Recent Insights into Long Bone Development: Central Role of Hedgehog Signaling Pathway in Regulating Growth Plate

 , and
, and 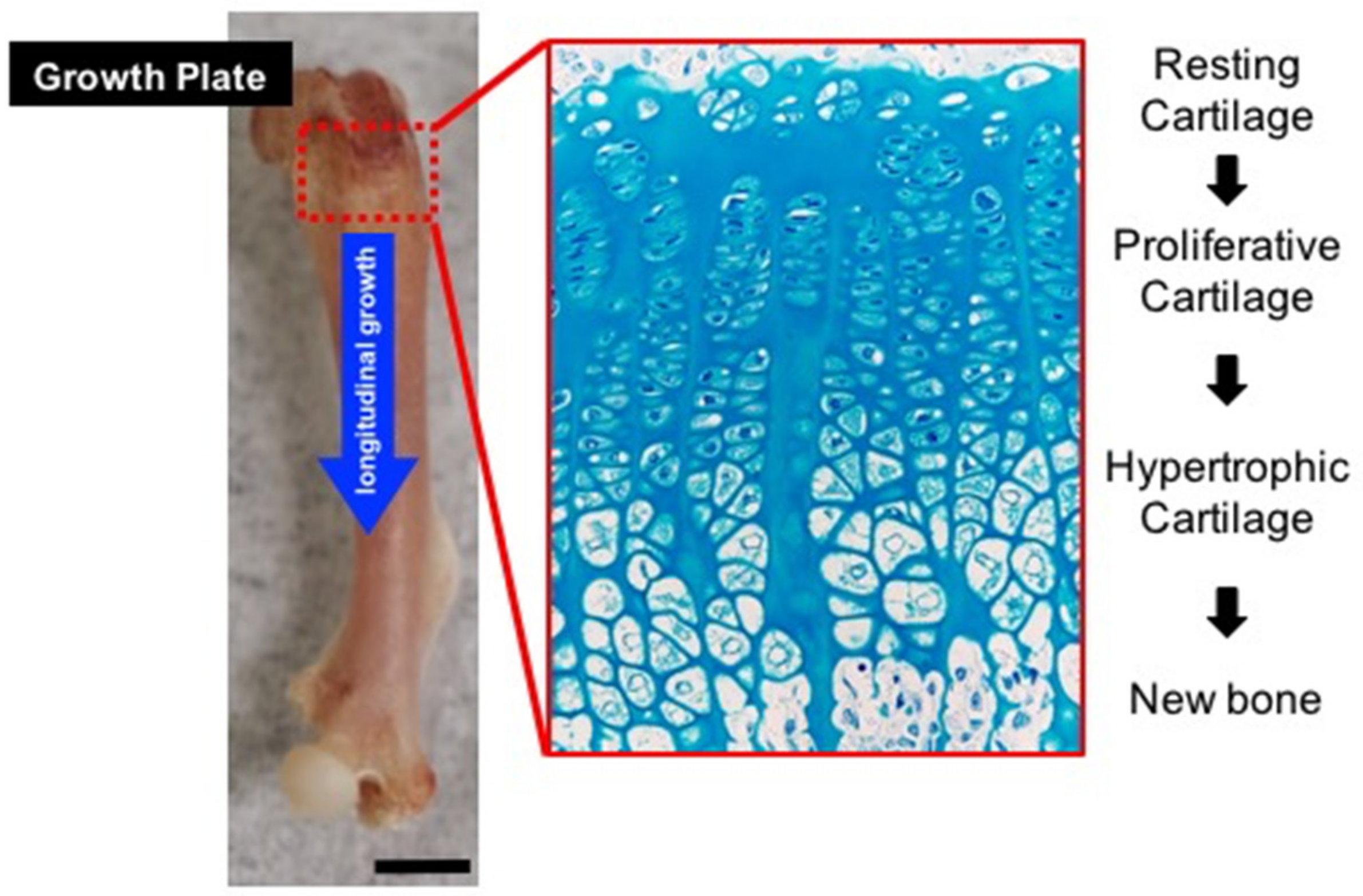{kind=link}
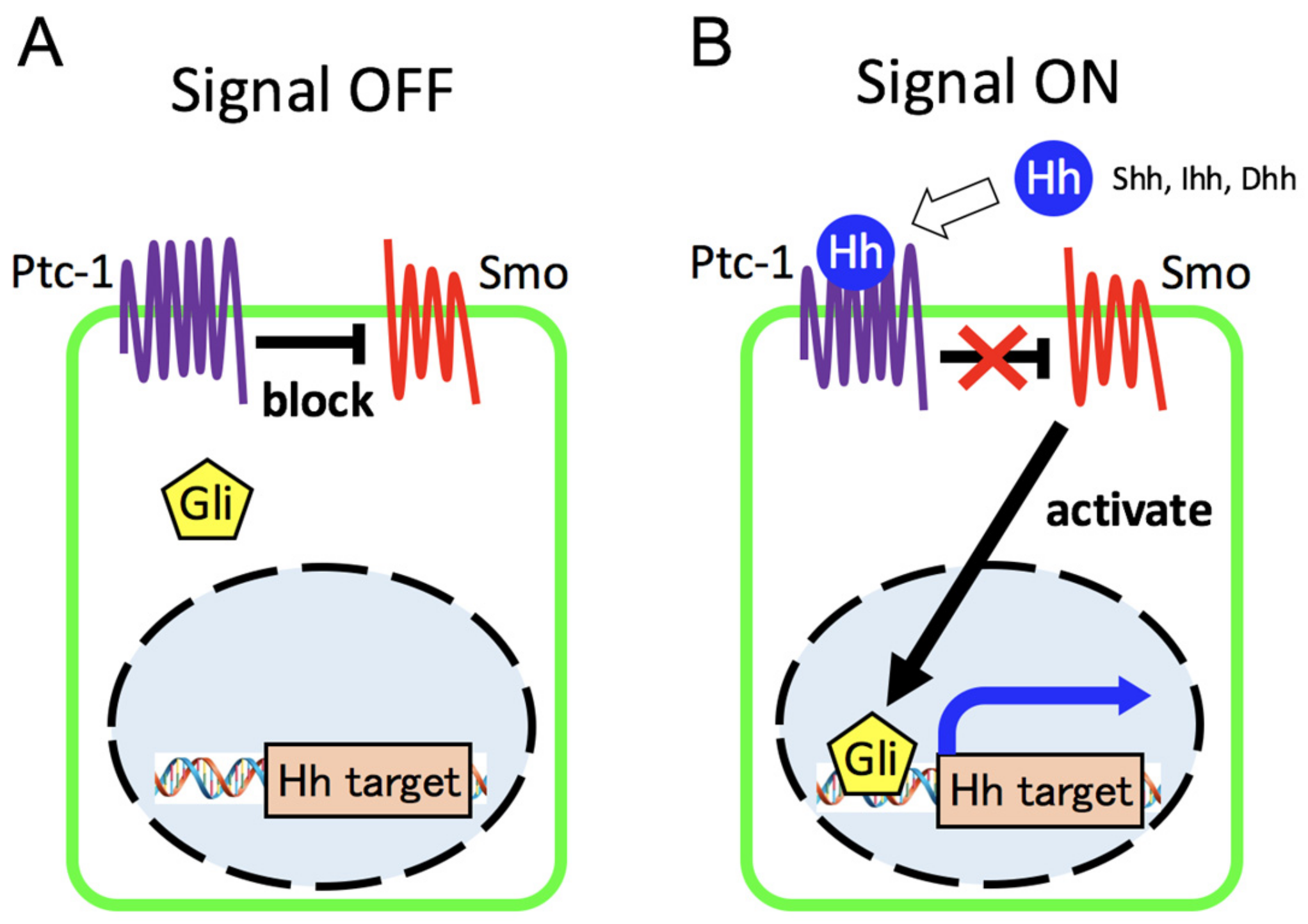{kind=link}
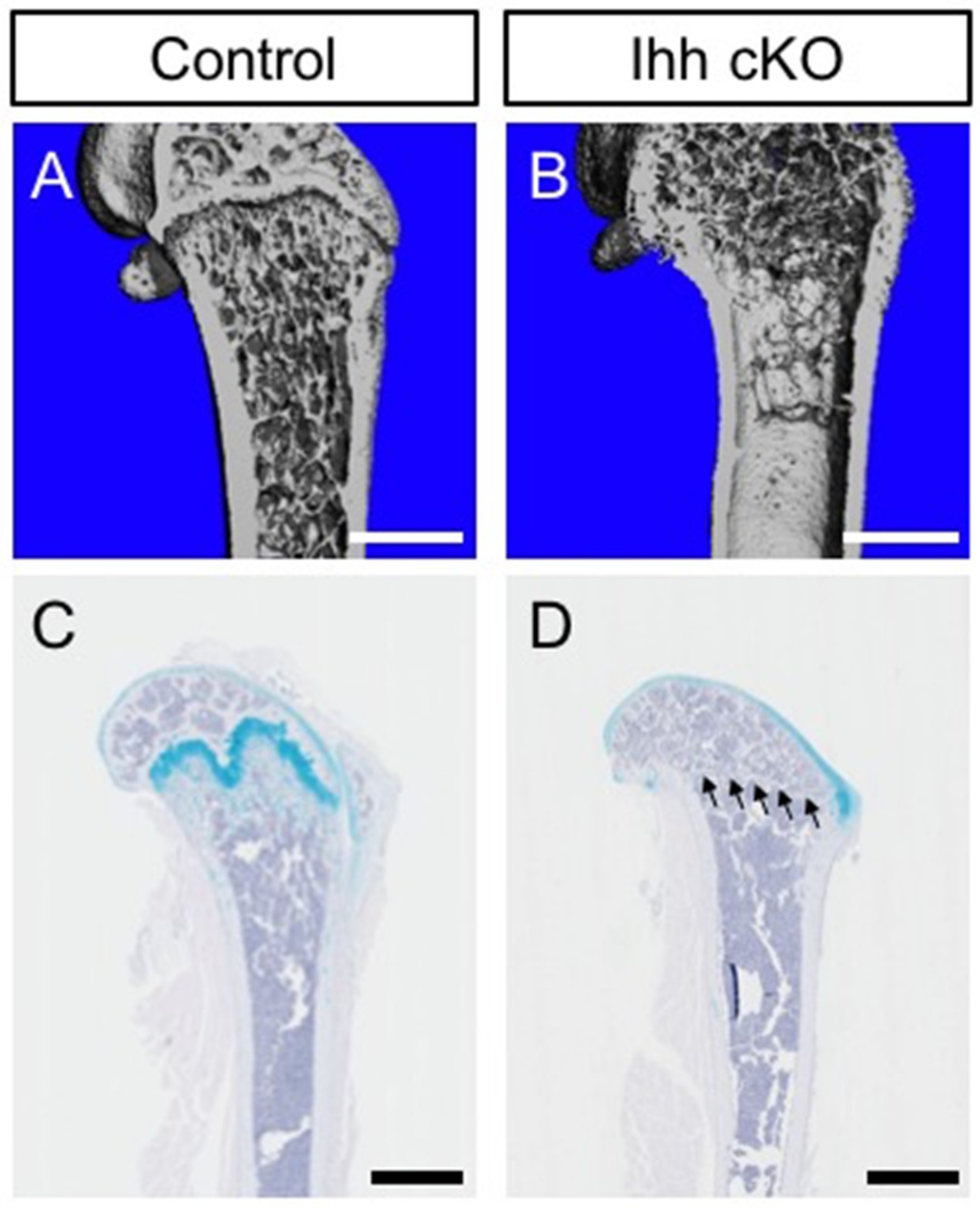{kind=link}
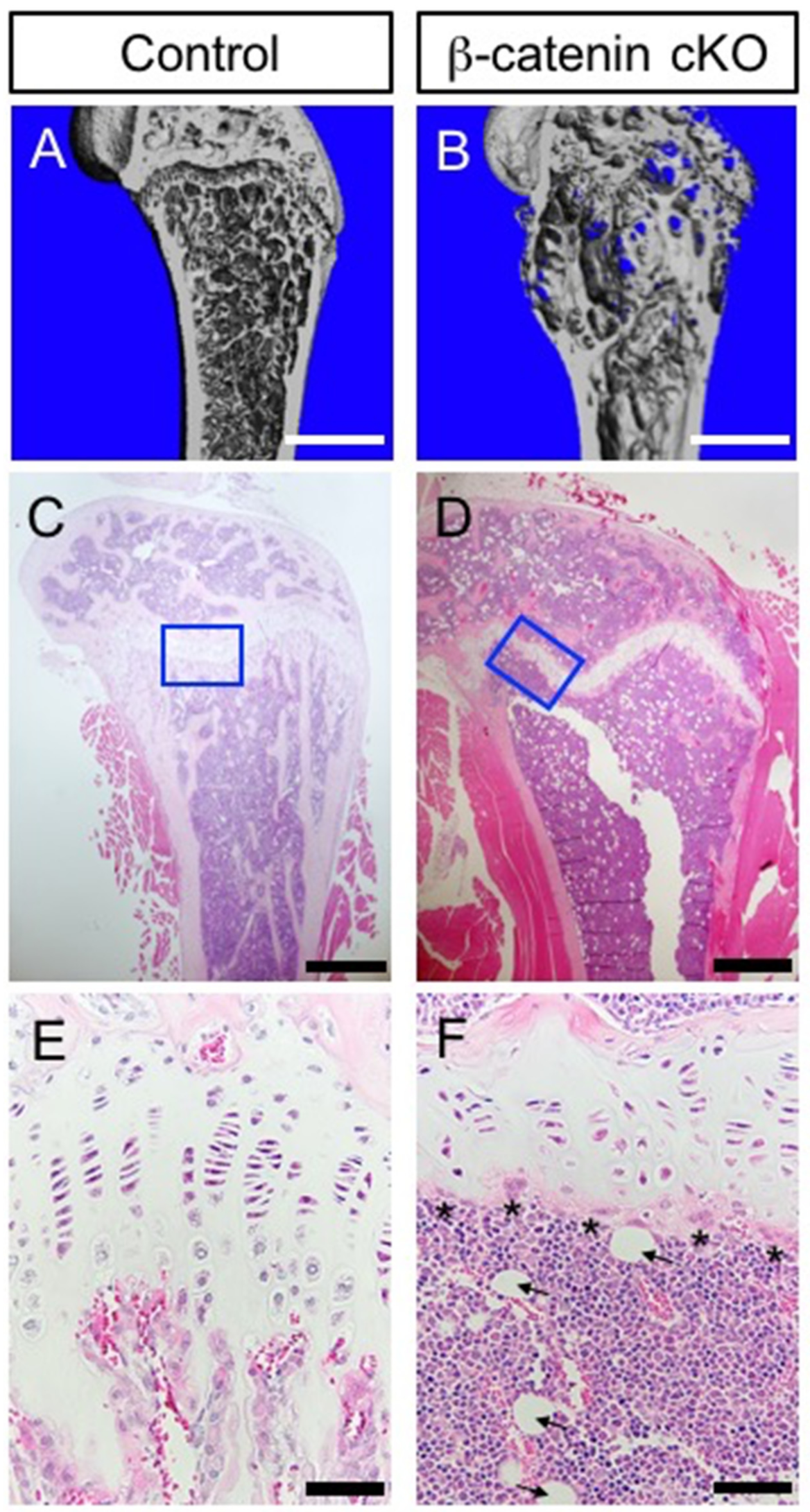{kind=link}
Abstract
1. Introduction
2. Hedgehog Signal Is a Critical Regulator of Early Chondrogenesis
3. Central Role of Hedgehog Signaling in Regulation of Growth Plate
3.1. Hh Pathway and Growth Plate Formation
3.2. Hh Pathway Controls Regulation of Growth Plate Differentiation through Interaction with PTH-PTHrP Signaling
3.3. Crosstalk between Hh and Other Signaling Pathways as a Basis for Regulatory Mechanisms of Growth Plate Development and Function
3.3.1. Hh and Wnt/β-Catenin Signaling
3.3.2. Hh and FGF Signaling
3.3.3. Hh and BMP Signaling
3.3.4. Hh Signaling and Angiogenic Factors
3.4. Coupling Role of Hh Signaling Pathway and Intracellular Cholesterol Production in Growth Plate Development
3.5. Hh Pathway and Developmental Contribution of Growth Plate Chondrocytes to Skeletal Bone Formation
4. Aberrant Hedgehog Signaling in Skeletal Disease
4.1. Hedgehog Signalling and Brachydactyly Syndrome
4.2. Hedgehog Signaling and Cartilage Tumorigenesis
4.3. Hedgehog Signaling and Heterotopic Ossification
5. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Abad, V.; Meyers, J.L.; Weise, M.; Gafni, R.I.; Barnes, K.M.; Nilsson, O.; Bacher, J.D.; Baron, J. The role of the resting zone in growth plate chondrogenesis. Endocrinology 2002, 143, 1851–1857. [Google Scholar] [CrossRef]
- Prein, C.; Warmbold, N.; Farkas, Z.; Schieker, M.; Aszodi, A.; Clausen-Schaumann, H. Structural and mechanical properties of the proliferative zone of the developing murine growth plate cartilage assessed by atomic force microscopy. Matrix Biol. 2016, 50, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Breur, G.J.; VanEnkevort, B.A.; Farnum, C.E.; Wilsman, N.J. Linear relationship between the volume of hypertrophic chondrocytes and the rate of longitudinal bone growth in growth plates. J. Orthop. Res. 1991, 9, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Farnum, C.E.; Lee, R.; O’Hara, K.; Urban, J.P. Volume increase in growth plate chondrocytes during hypertrophy: The contribution of organic osmolytes. Bone 2002, 30, 574–581. [Google Scholar] [CrossRef]
- Gerber, H.P.; Vu, T.H.; Ryan, A.M.; Kowalski, J.; Werb, Z.; Ferrara, N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 1999, 5, 623–628. [Google Scholar] [CrossRef]
- Kojima, T.; Hasegawa, T.; de Freitas, P.H.; Yamamoto, T.; Sasaki, M.; Horiuchi, K.; Hongo, H.; Yamada, T.; Sakagami, N.; Saito, N.; et al. Histochemical aspects of the vascular invasion at the erosion zone of the epiphyseal cartilage in MMP-9-deficient mice. Biomed. Res. 2013, 34, 119–128. [Google Scholar] [CrossRef]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef]
- Aghajanian, P.; Mohan, S. The art of building bone: Emerging role of chondrocyte-to-osteoblast transdifferentiation in endochondral ossification. Bone Res. 2018, 6, 19. [Google Scholar] [CrossRef]
- Alman, B.A. The role of hedgehog signalling in skeletal health and disease. Nat. Rev. Rheumatol. 2015, 11, 552–560. [Google Scholar] [CrossRef]
