Role of Nicotinamide in Genomic Stability and Skin Cancer Chemoprevention



Abstract
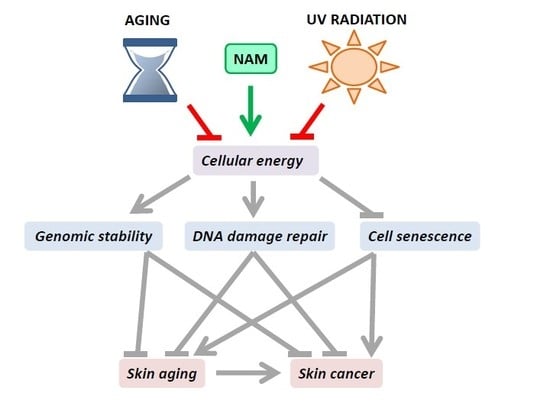
{kind=link}
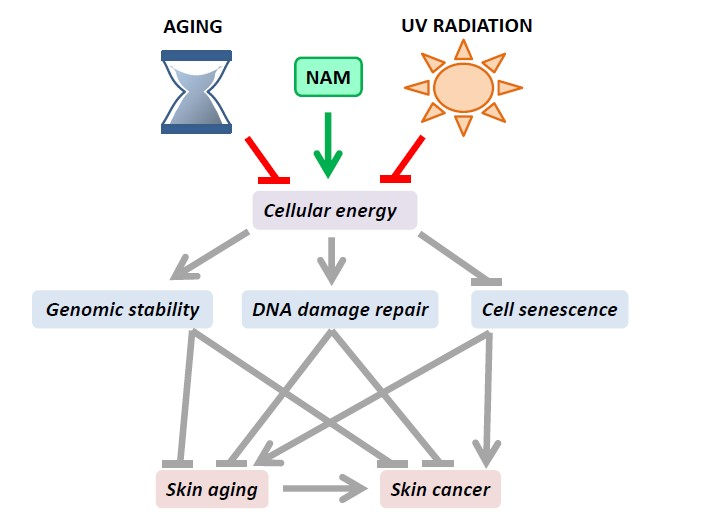
{kind=link}
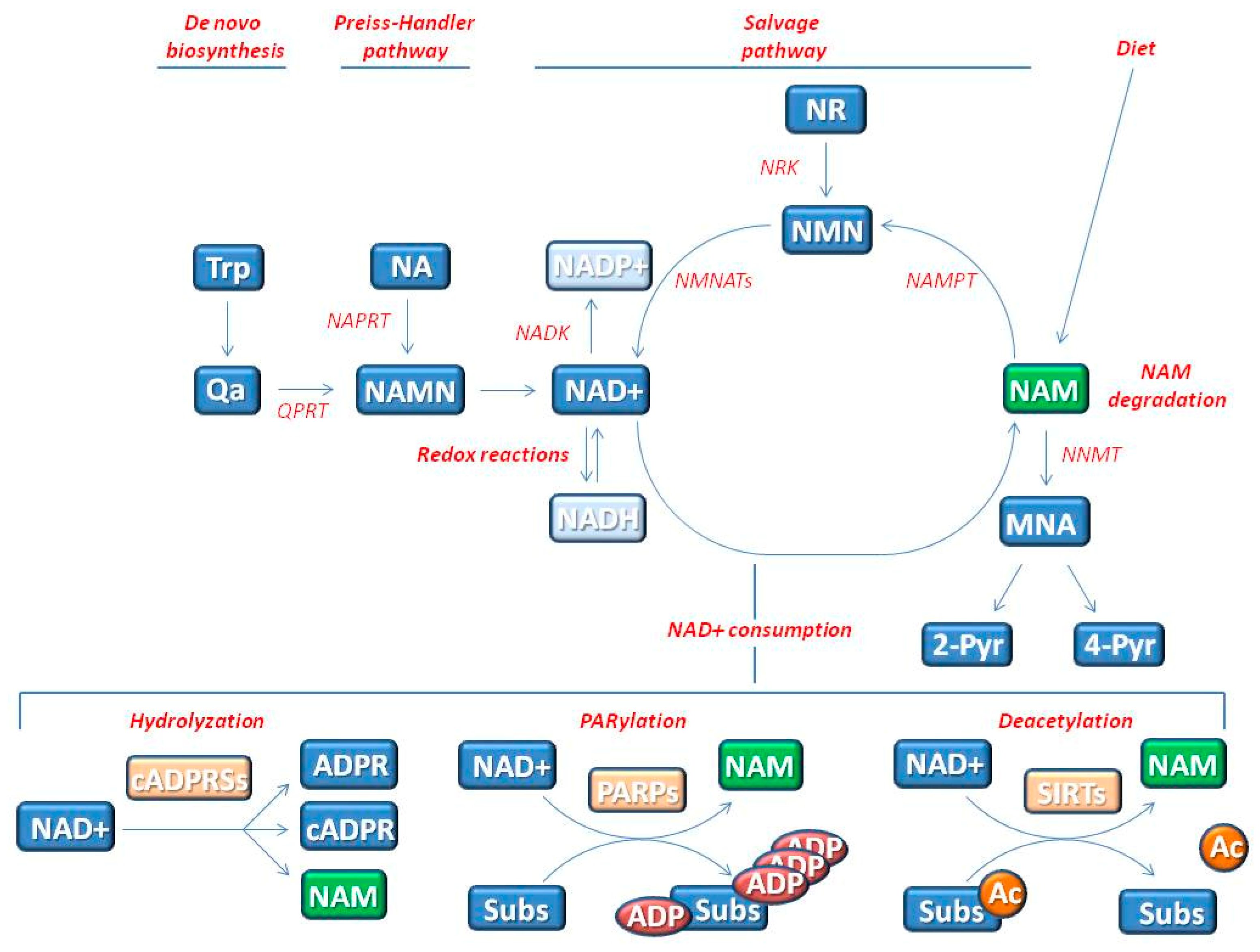
{kind=link}
1. Introduction
2. The Influence of NAD+ Status on Genomic Stability and Cell Senescence
2.1. NAD+ Influences Energy Production by Acting as Co-Enzyme
2.2. NAD+ Influences Cell Signaling by Acting as Substrate of NAD+-Consuming Enzymes
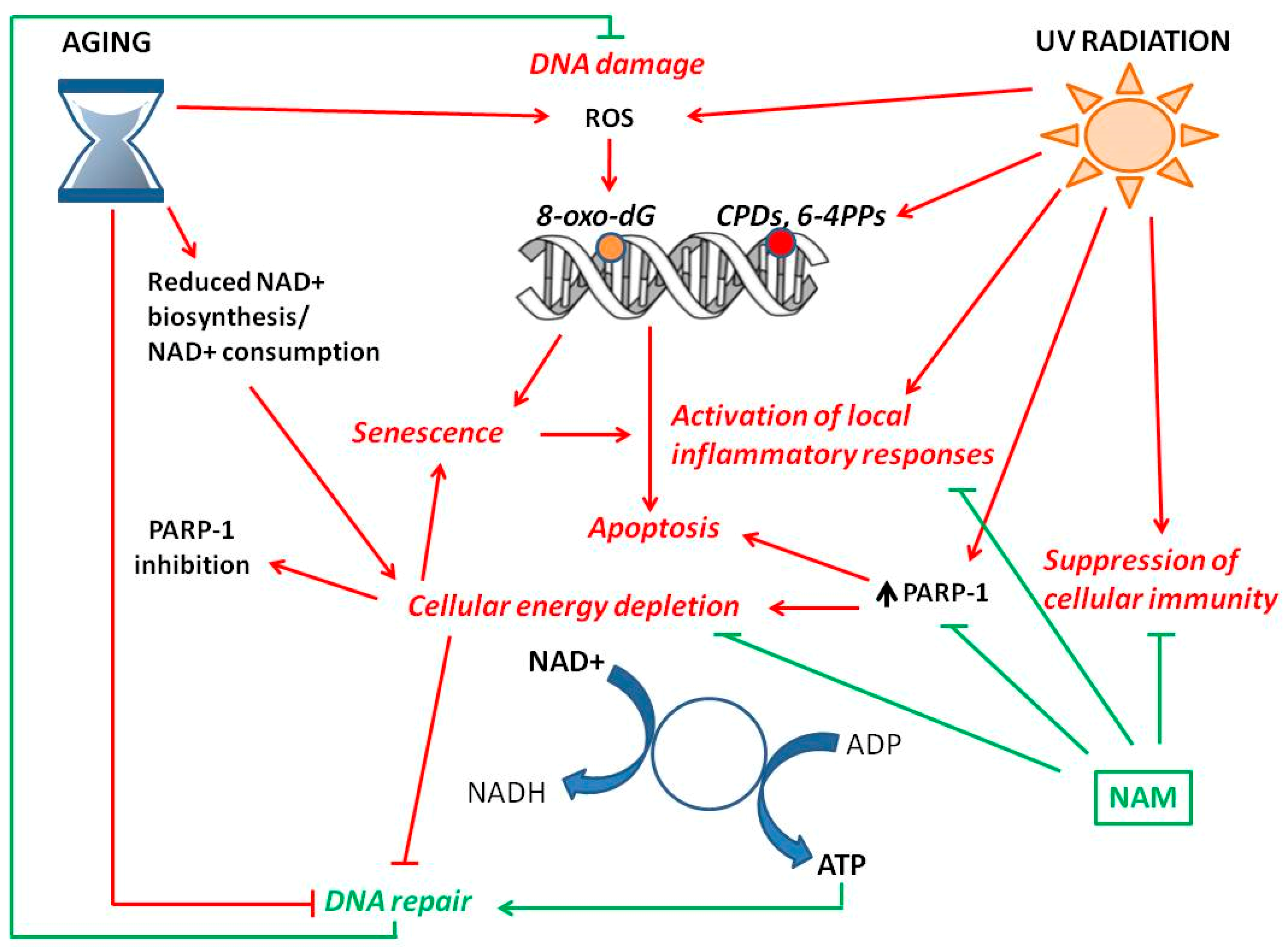
3. Genomic Instability—Hallmark of Aging and Cancer
4. NAD+ Status and Skin Aging
4.1. NAD+ and Chronological Skin Aging
4.2. NAD+ and Photo-Aging
4.3. NAM and Prevention of Aging Signs
5. NAD+ Status and Skin Cancer
5.1. Cutaneous Squamous-Cell Carcinoma
5.2. NAM and Chemoprevention
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NAM | nicotinamide |
| NAD+ | nicotinamide adenine dinucleotide |
| UV | ultraviolet |
| ATP | Adenosine triphosphate |
References
- Damian, D.L. Nicotinamide for skin cancer chemoprevention. Australas. J. Dermatol. 2017, 58, 174–180. [Google Scholar] [CrossRef]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD+ in Aging: Molecular Mechanisms and Translational Implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Benavente, C.A.; Jacobson, M.K.; Jacobson, E.L. NAD in skin: Therapeutic approaches for niacin. Curr. Pharm. Des. 2009, 15, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Surjana, D.; Halliday, G.M.; Damian, D.L. Role of nicotinamide in DNA damage, mutagenesis, and DNA repair. J. Nucleic Acids 2010, 2010, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Aman, Y.; Qiu, Y.; Tao, J.; Fang, E.F. Therapeutic potential of boosting NAD+ in aging and age-related diseases. Transl. Med. Aging 2018, 2, 30–37. [Google Scholar] [CrossRef]
- Jacobson, E.L.; Shieh, W.M.; Huang, A.C. Mapping the role of NAD metabolism in prevention and treatment of carcinogenesis. Mol. Cell. Biochem. 1999, 193, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.; Damian, D.L. Nicotinamide and the skin. Australas. J. Dermatol. 2014, 55, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H.J. DNA Damage, Aging, and Cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Osley, M.A.; Tsukuda, T.; Nickoloff, J.A. ATP-dependent chromatin remodeling factors and DNA damage repair. Mutat. Res. 2007, 618, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Robert, L. Cell senescence: Role in aging and age-related diseases. Interdiscip. Top. Gerontol. 2014, 39, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Campisi, J. From ancient pathways to aging cells -Connecting metabolism and cellular senescence. Cell Metab. 2017, 23, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Benavente, C.A.; Jacobson, E.L. Niacin restriction upregulates NADPH oxidase and reactive oxygen species (ROS) in human keratinocytes. Free Radic. Biol. Med. 2008, 44, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]
