Autophagy in the Central Nervous System and Effects of Chloroquine in Mucopolysaccharidosis Type II Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
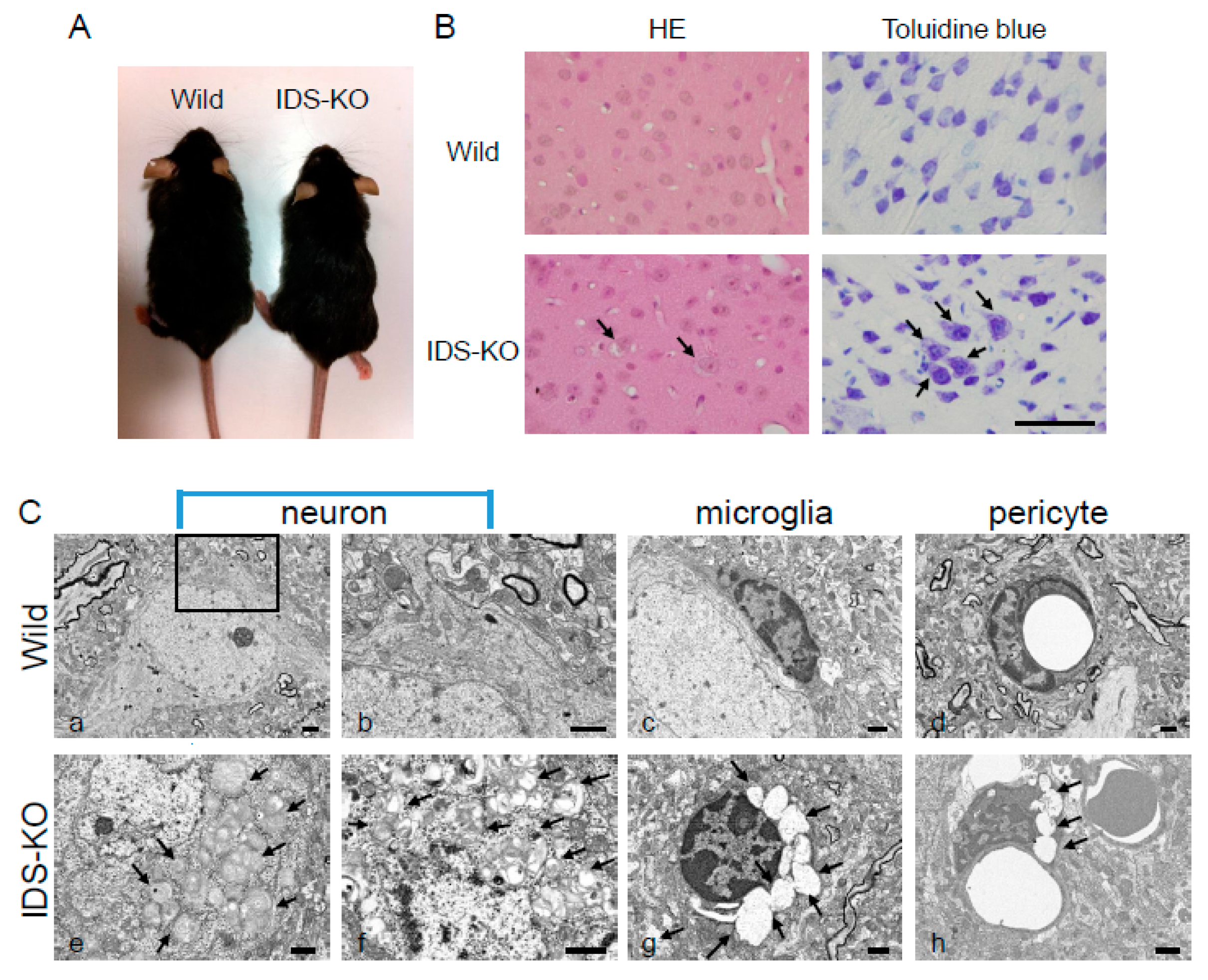
2.1. Vacuoles in Neurons, Microglia, and Pericytes in the CNS of IDS-KO Mice
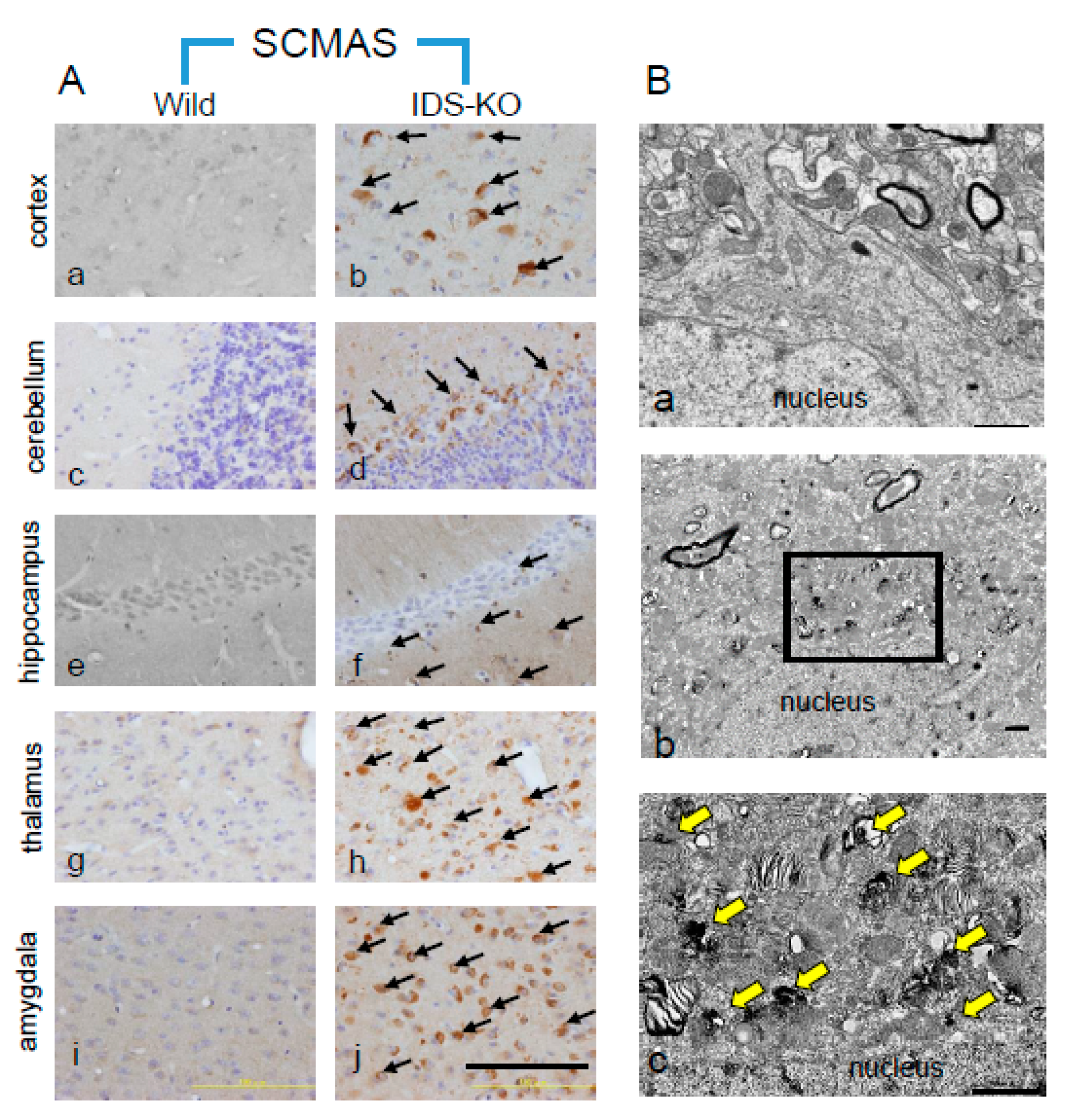
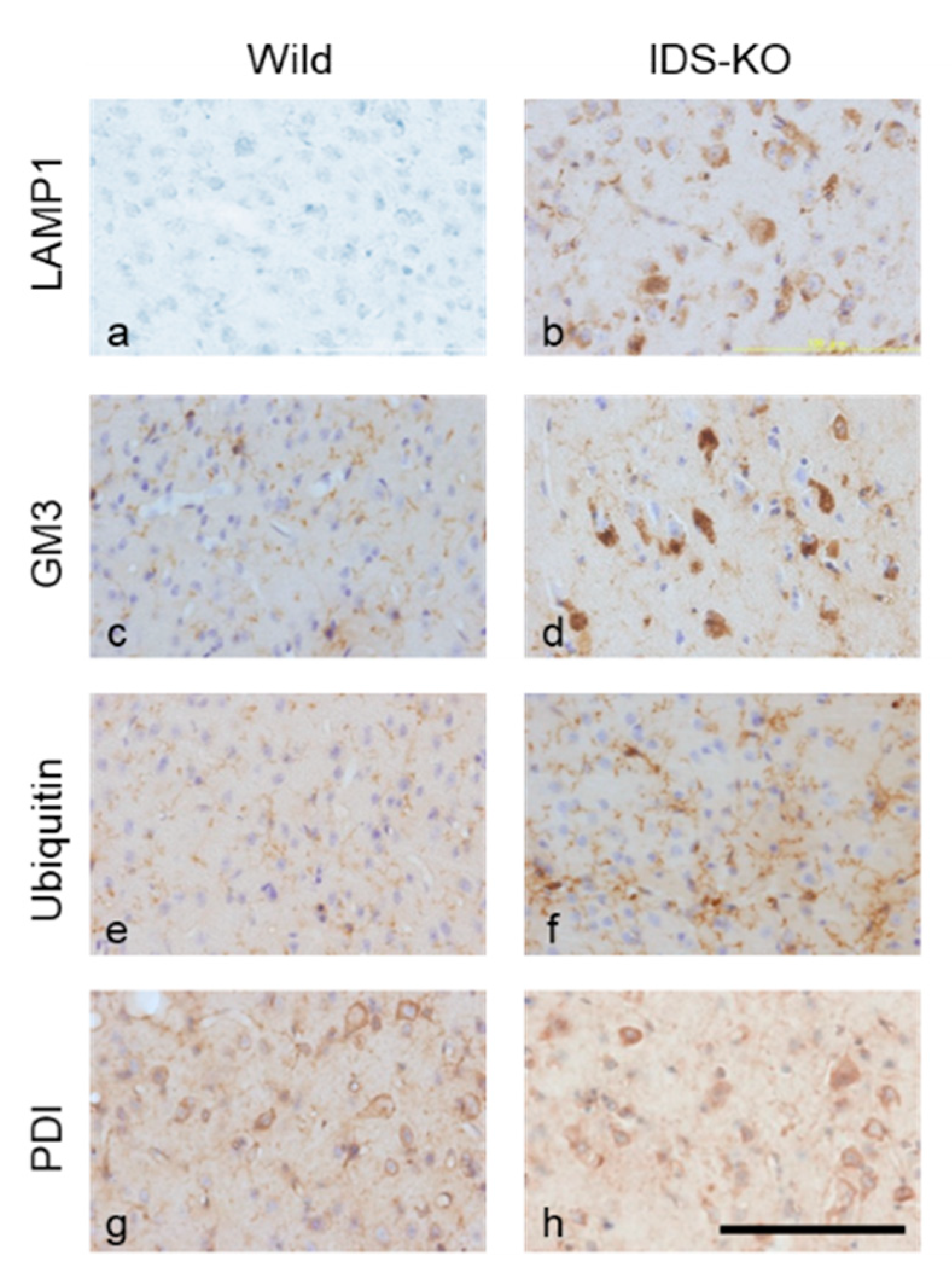
2.2. Autophagy-Like Changes in the Cerebral Cortex of IDS-KO Mice
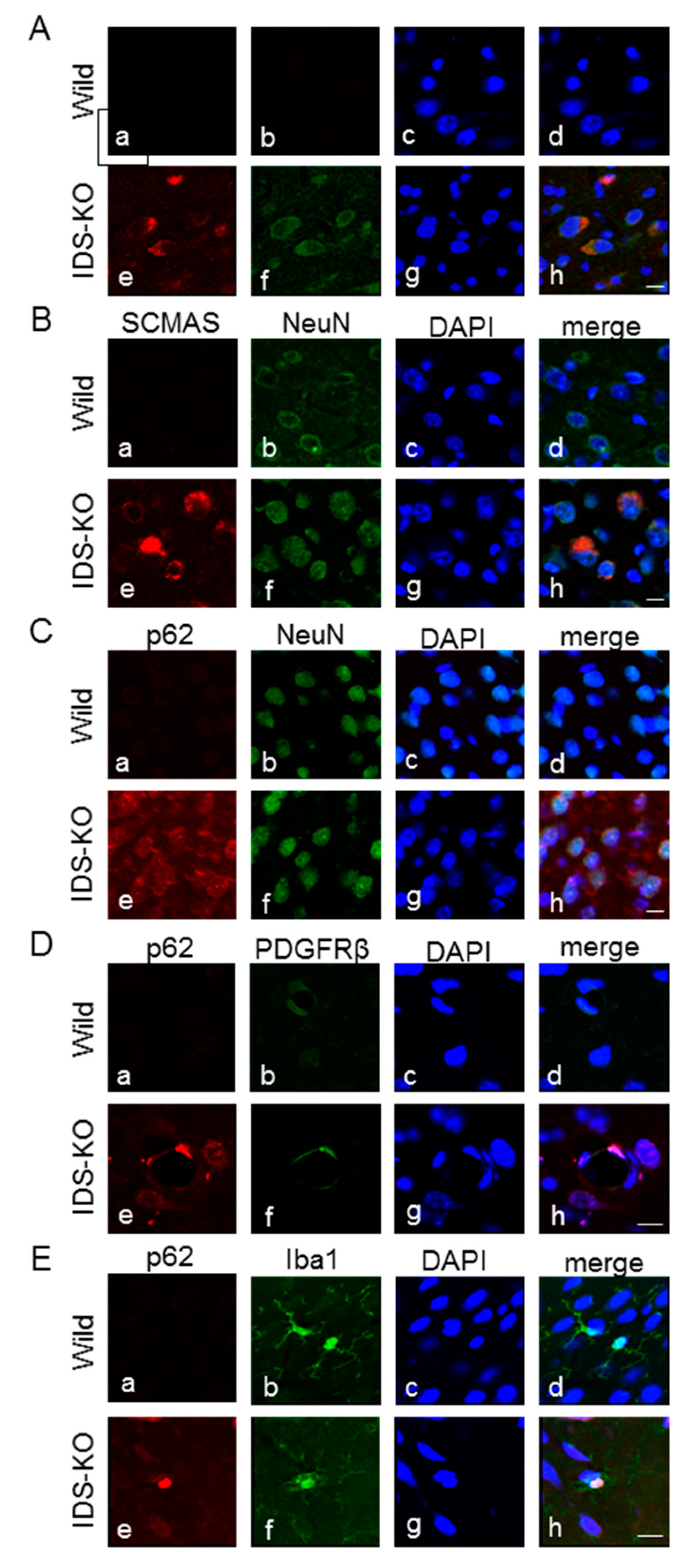
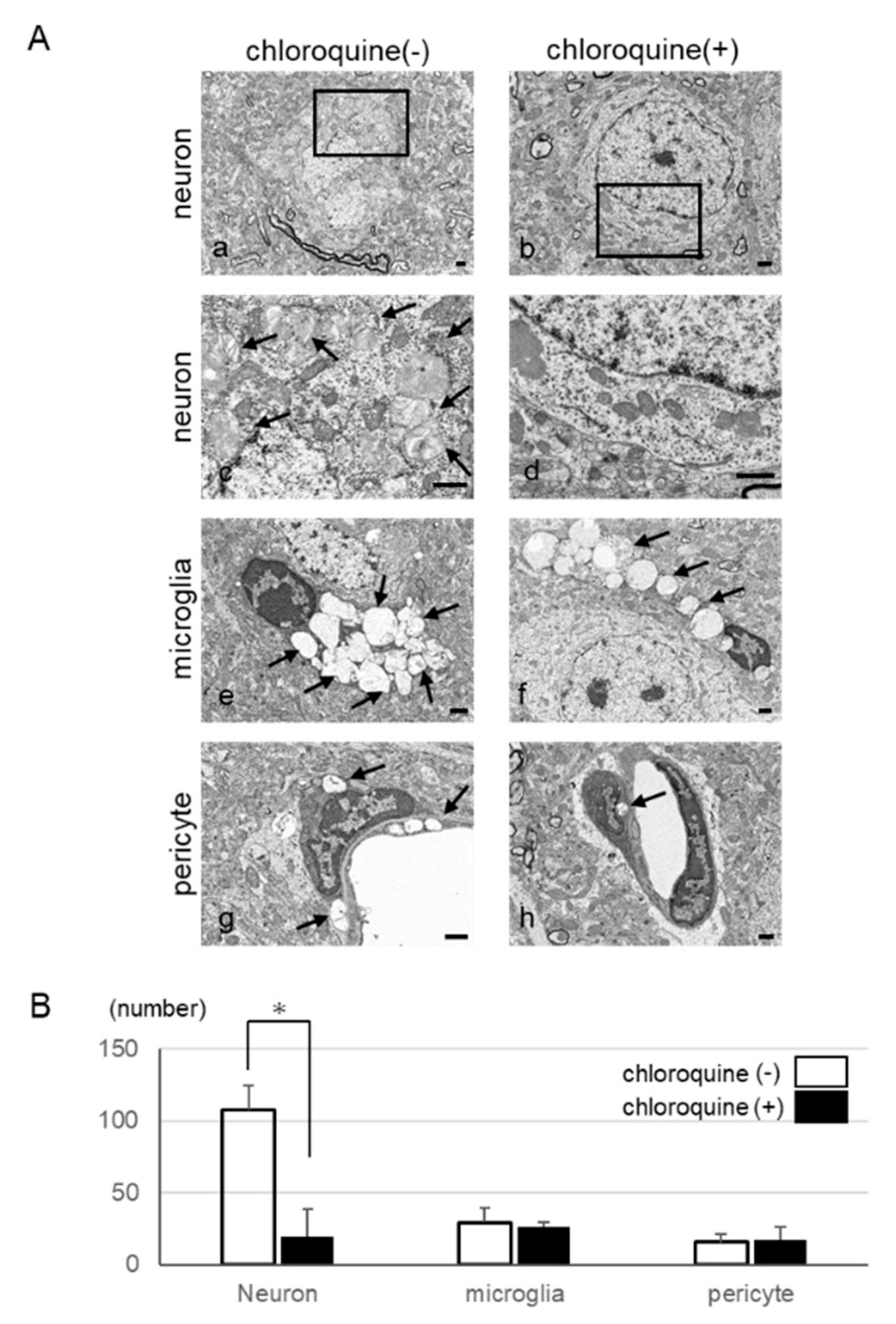
2.3. Autophagy-Like Changes in Neurons, Microglia, and Pericytes of IDS-KO Mice
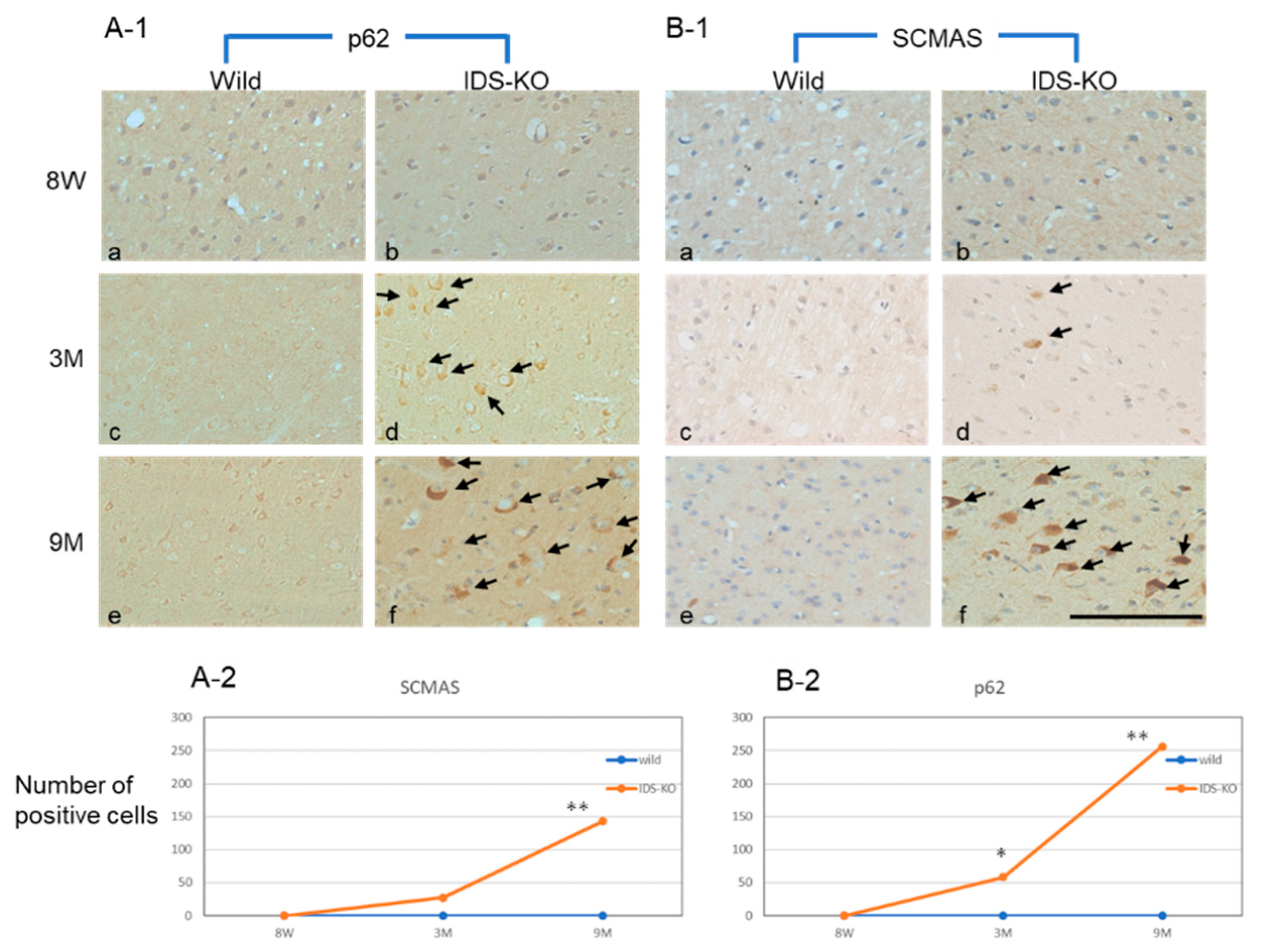
2.4. Increase in Autophagy-Related Proteins in the Cerebral Cortex
2.5. Microstructural Changes Observed by Electron Microscopy
2.6. Inhibition of Autophagy in Neurons by Using Chloroquine
3. Discussion
3.1. Limitations
3.2. Conclusions
4. Methods
4.1. Animal Model
4.2. Immunohistochemistry and Light Microscopy
4.3. Electron Microscopy
4.4. Immunoelectron Microscopy for SCMAS
4.5. Administration of Chloroquine to IDS-KO Mice
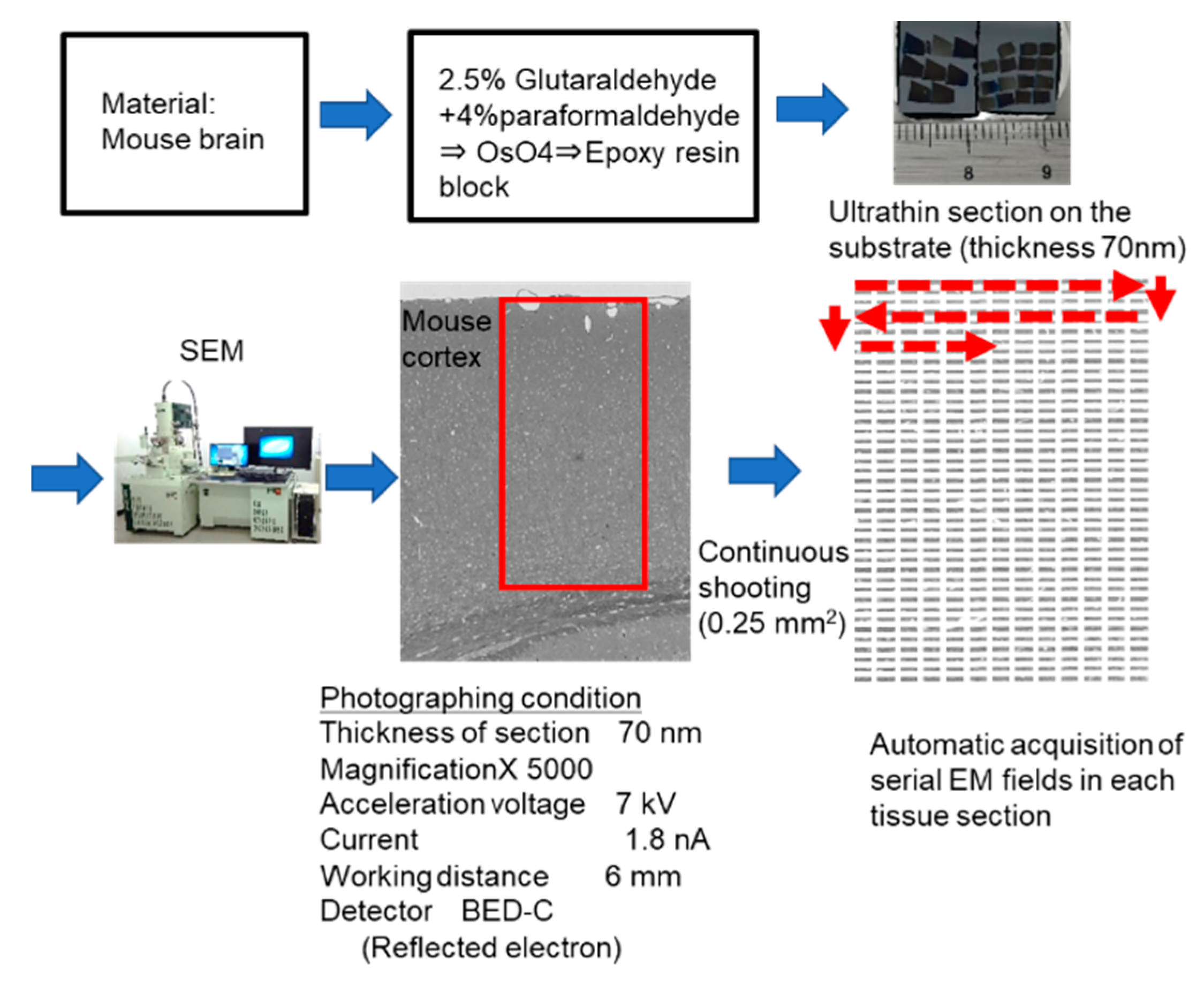
4.6. Cell Counting by Automatic Acquisition System of Sequential EM Images in Wider Tissue Fields and Statistical Analysis
Author Contributions
Funding
Acknowledgments
Ethical Issues
Conflicts of Interest
References
- Parini, R.; Deodato, F.; Di Rocco, M.; Lanino, E.; Locatelli, F.; Messina, C.; Rovelli, A.; Scarpa, M. Open issues in Mucopolysaccharidosis type I-Hurler. Orphanet. J. Rare Dis. 2017, 12, 112. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M.; Almassy, Z.; Beck, M.; Bodamer, O.; Bruce, I.A.; De Meirleir, L.; Guffon, N.; Guillen-Navarro, E.; Hensman, P.; Jones, S.; et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet. J. Rare Dis. 2011, 6, 72. [Google Scholar] [CrossRef]
- Suarez-Guerrero, J.L.; Gomez Higuera, P.J.; Arias Florez, J.S.; Contreras-Garcia, G.A. Mucopolysaccharidosis: Clinical features, diagnosis and management. Rev. Chil. Pediatr. 2016, 87, 295–304. [Google Scholar] [CrossRef]
- Okuyama, T.; Tanaka, A.; Suzuki, Y.; Ida, H.; Tanaka, T.; Cox, G.F.; Eto, Y.; Orii, T. Japan Elaprase Treatment (JET) study: Idursulfase enzyme replacement therapy in adult patients with attenuated Hunter syndrome (Mucopolysaccharidosis II, MPS II). Mol. Genet. Metab. 2010, 99, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Papadia, F.; Lozupone, M.S.; Gaeta, A.; Capodiferro, D.; Lacalendola, G. Long-term enzyme replacement therapy in a severe case of mucopolysaccharidosis type II (Hunter syndrome). Eur. Rev. Med. Pharmacol. Sci. 2011, 15, 253–258. [Google Scholar] [PubMed]
- Glamuzina, E.; Fettes, E.; Bainbridge, K.; Crook, V.; Finnegan, N.; Abulhoul, L.; Vellodi, A. Treatment of mucopolysaccharidosis type II (Hunter syndrome) with idursulfase: The relevance of clinical trial end points. J. Inherit. Metab. Dis. 2011, 34, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Al Sawaf, S.; Mayatepek, E.; Hoffmann, B. Neurological findings in Hunter disease: Pathology and possible therapeutic effects reviewed. J. Inherit. Metab. Dis. 2008, 31, 473–480. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Rubinsztein, D.C.; Ballabio, A. Lysosomal storage diseases as disorders of autophagy. Autophagy 2008, 4, 113–114. [Google Scholar] [CrossRef]