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; Spohr, T. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.T.; Zhao, Z.; Ingham, P.W. Hedgehog signalling. Development 2016, 143, 367–372. [Google Scholar] [CrossRef] [PubMed]
- McMahon, A.P. More surprises in the Hedgehog signaling pathway. Cell 2000, 100, 185–188. [Google Scholar] [CrossRef]
- Ryan, K.E.; Chiang, C. Hedgehog secretion and signal transduction in vertebrates. J. Biol. Chem. 2012, 287, 17905–17913. [Google Scholar] [CrossRef]
- Yang, J.; Andre, P.; Ye, L.; Yang, Y.Z. The Hedgehog signalling pathway in bone formation. Int. J. Oral Sci. 2015, 7, 73–79. [Google Scholar] [CrossRef]
- Bitgood, M.J.; McMahon, A.P. Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev. Biol. 1995, 172, 126–138. [Google Scholar] [CrossRef]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef]
- Ohba, S. Hedgehog Signaling in Endochondral Ossification. J. Dev. Biol. 2016, 4, 20. [Google Scholar] [CrossRef]
- Goldring, M.B.; Tsuchimochi, K.; Ijiri, K. The control of chondrogenesis. J. Cell. Biochem. 2006, 97, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Ornitz, D.M. Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 2013, 5, a008334. [Google Scholar] [CrossRef] [PubMed]
- Quintana, L.; zur Nieden, N.I.; Semino, C.E. Morphogenetic and regulatory mechanisms during developmental chondrogenesis: New paradigms for cartilage tissue engineering. Tissue Eng. Part B Rev. 2009, 15, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Bruce, S.J.; Butterfield, N.C.; Metzis, V.; Town, L.; McGlinn, E.; Wicking, C. Inactivation of Patched1 in the mouse limb has novel inhibitory effects on the chondrogenic program. J. Biol. Chem. 2010, 285, 27967–27981. [Google Scholar] [CrossRef]
- Chuang, P.T.; McMahon, A.P. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature 1999, 397, 617–621. [Google Scholar] [CrossRef]
- Pan, A.; Chang, L.; Nguyen, A.; James, A.W. A review of hedgehog signaling in cranial bone development. Front. Physiol. 2013, 4, 61. [Google Scholar] [CrossRef]
- Zhulyn, O.; Hui, C.C. Sufu and Kif7 in limb patterning and development. Dev. Dyn. 2015, 244, 468–478. [Google Scholar] [CrossRef]
- Kozhemyakina, E.; Lassar, A.B.; Zelzer, E. A pathway to bone: Signaling molecules and transcription factors involved in chondrocyte development and maturation. Development 2015, 142, 817–831. [Google Scholar] [CrossRef]
- Amano, K.; Densmore, M.; Fan, Y.; Lanske, B. Ihh and PTH1R signaling in limb mesenchyme is required for proper segmentation and subsequent formation and growth of digit bones. Bone 2016, 83, 256–266. [Google Scholar] [CrossRef]
- Amano, K.; Densmore, M.J.; Lanske, B. Conditional Deletion of Indian Hedgehog in Limb Mesenchyme Results in Complete Loss of Growth Plate Formation but Allows Mature Osteoblast Differentiation. J. Bone Miner. Res. 2015, 30, 2262–2272. [Google Scholar] [CrossRef]
- Maeda, Y.; Nakamura, E.; Nguyen, M.T.; Suva, L.J.; Swain, F.L.; Razzaque, M.S.; Mackem, S.; Lanske, B. Indian Hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc. Natl. Acad. Sci. USA 2007, 104, 6382–6387. [Google Scholar] [CrossRef] [PubMed]