- Van der Veer, E.; Ho, C.; O’Neil, C.; Barbosa, N.; Scott, R.; Cregan, S.P.; Pickering, J.G. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J. Biol. Chem. 2007, 282, 10841–10845. [Google Scholar] [CrossRef]
- Luna, A.; Aladjem, M.I.; Kohn, K.W. SIRT1/PARP1 crosstalk: Connecting DNA damage and metabolism. Genome Integr. 2013, 4, 6. [Google Scholar] [CrossRef]
- Hou, W.-H.; Chen, S.-H.; Yu, X. Poly-ADP ribosylation in DNA damage response and cancer therapy. Mutat. Res. 2016, 780, 82–91. [Google Scholar] [CrossRef]
- Ke, Y.; Wang, C.; Zhang, J.; Zhong, X.; Wang, R.; Zeng, X.; Ba, X. The Role of PARPs in Inflammation-and Metabolic-Related Diseases: Molecular Mechanisms and Beyond. Cells 2019, 8, 1047. [Google Scholar] [CrossRef]
- Kim, M.Y.; Mauro, S.; Gévry, N.; Lis, J.T.; Kraus, W.L. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell 2004, 119, 803–814. [Google Scholar] [CrossRef]
- Chung, H.T.; Joe, Y. Antagonistic crosstalk between SIRT1, PARP-1, and -2 in the regulation of chronic inflammation associated with aging and metabolic diseases. Integr. Med. Res. 2014, 3, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Vodenicharov, M.D.; Ghodgaonkar, M.M.; Halappanavar, S.S.; Shah, R.G.; Shah, G.M. Mechanism of early biphasic activation of poly(ADP-ribose) polymerase-1 in response to ultraviolet B radiation. J. Cell Sci. 2005, 118, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Peterson, L.M.; Wilking-Busch, M.J.; Ndiaye, M.A.; Philippe, C.G.A.; Setaluri, V.; Ahmad, N. Sirtuins in Skin and Skin Cancers. Ski. Pharmacol. Physiol. 2017, 30, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Orioli, D.; Dellambra, E. Epigenetic Regulation of Skin Cells in Natural Aging and Premature Aging Diseases. Cells 2018, 7, 268. [Google Scholar] [CrossRef]
- German, N.J.; Haigis, M.C. Sirtuins and the Metabolic Hurdles in Cancer. Curr. Biol. 2015, 25, R569–R583. [Google Scholar] [CrossRef]
- Poljsak, B. NAD+ in Cancer Prevention and Treatment: Pros and Cons. J. Clin. Exp. Oncol. 2016, 4, 2. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Kompaniez, K.; Barnes, J.; Lohani, A.; Evans, M.K. Poly(ADP-ribose) polymerase 1 (PARP-1) binds to 8-oxoguanine-DNA glycosylase (OGG1). J. Biol. Chem. 2011, 286, 44679–44690. [Google Scholar] [CrossRef]
- Shah, A.; Gray, K.; Figg, N.; Finigan, A.; Starks, L.; Bennett, M. Defective Base Excision Repair of Oxidative DNA Damage in Vascular Smooth Muscle Cells Promotes Atherosclerosis. Circulation 2018, 138, 1446–1462. [Google Scholar] [CrossRef]