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017, 61, 733–749. [Google Scholar] [CrossRef]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef]
- Raben, N.; Schreiner, C.; Baum, R.; Takikita, S.; Xu, S.; Xie, T.; Myerowitz, R.; Komatsu, M.; Van der Meulen, J.H.; Nagaraju, K.; et al. Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder--murine Pompe disease. Autophagy 2010, 6, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Spampanato, C.; Feeney, E.; Li, L.; Cardone, M.; Lim, J.A.; Annunziata, F.; Zare, H.; Polishchuk, R.; Puertollano, R.; Parenti, G.; et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol. Med. 2013, 5, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Takamura, A.; Higaki, K.; Kajimaki, K.; Otsuka, S.; Ninomiya, H.; Matsuda, J.; Ohno, K.; Suzuki, Y.; Nanba, E. Enhanced autophagy and mitochondrial aberrations in murine G(M1)-gangliosidosis. Biochem. Biophys. Res. Commun. 2008, 367, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G. The murine model of mucopolysaccharidosis IIIB develops cardiopathies over time leading to heart failure. PLoS ONE 2015, 10, e0131662. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Imarisio, S.; Sarkar, S.; O’Kane, C.J.; Rubinsztein, D.C. Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J. Cell Sci. 2008, 121, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.G.; Sun, L.; Miao, G.; Ji, C.; Zhao, H.; Sun, H.; Miao, L.; Yoshii, S.R.; Mizushima, N.; Wang, X.; et al. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy 2015, 11, 881–890. [Google Scholar] [CrossRef]