- Razzaque, M.S.; Soegiarto, D.W.; Chang, D.; Long, F.; Lanske, B. Conditional deletion of Indian hedgehog from collagen type 2alpha1-expressing cells results in abnormal endochondral bone formation. J. Pathol. 2005, 207, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Zhang, X.M.; Karp, S.; Yang, Y.; McMahon, A.P. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 2001, 128, 5099–5108. [Google Scholar] [PubMed]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 1996, 273, 613–622. [Google Scholar] [CrossRef]
- Karaplis, A.C.; Luz, A.; Glowacki, J.; Bronson, R.T.; Tybulewicz, V.L.; Kronenberg, H.M.; Mulligan, R.C. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994, 8, 277–289. [Google Scholar] [CrossRef]
- Lee, K.; Lanske, B.; Karaplis, A.C.; Deeds, J.D.; Kohno, H.; Nissenson, R.A.; Kronenberg, H.M.; Segre, G.V. Parathyroid hormone-related peptide delays terminal differentiation of chondrocytes during endochondral bone development. Endocrinology 1996, 137, 5109–5118. [Google Scholar] [CrossRef]
- Lanske, B.; Karaplis, A.C.; Lee, K.; Luz, A.; Vortkamp, A.; Pirro, A.; Karperien, M.; Defize, L.H.; Ho, C.; Mulligan, R.C.; et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 1996, 273, 663–666. [Google Scholar] [CrossRef]
- Weir, E.C.; Philbrick, W.M.; Amling, M.; Neff, L.A.; Baron, R.; Broadus, A.E. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc. Natl. Acad. Sci. USA 1996, 93, 10240–10245. [Google Scholar] [CrossRef]
- Hilton, M.J.; Tu, X.; Long, F. Tamoxifen-inducible gene deletion reveals a distinct cell type associated with trabecular bone, and direct regulation of PTHrP expression and chondrocyte morphology by Ihh in growth region cartilage. Dev. Biol. 2007, 308, 93–105. [Google Scholar] [CrossRef]
- Kronenberg, H.M. PTHrP and skeletal development. Ann. N. Y. Acad. Sci. 2006, 1068, 1–13. [Google Scholar] [CrossRef]
- Karp, S.J.; Schipani, E.; St-Jacques, B.; Hunzelman, J.; Kronenberg, H.; McMahon, A.P. Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone related-protein-dependent and -independent pathways. Development 2000, 127, 543–548. [Google Scholar] [PubMed]
- Maeda, Y.; Schipani, E.; Densmore, M.J.; Lanske, B. Partial rescue of postnatal growth plate abnormalities in Ihh mutants by expression of a constitutively active PTH/PTHrP receptor. Bone 2010, 46, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.P.; Mitchell, J. Indian hedgehog: Its roles and regulation in endochondral bone development. J. Cell. Biochem. 2005, 96, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Marino, R. Growth plate biology: New insights. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 9–13. [Google Scholar] [CrossRef]
- Mak, K.K.; Chen, M.H.; Day, T.F.; Chuang, P.T.; Yang, Y. Wnt/beta-catenin signaling interacts differentially with Ihh signaling in controlling endochondral bone and synovial joint formation. Development 2006, 133, 3695–3707. [Google Scholar] [CrossRef]
- Xie, Y.; Zhou, S.; Chen, H.; Du, X.; Chen, L. Recent research on the growth plate: Advances in fibroblast growth factor signaling in growth plate development and disorders. J. Mol. Endocrinol. 2014, 53, T11–T34. [Google Scholar] [CrossRef]
