Current Approaches in NSCLC Targeting K-RAS and EGFR


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. K-RAS
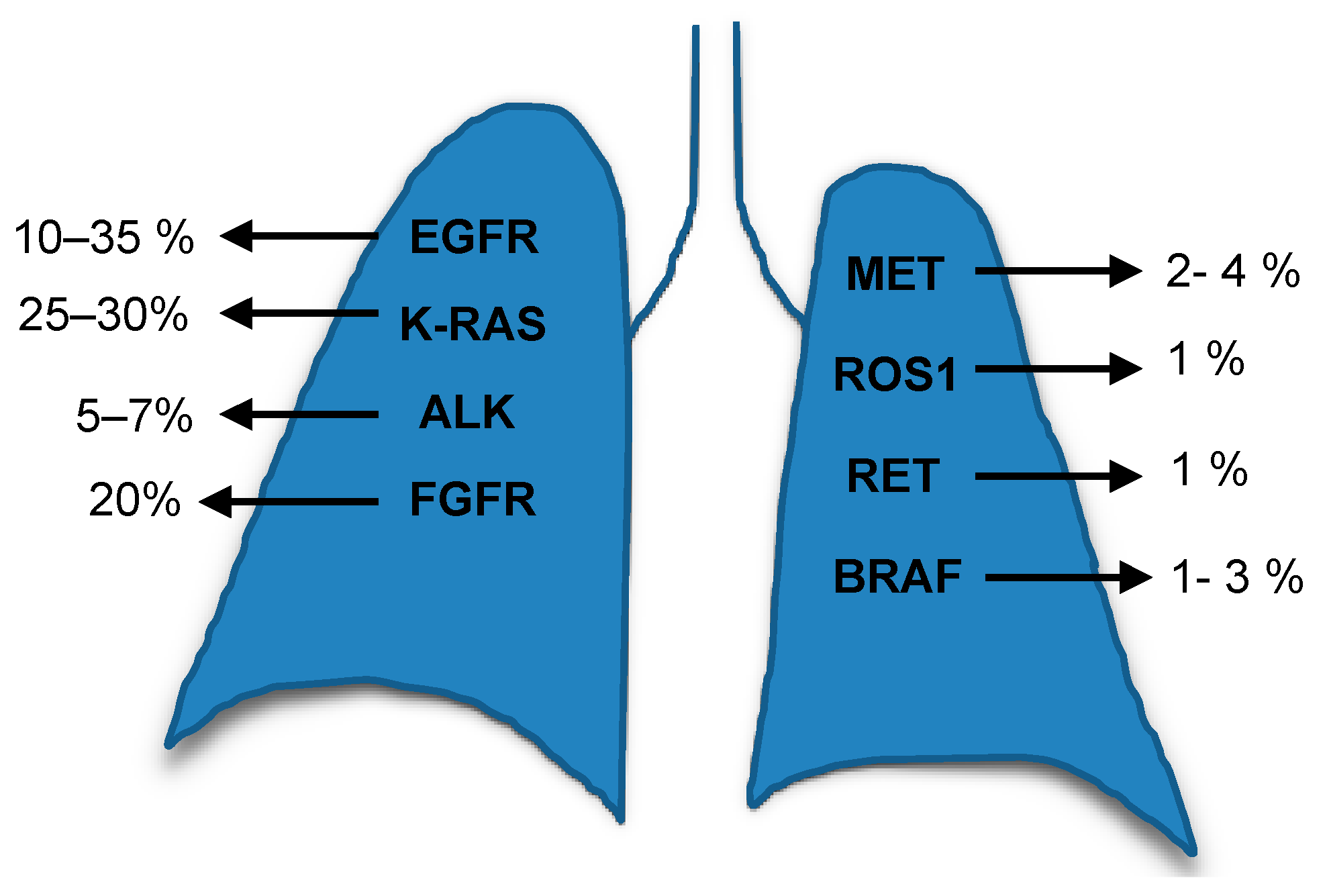