- Elleder, M.; Sokolova, J.; Hrebicek, M. Follow-up study of subunit c of mitochondrial ATP synthase (SCMAS) in Batten disease and in unrelated lysosomal disorders. Acta Neuropathol. 1997, 93, 379–390. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell. 2013, 51, 618–631. [Google Scholar] [CrossRef]
- Bhaumik, M.; Muller, V.J.; Rozaklis, T.; Johnson, L.; Dobrenis, K.; Bhattacharyya, R.; Wurzelmann, S.; Finamore, P.; Hopwood, J.J.; Walkley, S.U.; et al. A mouse model for mucopolysaccharidosis type III A (Sanfilippo syndrome). Glycobiology 1999, 9, 389–1396. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Li, H.H.; Zhao, H.Z.; Neufeld, E.F.; Cai, Y.; Gomez-Pinilla, F. Attenuated plasticity in neurons and astrocytes in the mouse model of Sanfilippo syndrome type B. J. Neurosci. Res. 2002, 69, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Ohmi, K.; Kudo, L.C.; Ryazantsev, S.; Zhao, H.Z.; Karsten, S.L.; Neufeld, E.F. Sanfilippo syndrome type B, a lysosomal storage disease, is also a tauopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 8332–8337. [Google Scholar] [CrossRef] [PubMed]
