li, Y.; Zhou, X.; Chen, S.; Li, J. Comparative Proteomic Analysis of Soybean Leaves and Roots by iTRAQ Provides Insights into Response Mechanisms to Short-Term Salt Stress. Front Plant Sci. 2016, 7, 573. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).