- Foldynova-Trantirkova, S.; Wilcox, W.R.; Krejci, P. Sixteen years and counting: The current understanding of fibroblast growth factor receptor 3 (FGFR3) signaling in skeletal dysplasias. Hum. Mutat. 2012, 33, 29–41. [Google Scholar] [CrossRef]
- Peters, K.; Ornitz, D.; Werner, S.; Williams, L. Unique expression pattern of the FGF receptor 3 gene during mouse organogenesis. Dev. Biol. 1993, 155, 423–430. [Google Scholar] [CrossRef]
- Colvin, J.S.; Bohne, B.A.; Harding, G.W.; McEwen, D.G.; Ornitz, D.M. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat. Genet. 1996, 12, 390–397. [Google Scholar] [CrossRef]
- Deng, C.; Wynshaw-Boris, A.; Zhou, F.; Kuo, A.; Leder, P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 1996, 84, 911–921. [Google Scholar] [CrossRef]
- Chen, L.; Adar, R.; Yang, X.; Monsonego, E.O.; Li, C.; Hauschka, P.V.; Yayon, A.; Deng, C.X. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J. Clin. Investig. 1999, 104, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.; Chen, L.; Li, C.; Ovchinnikov, D.A.; Behringer, R.R.; Francomano, C.A.; Deng, C.X. A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Hum. Mol. Genet. 2000, 9, 1603–1613. [Google Scholar] [CrossRef] [PubMed]
- Naski, M.C.; Colvin, J.S.; Coffin, J.D.; Ornitz, D.M. Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development 1998, 125, 4977–4988. [Google Scholar] [PubMed]
- Segev, O.; Chumakov, I.; Nevo, Z.; Givol, D.; Madar-Shapiro, L.; Sheinin, Y.; Weinreb, M.; Yayon, A. Restrained chondrocyte proliferation and maturation with abnormal growth plate vascularization and ossification in human FGFR-3(G380R) transgenic mice. Hum. Mol. Genet. 2000, 9, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Spatz, M.K.; Kannan, K.; Hayk, H.; Avivi, A.; Gorivodsky, M.; Pines, M.; Yayon, A.; Lonai, P.; Givol, D. A mouse model for achondroplasia produced by targeting fibroblast growth factor receptor 3. Proc. Natl. Acad. Sci. USA 1999, 96, 4455–4460. [Google Scholar] [CrossRef]
- Minina, E.; Kreschel, C.; Naski, M.C.; Ornitz, D.M.; Vortkamp, A. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev. Cell 2002, 3, 439–449. [Google Scholar] [CrossRef]
- Zhou, S.; Xie, Y.; Tang, J.; Huang, J.; Huang, Q.; Xu, W.; Wang, Z.; Luo, F.; Wang, Q.; Chen, H.; et al. FGFR3 Deficiency Causes Multiple Chondroma-like Lesions by Upregulating Hedgehog Signaling. PLoS Genet. 2015, 11, e1005214. [Google Scholar] [CrossRef]
- Garrison, P.; Yue, S.; Hanson, J.; Baron, J.; Lui, J.C. Spatial regulation of bone morphogenetic proteins (BMPs) in postnatal articular and growth plate cartilage. PLoS ONE 2017, 12, e0176752. [Google Scholar] [CrossRef]
- Jing, Y.; Jing, J.; Ye, L.; Liu, X.; Harris, S.E.; Hinton, R.J.; Feng, J.Q. Chondrogenesis and osteogenesis are one continuous developmental and lineage defined biological process. Sci. Rep. 2017, 7, 10020. [Google Scholar] [CrossRef]
- Salazar, V.S.; Gamer, L.W.; Rosen, V. BMP signalling in skeletal development, disease and repair. Nat. Rev. Endocrinol. 2016, 12, 203–221. [Google Scholar] [CrossRef]
- Minina, E.; Wenzel, H.M.; Kreschel, C.; Karp, S.; Gaffield, W.; McMahon, A.P.; Vortkamp, A. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development 2001, 128, 4523–4534. [Google Scholar] [PubMed]