- D’Azzo, A.; Tessitore, A.; Sano, R. Gangliosides as apoptotic signals in ER stress response. Cell Death Differ. 2006, 13, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Ryazantsev, S.; Yu, W.H.; Zhao, H.Z.; Neufeld, E.F.; Ohmi, K. Lysosomal accumulation of SCMAS (subunit c of mitochondrial ATP synthase) in neurons of the mouse model of mucopolysaccharidosis III B. Mol. Genet. Metab. 2007, 90, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Martine, W.; Timmermans, J.P.; Guido, R.Y.; Meyer, D. Methods to Assess autophagy in situ-transmission electron microscopy versus immunohistochemistry. Methods Enzymol. 2014, 543, 89–114. [Google Scholar]
- Peters, S.; Reinthal, E.; Blitgen-Heinecke, P.; Bartz-Schmidt, K.U.; Schraermeyer, U. Inhibition of lysosomal degradation in retinal pigment epithelium cells induces exocytosis of phagocytic residual material at the basolateral plasma membrane. Ophthalmic. Res. 2006, 38, 83–88. [Google Scholar] [CrossRef]
- Chen, X.; Clark, J.; Wunderlich, M.; Fan, C.; Davis, A.; Chen, S.; Guan, J.L.; Mulloy, J.C.; Kumar, A.; Zheng, Y. Autophagy is dispensable for Kmt2a/Mll-Mllt3/Af9 AML maintenance and anti-leukemic effect of chloroquine. Autophagy 2017, 13, 955–966. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Solakyildirim, K.; Zhang, Z.; Linhardt, R.J.; Tobacman, J.K. Chloroquine reduces arylsulphatase B activity and increases chondroitin-4-sulphate: Implications for mechanisms of action and resistance. Malaria J. 2009, 8, 303. [Google Scholar] [CrossRef]
- Alim, M.A.; Al-Bari, A. Chloroquine analogues in drug discovery: New directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J. Antimicrob. Chemother. 2015, 70, 1608–1621. [Google Scholar]