- Dotto, G.P. Multifocal epithelial tumors and field cancerization: Stroma as a primary determinant. J. Clin. Investig. 2014, 124, 1446–1453. [Google Scholar] [CrossRef]
- Loaiza, N.; Demaria, M. Cellular senescence and tumor promotion: Is aging the key? Biochim. Biophys. Acta 2016, 1865, 155–167. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Keyes, W.M.; Wu, Y.; Vogel, H.; Guo, X.; Lowe, S.W.; Mills, A.A. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev. 2005, 19, 1986–1999. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef]
- Wang, A.S.; Dreesen, O. Biomarkers of cellular senescence and skin aging. Front. Genet. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Abbadie, C.; Pluquet, O.; Pourtier, A. Epithelial cell senescence: An adaptive response to pre-carcinogenic stresses? Cell Mol. Life Sci. 2017, 74, 4471–4509. [Google Scholar] [CrossRef]
- Stratigos, A.; Garbe, C.; Lebbe, C.; Malvehy, J.; del Marmol, V.; Pehamberger, H.; Peris, K.; Becker, J.C.; Zalaudek, I.; Saiag, P.; et al. European Dermatology Forum (EDF); European Association of Dermato-Oncology (EADO); European Organization for Research and Treatment of Cancer (EORTC) Diagnosis and treatment of invasive squamous cell carcinoma of the skin: European consensus-based interdisciplinary guideline. Eur. J. Cancer 2015, 51, 1989–2007. [Google Scholar] [CrossRef]
- Rinnerthaler, M.; Bischof, J.; Streubel, M.; Trost, A.; Richter, K. Oxidative Stress in Aging Human Skin. Biomolecules 2015, 5, 545–589. [Google Scholar] [CrossRef]
- Fuchs, E. Epithelial skin biology: three decades of developmental biology, a hundred questions answered and a thousand new ones to address. In Current Topics in Developmental Biology; Academic Press: London, UK, 2016; pp. 357–374. [Google Scholar]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G.J. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS ONE 2012, 7, e42357. [Google Scholar] [CrossRef]
- Tinaburri, L.; D’Errico, M.; Sileno, S.; Maurelli, R.; Degan, P.; Magenta, A.; Dellambra, E. miR-200a Modulates the Expression of the DNA Repair Protein OGG1 Playing a Role in Aging of Primary Human Keratinocytes. Oxid. Med. Cell. Longev. 2018, 2018, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.L.; Chin, T.; Yan, C.; Tan, R.; Rovito, H.A.; Quek, L.S.; Oblong, J.E.; Bellanger, S. Nicotinamide Metabolism Modulates the Proliferation/Differentiation Balance and Senescence of Human Primary Keratinocytes. J. Investig. Dermatol. 2019, 139, 1638–1647. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, M.; Parlanti, E.; Teson, M.; de Jesus, B.M.B.; Degan, P.; Calcagnile, A.; Jaruga, P.; Bjørås, M.; Crescenzi, M.; Pedrini, A.M.; et al. New functions of XPC in the protection of human skin cells from oxidative damage. EMBO J. 2006, 25, 4305–4315. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.; Mahfouf, W.; Serrano-Sanchez, M.; Raad, H.; Harfouche, G.; Bonneu, M.; Claverol, S.; Mazurier, F.; Rossignol, R.; Taieb, A.; et al. Premature Skin Aging Features Rescued by Inhibition of NADPH Oxidase Activity in XPC-Deficient Mice. J. Investig. Dermatol. 2014, 135, 1–23. [Google Scholar] [CrossRef]