- Shu, B.; Zhang, M.; Xie, R.; Wang, M.; Jin, H.; Hou, W.; Tang, D.; Harris, S.E.; Mishina, Y.; O’Keefe, R.J.; et al. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J. Cell. Sci. 2011, 124 Pt 20, 3428–3440. [Google Scholar] [CrossRef]
- Jing, J.; Ren, Y.; Zong, Z.; Liu, C.; Kamiya, N.; Mishina, Y.; Liu, Y.; Zhou, X.; Feng, J.Q. BMP receptor 1A determines the cell fate of the postnatal growth plate. Int. J. Biol. Sci. 2013, 9, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Retting, K.N.; Song, B.; Yoon, B.S.; Lyons, K.M. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 2009, 136, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.S.; Pogue, R.; Ovchinnikov, D.A.; Yoshii, I.; Mishina, Y.; Behringer, R.R.; Lyons, K.M. BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development 2006, 133, 4667–4678. [Google Scholar] [CrossRef] [PubMed]
- Ortega, N.; Wang, K.; Ferrara, N.; Werb, Z.; Vu, T.H. Complementary interplay between matrix metalloproteinase-9, vascular endothelial growth factor and osteoclast function drives endochondral bone formation. Dis. Model. Mech. 2010, 3, 224–235. [Google Scholar] [CrossRef]
- Haimov, H.; Shimoni, E.; Brumfeld, V.; Shemesh, M.; Varsano, N.; Addadi, L.; Weiner, S. Mineralization pathways in the active murine epiphyseal growth plate. Bone 2019, 130, 115086. [Google Scholar] [CrossRef]
- Stickens, D.; Behonick, D.J.; Ortega, N.; Heyer, B.; Hartenstein, B.; Yu, Y.; Fosang, A.J.; Schorpp-Kistner, M.; Angel, P.; Werb, Z. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development 2004, 131, 5883–5895. [Google Scholar] [CrossRef]
- Colnot, C.; de la Fuente, L.; Huang, S.; Hu, D.; Lu, C.; St-Jacques, B.; Helms, J.A. Indian hedgehog synchronizes skeletal angiogenesis and perichondrial maturation with cartilage development. Development 2005, 132, 1057–1067. [Google Scholar] [CrossRef]
- Yoshida, C.A.; Yamamoto, H.; Fujita, T.; Furuichi, T.; Ito, K.; Inoue, K.; Yamana, K.; Zanma, A.; Takada, K.; Ito, Y.; et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004, 18, 952–963. [Google Scholar] [CrossRef]
- Zelzer, E.; Glotzer, D.J.; Hartmann, C.; Thomas, D.; Fukai, N.; Soker, S.; Olsen, B.R. Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech. Dev. 2001, 106, 97–106. [Google Scholar] [CrossRef]
- Rossi, M.; Hall, C.M.; Bouvier, R.; Collardeau-Frachon, S.; Le Breton, F.; Bucourt, M.; Cordier, M.P.; Vianey-Saban, C.; Parenti, G.; Andria, G.; et al. Radiographic features of the skeleton in disorders of post-squalene cholesterol biosynthesis. Pediatric Radiol. 2015, 45, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Gofflot, F.; Hars, C.; Illien, F.; Chevy, F.; Wolf, C.; Picard, J.J.; Roux, C. Molecular mechanisms underlying limb anomalies associated with cholesterol deficiency during gestation: Implications of Hedgehog signaling. Hum. Mol. Genet. 2003, 12, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; De Luca, F. Role of cholesterol in the regulation of growth plate chondrogenesis and longitudinal bone growth. J. Biol. Chem. 2004, 279, 4642–4647. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. USA 1999, 96, 11041–11048. [Google Scholar] [CrossRef]