- Scionti, F.; Di Martino, M.T.; Sestito, S.; Nicoletti, A.; Falvo, F.; Roppa, K.; Arbitrio, M.; Guzzi, P.H.; Agapito, G.; Pisani, A.; et al. Genetic variants associated with Fabry disease progression despite enzyme replacement therapy. Oncotarget 2017, 8, 107558–107564. [Google Scholar] [CrossRef] [PubMed]
- Yesilipek, A.M.; Akcan, M.; Karasu, G.; Uygun, V.; Kupesiz, A.; Hazar, V. Successful unrelated bone marrow transplantation in two siblings with alpha-mannosidosis. Pediatr. Transplant. 2012, 16, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Sands, M.S.; Haskins, M.E. CNS-directed gene therapy for lysosomal storage diseases. Acta Paediatr. 2008, 97, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.M.; Gurda, B.L.; Casal, M.L.; Ponder, K.P.; Vite, C.H.; Haskins, M.E. A review of gene therapy in canine and feline models of lysosomal storage disorders. Hum. Gene. Ther. Clin. Dev. 2015, 26, 27–37. [Google Scholar] [CrossRef]
- Whiteman, D.A.; Kimura, A. Development of idursulfase therapy for mucopolysaccharidosis type II (Hunter syndrome): The past, the present and the future. Drug Des. Devel. Ther. 2017, 11, 2467–2480. [Google Scholar] [CrossRef]
- Seto, T.; Kono, K.; Morimoto, K.; Inoue, Y.; Shintaku, H.; Hattori, H.; Matsuoka, O.; Yamano, T.; Tanaka, A. Brain magnetic resonance imaging in 23 patients with mucopolysaccharidoses and the effect of bone marrow transplantation. Ann. Neurol. 2001, 50, 79–92. [Google Scholar] [CrossRef]