- Rezvani, H.R.; Rossignol, R.; Ali, N.; Benard, G.; Tang, X.; Yang, H.S.; Jouary, T.; De Verneuil, H.; Taïeb, A.; Kim, A.L.; et al. XPC silencing in normal human keratinocytes triggers metabolic alterations through NOX-1 activation-mediated reactive oxygen species. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 609–619. [Google Scholar] [CrossRef]
- Oblong, J.E. The evolving role of the NAD+/nicotinamide metabolome in skin homeostasis, cellular bioenergetics, and aging. DNA Repair 2014, 23, 59–63. [Google Scholar] [CrossRef]
- Mendelsohn, A.R.; Larrick, J.W. The NAD+/PARP1/SIRT1 Axis in Aging. Rejuvenation Res. 2017, 20, 244–247. [Google Scholar] [CrossRef]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD+ and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef]
- Li, J.; Bonkowski, M.S.; Moniot, S.; Zhang, D.; Hubbard, B.P.; Ling, A.J.Y.; Rajman, L.A.; Qin, B.; Lou, Z.; Gorbunova, V.; et al. A conserved NAD+ binding pocket that regulates protein-protein interactions during aging. Science 2017, 355, 1312–1317. [Google Scholar] [CrossRef]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef]
- Ming, M.; Zhao, B.; Shea, C.R.; Shah, P.; Qiang, L.; White, S.R.; Sims, D.M.; He, Y.-Y. Loss of sirtuin 1 (SIRT1) disrupts skin barrier integrity and sensitizes mice to epicutaneous allergen challenge. J. Allergy Clin. Immunol. 2015, 135, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Zhang, X.; Yi, J.; Huang, J.; He, J.; Tao, Y. Sirtuins in metabolism, DNA repair and cancer. J. Exp. Clin. Cancer Res. 2016, 35, 182. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, H.J. Molecular Mechanisms of Skin Aging and Rejuvenation. In Molecular Mechanisms of the Aging Process and Rejuvenation; Naofumi Shiomi; IntechOpen Limited: London, UK, 2016; p. 450. ISBN 9789533077826. [Google Scholar]
- Kammeyer, A.; Luiten, R.M. Oxidation events and skin aging. Ageing Res. Rev. 2015, 21, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Dotto, G.P.; Rustgi, A.K. Squamous Cell Cancers: A Unified Perspective on Biology and Genetics. Cancer Cell 2016, 29, 622–637. [Google Scholar] [CrossRef]
- Benavente, C.A.; Schnell, S.A.; Jacobson, E.L. Effects of niacin restriction on sirtuin and PARP responses to photodamage in human skin. PLoS ONE 2012, 7, e42276. [Google Scholar] [CrossRef]
- Park, J.; Halliday, G.M.; Surjana, D.; Damian, D.L. Nicotinamide Prevents Ultraviolet Radiation-induced Cellular Energy Loss. Photochem. Photobiol. 2010, 86, 942–948. [Google Scholar] [CrossRef]