- Tsushima, H.; Tang, Y.J.; Puviindran, V.; Hsu, S.C.; Nadesan, P.; Yu, C.; Zhang, H.; Mirando, A.J.; Hilton, M.J.; Alman, B.A. Intracellular biosynthesis of lipids and cholesterol by Scap and Insig in mesenchymal cells regulates long bone growth and chondrocyte homeostasis. Development 2018, 145, 162396. [Google Scholar] [CrossRef]
- Ali, S.A.; Al-Jazrawe, M.; Ma, H.; Whetstone, H.; Poon, R.; Farr, S.; Naples, M.; Adeli, K.; Alman, B.A. Regulation of Cholesterol Homeostasis by Hedgehog Signaling in Osteoarthritic Cartilage. Arthritis Rheumatol. 2016, 68, 127–137. [Google Scholar] [CrossRef]
- Buglino, J.A.; Resh, M.D. Palmitoylation of Hedgehog proteins. Vitam. Horm. 2012, 88, 229–252. [Google Scholar]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 2016, 166, 1176–1187. [Google Scholar] [CrossRef]
- Ono, N.; Ono, W.; Nagasawa, T.; Kronenberg, H.M. A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat. Cell Biol. 2014, 16, 1157–1167. [Google Scholar] [CrossRef]
- Yang, G.; Zhu, L.; Hou, N.; Lan, Y.; Wu, X.M.; Zhou, B.; Teng, Y.; Yang, X. Osteogenic fate of hypertrophic chondrocytes. Cell Res. 2014, 24, 1266–1269. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Tsang, K.Y.; Tang, H.C.; Chan, D.; Cheah, K.S. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 2014, 111, 12097–12102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; von der Mark, K.; Henry, S.; Norton, W.; Adams, H.; de Crombrugghe, B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 2014, 10, e1004820. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, R.; Kitazawa, R.; Imai, Y.; Kitazawa, S. Growth plate-derived hedgehog-signal-responsive cells provide skeletal tissue components in growing bone. Histochem. Cell Biol. 2018, 149, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; He, G.; Lee, W.C.; McKenzie, J.A.; Silva, M.J.; Long, F. Gli1 identifies osteogenic progenitors for bone formation and fracture repair. Nat. Commun. 2017, 8, 2043. [Google Scholar] [CrossRef]
- Karimian, E.; Chagin, A.S.; Savendahl, L. Genetic regulation of the growth plate. Front. Endocrinol. 2011, 2, 113. [Google Scholar] [CrossRef]
- Makela, E.A.; Vainionpaa, S.; Vihtonen, K.; Mero, M.; Rokkanen, P. The effect of trauma to the lower femoral epiphyseal plate. An experimental study in rabbits. J. Bone Jt. Surg. Br. 1988, 70, 187–191. [Google Scholar] [CrossRef]
- Gao, B.; Guo, J.; She, C.; Shu, A.; Yang, M.; Tan, Z.; Yang, X.; Guo, S.; Feng, G.; He, L. Mutations in IHH, encoding Indian hedgehog, cause brachydactyly type A-1. Nat. Genet. 2001, 28, 386–388. [Google Scholar] [CrossRef]
- Hellemans, J.; Coucke, P.J.; Giedion, A.; De Paepe, A.; Kramer, P.; Beemer, F.; Mortier, G.R. Homozygous mutations in IHH cause acrocapitofemoral dysplasia, an autosomal recessive disorder with cone-shaped epiphyses in hands and hips. Am. J. Hum. Genet. 2003, 72, 1040–1046. [Google Scholar] [CrossRef]
- Mortier, G.R.; Kramer, P.P.; Giedion, A.; Beemer, F.A. Acrocapitofemoral dysplasia: An autosomal recessive skeletal dysplasia with cone shaped epiphyses in the hands and hips. J. Med. Genet. 2003, 40, 201–207. [Google Scholar] [CrossRef]
- Gao, B.; Hu, J.; Stricker, S.; Cheung, M.; Ma, G.; Law, K.F.; Witte, F.; Briscoe, J.; Mundlos, S.; He, L.; et al. A mutation in Ihh that causes digit abnormalities alters its signalling capacity and range. Nature 2009, 458, 1196–1200. [Google Scholar] [CrossRef] [PubMed]