- Kubaski, F.; Yabe, H.; Suzuki, Y.; Seto, T.; Hamazaki, T.; Mason, R.W.; Xie, L.; Onsten, T.G.H.; Leistner-Segal, S.; Giugliani, R.; et al. Hematopoietic Stem Cell Transplantation for Patients with Mucopolysaccharidosis II. Biol. Blood Marrow Transplant. 2017, 23, 1795–1803. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Dickson, P.I.; Muldowney, L.; Lee, J.J.; Rosenberg, A.; Abichandani, R.; Bluestone, J.A.; Burton, B.K.; Dewey, M.; Freitas, A.; et al. Immune response to enzyme replacement therapies in lysosomal storage diseases and the role of immune tolerance induction. Mol. Genet. Metab. 2016, 117, 66–83. [Google Scholar] [CrossRef]
- Valayannopoulos, V. Enzyme replacement therapy and substrate reduction therapy in lysosomal storage disorders with neurological expression. Handb. Clin. Neurol. 2013, 113, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- Hoshina, H.; Shimada, Y.; Higuchi, T.; Kobayashi, H.; Ida, H.; Ohashi, T. Chaperone effect of sulfated disaccharide from heparin on mutant iduronate-2-sulfatase in mucopolysaccharidosis type II. Mol. Genet. Metab. 2017, 123, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Yanos, M.; Kayser, E.B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde Hui, J.; Wall, V.Z.; Gagnidze, A. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, F.; Meroni, P.; McCubrey, J.; Bendtzen, K. mTOR as a multifunctional therapeutic target in HIV infection. Drug Discov. Today 2011, 16, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Shimizu, H.; Fukuda, T.; Kawagoe, S.; Matsumoto, J.; Shimada, Y.; Kobayashi, H.; Ida, H.; Ohashi, T.; Morimoto, H.; et al. Enzyme replacement therapy (ERT) procedure for mucopolysaccharidosis type II (MPS II) by intraventricular administration (IVA) in murine MPS II. Mol. Genet. Metab. 