- Surjana, D.; Halliday, G.M.; Damian, D.L. Nicotinamide enhances repair of ultraviolet radiation-induced DNA damage in human keratinocytes and ex vivo skin. Carcinogenesis 2013, 34, 1144–1149. [Google Scholar] [CrossRef]
- Feldmeyer, L.; Keller, M.; Niklaus, G.; Hohl, D.; Werner, S.; Beer, H.D. The Inflammasome Mediates UVB-Induced Activation and Secretion of Interleukin-1β by Keratinocytes. Curr. Biol. 2007, 17, 1140–1145. [Google Scholar] [CrossRef]
- Fagot, D.; Asselineau, D.; Bernerd, F. Matrix metalloproteinase-1 production observed after solar-simulated radiation exposure is assumed by dermal fibroblasts but involves a paracrine activation through epidermal keratinocytes. Photochem. Photobiol. 2004, 79, 499–505. [Google Scholar] [CrossRef]
- Bernerd, F.; Marionnet, C.; Duval, C. Solar ultraviolet radiation induces biological alterations in human skin in vitro: Relevance of a well-balanced UVA/UVB protection. Indian J. Dermatol. Venereol. Leprol. 2012, 78, S15–S23. [Google Scholar] [CrossRef]
- Imokawa, G.; Ishida, K. Biological mechanisms underlying the ultraviolet radiation-induced formation of skin wrinkling and sagging I: Reduced skin elasticity, highly associated with enhanced dermal elastase activity, triggers wrinkling and sagging. Int. J. Mol. Sci. 2015, 16, 7753–7775. [Google Scholar] [CrossRef] [PubMed]
- Bernerd, F.; Asselineau, D. An organotypic model of skin to study photodamage and photoprotection in vitro. J. Am. Acad. Dermatol. 2008, 58, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Duval, C.; Cohen, C.; Chagnoleau, C.; Flouret, V.; Bourreau, E.; Bernerd, F. Key Regulatory Role of Dermal Fibroblasts in Pigmentation as Demonstrated Using a Reconstructed Skin Model: Impact of Photo-Aging. PLoS ONE 2014, 9, e114182. [Google Scholar] [CrossRef]
- Martin-oliva, D.; Valle, F.O.; Mun, J.A.; Garcı, R.; Villuendas, R.; Piris, M.A.; Oliver, F.J. Inhibition of Poly (ADP-Ribose) Polymerase Modulates Tumor- Related Gene Expression, Including Hypoxia-Inducible Factor-1 Activation, during Skin Carcinogenesis. Cancer Res. 2006, 66, 5744–5757. [Google Scholar] [CrossRef] [PubMed]
- Yiasemides, E.; Sivapirabu, G.; Halliday, G.M.; Park, J.; Damian, D.L. Oral nicotinamide protects against ultraviolet radiation-induced immunosuppression in humans. Carcinogenesis 2009, 30, 101–105. [Google Scholar] [CrossRef]
- Snaidr, V.A.; Damian, D.L.; Halliday, G.M. Nicotinamide for photoprotection and skin cancer chemoprevention: A review of efficacy and safety. Exp. Dermatol. 2019, 28, 15–22. [Google Scholar] [CrossRef]
- Halliday, G.M.; Byrne, S.N.; Damian, D.L. Ultraviolet A radiation: Its role in immunosuppression and carcinogenesis. Semin. Cutan. Med. Surg. 2011, 30, 214–221. [Google Scholar] [CrossRef]