- Ho, R.; McIntyre, A.D.; Kennedy, B.A.; Hegele, R.A. Whole-exome sequencing identifies a novel IHH insertion in an Ontario family with brachydactyly type A1. SAGE Open Med. Case Rep. 2018, 6, 2050313X18818711. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Yu, J.; Xiao, Y.; Chan, D.; Gao, B.; Hu, J.; He, Y.; Guo, S.; Zhou, J.; Zhang, L.; et al. Indian hedgehog mutations causing brachydactyly type A1 impair Hedgehog signal transduction at multiple levels. Cell Res. 2011, 21, 1343–1357. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Ma, G.; Shi, Y.; Ruan, Y.; Yang, X.; Wu, X.; Xiong, Y.; Wan, C.; Yang, C.; Cai, L.; et al. p. E95K mutation in Indian hedgehog causing brachydactyly type A1 impairs IHH/Gli1 downstream transcriptional regulation. BMC Genet. 2019, 20, 10. [Google Scholar] [CrossRef] [PubMed]
- Bovee, J.V.; Hogendoorn, P.C.; Wunder, J.S.; Alman, B.A. Cartilage tumours and bone development: Molecular pathology and possible therapeutic targets. Nat. Rev. Cancer 2010, 10, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.A.; Inwards, C.Y.; Unni, K.K. Benign bone tumors—Recent developments. Semin. Diagn. Pathol. 2011, 28, 73–85. [Google Scholar] [CrossRef]
- Romeo, S.; Hogendoorn, P.C.; Dei Tos, A.P. Benign cartilaginous tumors of bone: From morphology to somatic and germ-line genetics. Adv. Anat. Pathol. 2009, 16, 307–315. [Google Scholar] [CrossRef]
- Bowen, M.E.; Boyden, E.D.; Holm, I.A.; Campos-Xavier, B.; Bonafe, L.; Superti-Furga, A.; Ikegawa, S.; Cormier-Daire, V.; Bovee, J.V.; Pansuriya, T.C.; et al. Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome. PLoS Genet. 2011, 7, e1002050. [Google Scholar] [CrossRef]
- Deng, Q.; Li, P.; Che, M.; Liu, J.; Biswas, S.; Ma, G.; He, L.; Wei, Z.; Zhang, Z.; Yang, Y.; et al. Activation of hedgehog signaling in mesenchymal stem cells induces cartilage and bone tumor formation via Wnt/beta-Catenin. Elife 2019, 8, e50208. [Google Scholar] [CrossRef]
- Hopyan, S.; Gokgoz, N.; Poon, R.; Gensure, R.C.; Yu, C.; Cole, W.G.; Bell, R.S.; Juppner, H.; Andrulis, I.L.; Wunder, J.S.; et al. A mutant PTH/PTHrP type I receptor in enchondromatosis. Nat. Genet. 2002, 30, 306–310. [Google Scholar] [CrossRef]
- Huegel, J.; Sgariglia, F.; Enomoto-Iwamoto, M.; Koyama, E.; Dormans, J.P.; Pacifici, M. Heparan sulfate in skeletal development, growth, and pathology: The case of hereditary multiple exostoses. Dev. Dyn. 2013, 242, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Jennes, I.; Pedrini, E.; Zuntini, M.; Mordenti, M.; Balkassmi, S.; Asteggiano, C.G.; Casey, B.; Bakker, B.; Sangiorgi, L.; Wuyts, W. Multiple osteochondromas: Mutation update and description of the multiple osteochondromas mutation database (MOdb). Hum. Mutat. 2009, 30, 1620–1627. [Google Scholar] [CrossRef] [PubMed]
- Sobreira, N.L.; Cirulli, E.T.; Avramopoulos, D.; Wohler, E.; Oswald, G.L.; Stevens, E.L.; Ge, D.; Shianna, K.V.; Smith, J.P.; Maia, J.M.; et al. Whole-genome sequencing of a single proband together with linkage analysis identifies a Mendelian disease gene. PLoS Genet. 2010, 6, e1000991. [Google Scholar] [CrossRef] [PubMed]