2012, 107, 122–128. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maeda, M.; Seto, T.; Kadono, C.; Morimoto, H.; Kida, S.; Suga, M.; Nakamura, M.; Kataoka, Y.; Hamazaki, T.; Shintaku, H. Autophagy in the Central Nervous System and Effects of Chloroquine in Mucopolysaccharidosis Type II Mice. Int. J. Mol. Sci. 2019, 20, 5829. https://doi.org/10.3390/ijms20235829
Maeda M, Seto T, Kadono C, Morimoto H, Kida S, Suga M, Nakamura M, Kataoka Y, Hamazaki T, Shintaku H. Autophagy in the Central Nervous System and Effects of Chloroquine in Mucopolysaccharidosis Type II Mice. International Journal of Molecular Sciences. 2019; 20(23):5829. https://doi.org/10.3390/ijms20235829
Chicago/Turabian StyleMaeda, Mitsuyo, Toshiyuki Seto, Chiho Kadono, Hideto Morimoto, Sachiho Kida, Mitsuo Suga, Motohiro Nakamura, Yosky Kataoka, Takashi Hamazaki, and Haruo Shintaku. 2019. "Autophagy in the Central Nervous System and Effects of Chloroquine in Mucopolysaccharidosis Type II Mice" International Journal of Molecular Sciences 20, no. 23: 5829. https://doi.org/10.3390/ijms20235829
APA StyleMaeda, M., Seto, T., Kadono, C., Morimoto, H., Kida, S., Suga, M., Nakamura, M., Kataoka, Y., Hamazaki, T., & Shintaku, H. (2019). Autophagy in the Central Nervous System and Effects of Chloroquine in Mucopolysaccharidosis Type II Mice. International Journal of Molecular Sciences, 20(23), 5829. https://doi.org/10.3390/ijms20235829