- Cooke, M.S.; Harry, E.L.; Liljendahl, T.S.; Segerbäck, D. DNA nucleotide excision repair, where do all the cyclobutane pyrimidine dimers go? Cell Cycle 2013, 12, 1642. [Google Scholar] [CrossRef][Green Version]
- Prasad, R.; Katiyar, S.K. Crosstalk Among UV-Induced Inflammatory Mediators, DNA Damage and Epigenetic Regulators Facilitates Suppression of the Immune System. Photochem. Photobiol. 2017, 93, 930–936. [Google Scholar] [CrossRef]
- Gensler, H.L. Prevention of photoimmunosuppression and photocarcinogenesis by topical nicotinamide. Nutr. Cancer 1997, 29, 157–162. [Google Scholar] [CrossRef]
- Gensler, H.L.; Williams, T.; Huang, A.C.; Jacobson, E.L. Oral niacin prevents photocarcinogenesis and photoimmunosuppression in mice. Nutr. Cancer 1999, 34, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Sivapirabu, G.; Yiasemides, E.; Halliday, G.M.; Park, J.; Damian, D.L. Topical nicotinamide modulates cellular energy metabolism and provides broad-spectrum protection against ultraviolet radiation-induced immunosuppression in humans. Br. J. Dermatol. 2009, 161, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Damian, D.L.; Patterson, C.R.S.; Stapelberg, M.; Park, J.; Barnetson, R.S.C.; Halliday, G.M. UV radiation-induced immunosuppression is greater in men and prevented by topical nicotinamide. J. Investig. Dermatol. 2008, 128, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.; Martin, A.J.; Dalziell, R.A.; Halliday, G.M.; Damian, D.L. Oral nicotinamide reduces transepidermal water loss: A randomized controlled trial. Br. J. Dermatol. 2016, 175, 1363–1365. [Google Scholar] [CrossRef] [PubMed]
- Bissett, D.L.; Oblong, J.E.; Berge, C.A. Niacinamide: A B vitamin that improves aging facial skin appearance. Dermatol. Surg. 2005, 31, 860–865. [Google Scholar] [CrossRef]
- Kawada, A.; Konishi, N.; Oiso, N.; Kawara, S.; Date, A. Evaluation of anti-wrinkle effects of a novel cosmetic containing niacinamide. J. Dermatol. 2008, 35, 637–642. [Google Scholar] [CrossRef]
- Hakozaki, T.; Minwalla, L.; Zhuang, J.; Chhoa, M.; Matsubara, A.; Miyamoto, K.; Greatens, A.; Hillebrand, G.G.; Bissett, D.L.; Boissy, R.E. The effect of niacinamide on reducing cutaneous pigmentation and suppression of melanosome transfer. Br. J. Dermatol. 2002, 147, 20–31. [Google Scholar] [CrossRef]
- Kimball, A.B.; Kaczvinsky, J.R.; Li, J.; Robinson, L.R.; Matts, P.J.; Berge, C.A.; Miyamoto, K.; Bissett, D.L. Reduction in the appearance of facial hyperpigmentation after use of moisturizers with a combination of topical niacinamide and N-acetyl glucosamine: Results of a randomized, double-blind, vehicle-controlled trial. Br. J. Dermatol. 2010, 162, 435–441. [Google Scholar] [CrossRef]