- Tiet, T.D.; Hopyan, S.; Nadesan, P.; Gokgoz, N.; Poon, R.; Lin, A.C.; Yan, T.; Andrulis, I.L.; Alman, B.A.; Wunder, J.S. Constitutive hedgehog signaling in chondrosarcoma up-regulates tumor cell proliferation. Am. J. Pathol. 2006, 168, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Koziel, L.; Kunath, M.; Kelly, O.G.; Vortkamp, A. Ext1-dependent heparan sulfate regulates the range of Ihh signaling during endochondral ossification. Dev. Cell 2004, 6, 801–813. [Google Scholar] [CrossRef]
- Stickens, D.; Zak, B.M.; Rougier, N.; Esko, J.D.; Werb, Z. Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Development 2005, 132, 5055–5068. [Google Scholar] [CrossRef]
- Zak, B.M.; Schuksz, M.; Koyama, E.; Mundy, C.; Wells, D.E.; Yamaguchi, Y.; Pacifici, M.; Esko, J.D. Compound heterozygous loss of Ext1 and Ext2 is sufficient for formation of multiple exostoses in mouse ribs and long bones. Bone 2011, 48, 979–987. [Google Scholar] [CrossRef]
- Yang, W.; Wang, J.; Moore, D.C.; Liang, H.; Dooner, M.; Wu, Q.; Terek, R.; Chen, Q.; Ehrlich, M.G.; Quesenberry, P.J.; et al. Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature 2013, 499, 491–495. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Hahn, G.V.; Zasloff, M.A. Heterotopic Ossification: Two Rare Forms and What They Can Teach Us. J. Am. Acad. Orthop. Surg. 1994, 2, 288–296. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Shore, E.M. Progressive osseous heteroplasia. J. Bone Miner. Res. 2000, 15, 2084–2094. [Google Scholar] [CrossRef]
- Regard, J.B.; Cherman, N.; Palmer, D.; Kuznetsov, S.A.; Celi, F.S.; Guettier, J.M.; Chen, M.; Bhattacharyya, N.; Wess, J.; Coughlin, S.R.; et al. Wnt/beta-catenin signaling is differentially regulated by Galpha proteins and contributes to fibrous dysplasia. Proc. Natl. Acad. Sci. USA 2011, 108, 20101–20106. [Google Scholar] [CrossRef] [PubMed]
- Regard, J.B.; Malhotra, D.; Gvozdenovic-Jeremic, J.; Josey, M.; Chen, M.; Weinstein, L.S.; Lu, J.; Shore, E.M.; Kaplan, F.S.; Yang, Y. Activation of Hedgehog signaling by loss of GNAS causes heterotopic ossification. Nat. Med. 2013, 19, 1505–1512. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haraguchi, R.; Kitazawa, R.; Kohara, Y.; Ikedo, A.; Imai, Y.; Kitazawa, S. Recent Insights into Long Bone Development: Central Role of Hedgehog Signaling Pathway in Regulating Growth Plate. Int. J. Mol. Sci. 2019, 20, 5840. https://doi.org/10.3390/ijms20235840
Haraguchi R, Kitazawa R, Kohara Y, Ikedo A, Imai Y, Kitazawa S. Recent Insights into Long Bone Development: Central Role of Hedgehog Signaling Pathway in Regulating Growth Plate. International Journal of Molecular Sciences. 2019; 20(23):5840. https://doi.org/10.3390/ijms20235840
Chicago/Turabian StyleHaraguchi, Ryuma, Riko Kitazawa, Yukihiro Kohara, Aoi Ikedo, Yuuki Imai, and Sohei Kitazawa. 2019. "Recent Insights into Long Bone Development: Central Role of Hedgehog Signaling Pathway in Regulating Growth Plate" International Journal of Molecular Sciences 20, no. 23: 5840. https://doi.org/10.3390/ijms20235840
APA StyleHaraguchi, R., Kitazawa, R., Kohara, Y., Ikedo, A., Imai, Y., & Kitazawa, S. (2019). Recent Insights into Long Bone Development: Central Role of Hedgehog Signaling Pathway in Regulating Growth Plate. International Journal of Molecular Sciences, 20(23), 5840. https://doi.org/10.3390/ijms20235840