- Berardesca, E.; Ardigo, M.; Cameli, N.; Mariano, M.; Agozzino, M.; Matts, P.J. Randomized, double-blinded, vehicle-controlled, split-face study to evaluate the effects of topical application of a Gold Silk Sericin/Niacinamide/Signaline complex on biophysical parameters related to skin ageing. Int. J. Cosmet. Sci. 2015, 37, 606–612. [Google Scholar] [CrossRef]
- Xiang, F.; Lucas, R.; Hales, S.; Neale, R. Incidence of Nonmelanoma Skin Cancer in Relation to Ambient UV Radiation in White Populations, 1978-2012: Empirical Relationships. JAMA Dermatol. 2014, 150, 1–9. [Google Scholar] [CrossRef]
- Brantsch, K.D.; Meisner, C.; Schönfisch, B.; Trilling, B.; Wehner-Caroli, J.; Röcken, M.; Breuninger, H. Analysis of risk factors determining prognosis of cutaneous squamous-cell carcinoma: A prospective study. Lancet Oncol. 2008, 9, 713–720. [Google Scholar] [CrossRef]
- Werner, R.N.; Stockfleth, E.; Connolly, S.M.; Correia, O.; Erdmann, R.; Foley, P.; Gupta, A.K.; Jacobs, A.; Kerl, H.; Lim, H.W.; et al. Evidence- and consensus-based (S3) Guidelines for the Treatment of Actinic Keratosis-International League of Dermatological Societies in cooperation with the European Dermatology Forum-Short version. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 2069–2079. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.L. Actinic keratosis treatment as a key component of preventive strategies for nonmelanoma skin cancer. J. Clin. Aesthet. Dermatol. 2010, 3, 39–44. [Google Scholar] [PubMed]
- Alam, M.; Ratner, D. Cutaneous squamous-cell carcinoma. N. Engl. J. Med. 2001, 344, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Surjana, D.; Halliday, G.M.; Martin, A.J.; Moloney, F.J.; Damian, D.L. Oral nicotinamide reduces actinic keratoses in phase II double-blinded randomized controlled trials. J. Investig. Dermatol. 2012, 132, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.; Martin, A.J.; Choy, B.; Fernández-Peñas, P.; Dalziell, R.A.; McKenzie, C.A.; Scolyer, R.A.; Dhillon, H.M.; Vardy, J.L.; Kricker, A.; et al. A Phase 3 Randomized Trial of Nicotinamide for Skin-Cancer Chemoprevention. N. Engl. J. Med. 2015, 373, 1618–1626. [Google Scholar] [CrossRef]
- Minocha, R.; Martin, A.J.; Chen, A.C.; Scolyer, R.A.; Lyons, J.G.; McKenzie, C.A.; Madore, J.; Halliday, G.M.; Damian, D.L. A Reduction in Inflammatory Macrophages May Contribute to Skin Cancer Chemoprevention by Nicotinamide. J. Investig. Dermatol. 2019, 139, 467–469. [Google Scholar] [CrossRef]
- Martin, A.J.; Dhillon, H.M.; Vardy, J.L.; Dalziell, R.A.; Choy, B.; Fernández-Peñas, P.; Dixon, A.; Renton, C.; St George, G.; Chinniah, N.; et al. Neurocognitive Function and Quality of Life Outcomes in the ONTRAC Study for Skin Cancer Chemoprevention by Nicotinamide. Geriatrics 2019, 4, 31. [Google Scholar] [CrossRef]
- Drago, F.; Ciccarese, G.; Cogorno, L.; Calvi, C.; Marsano, L.A.; Parodi, A. Prevention of non-melanoma skin cancers with nicotinamide in transplant recipients: A case-control study. Eur. J. Dermatol. 2017, 27, 382–385. [Google Scholar] [CrossRef]
- Chen, A.C.; Martin, A.J.; Dalziell, R.A.; McKenzie, C.A.; Lowe, P.M.; Eris, J.M.; Scolyer, R.A.; Dhillon, H.M.; Vardy, J.L.; Bielski, V.A.; et al. A phase II randomized controlled trial of nicotinamide for skin cancer chemoprevention in renal transplant recipients. Br. J. Dermatol. 2016, 175, 1073–1075. [Google Scholar] [CrossRef]
- Niren, N.M. Pharmacologic doses of nicotinamide in the treatment of inflammatory skin conditions: A review. Cutis 2006, 77, 11–16. [Google Scholar] [PubMed]
- Bernier, J.; Stratford, M.R.L.; Denekamp, J.; Dennis, M.F.; Bieri, S.; Hagen, F.; Kocagöncü, O.; Bolla, M.; Rojas, A. Pharmacokinetics of nicotinamide in cancer patients treated with accelerated radiotherapy: The experience of the co-operative group of radiotherapy of the European organization for research and treatment of cancer. Radiother. Oncol. 1998, 48, 123–133. [Google Scholar] [CrossRef]
- Yilmaz, Z.; Piracha, F.; Anderson, L.; Mazzola, N. Supplements for Diabetes Mellitus: A Review of the Literature. J. Pharm. Pract. 2016, 30, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Final Report of the Safety Assessment of Niacinamide and Niacin1. Int. J. Toxicol. 2005, 24, 1–31. [CrossRef]
- Lenglet, A.; Liabeuf, S.; El Esper, N.; Brisset, S.; Mansour, J.; Lemaire-Hurtel, A.S.; Mary, A.; Brazier, M.; Kamel, S.; Mentaverri, R.; et al. Efficacy and safety of nicotinamide in haemodialysis patients: The NICOREN study. Nephrol. Dial. Transplant. 2017, 32, 870–879. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fania, L.; Mazzanti, C.; Campione, E.; Candi, E.; Abeni, D.; Dellambra, E. Role of Nicotinamide in Genomic Stability and Skin Cancer Chemoprevention. Int. J. Mol. Sci. 2019, 20, 5946. https://doi.org/10.3390/ijms20235946
Fania L, Mazzanti C, Campione E, Candi E, Abeni D, Dellambra E. Role of Nicotinamide in Genomic Stability and Skin Cancer Chemoprevention. International Journal of Molecular Sciences. 2019; 20(23):5946. https://doi.org/10.3390/ijms20235946
Chicago/Turabian StyleFania, Luca, Cinzia Mazzanti, Elena Campione, Eleonora Candi, Damiano Abeni, and Elena Dellambra. 2019. "Role of Nicotinamide in Genomic Stability and Skin Cancer Chemoprevention" International Journal of Molecular Sciences 20, no. 23: 5946. https://doi.org/10.3390/ijms20235946
APA StyleFania, L., Mazzanti, C., Campione, E., Candi, E., Abeni, D., & Dellambra, E. (2019). Role of Nicotinamide in Genomic Stability and Skin Cancer Chemoprevention. International Journal of Molecular Sciences, 20(23), 5946. https://doi.org/10.3390/ijms20235946