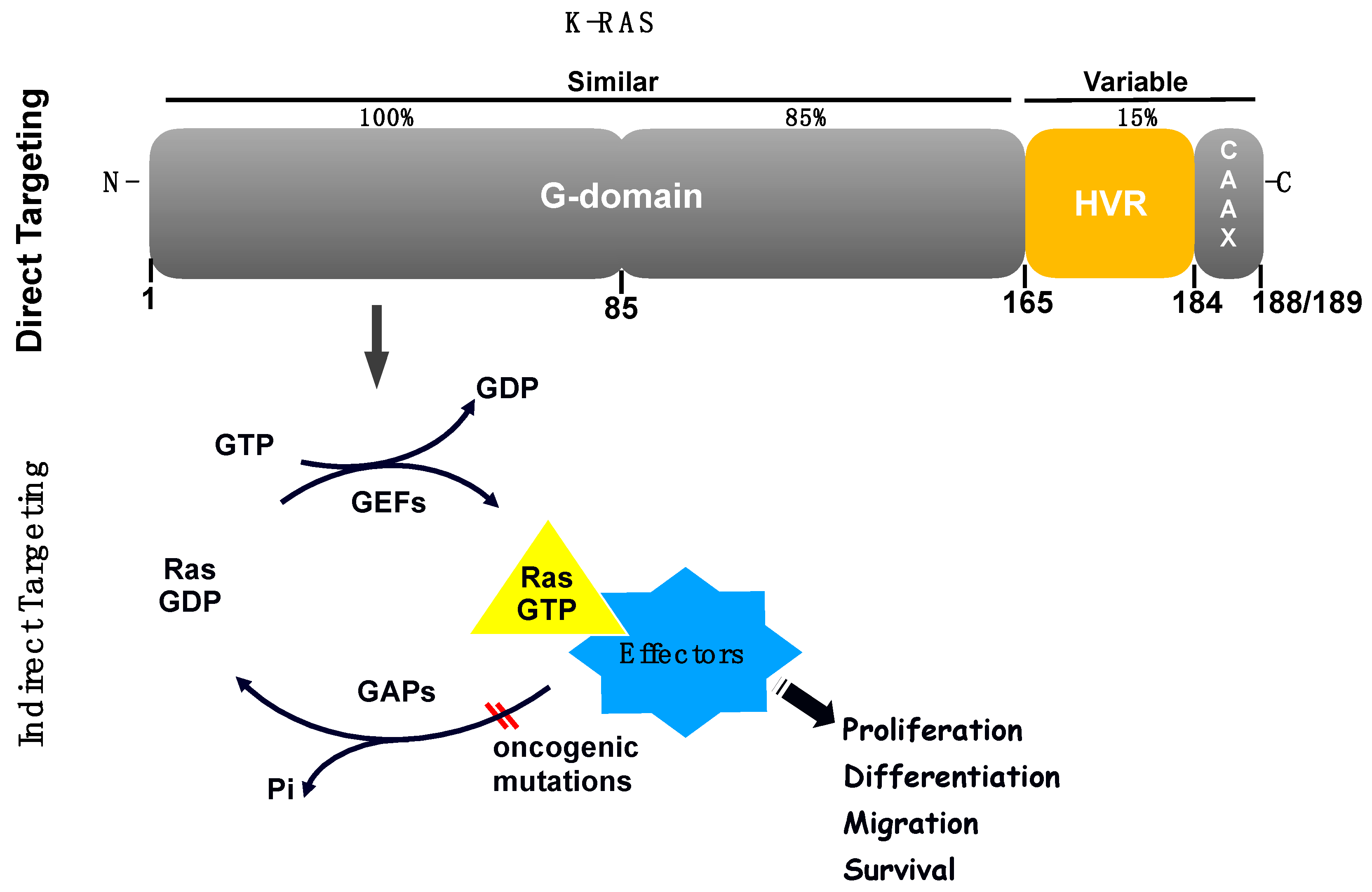
2.1. RAS Biomarkers
2.2. Examples of Direct Inhibition of RAS
2.3. Examples of Indirect Inhibition of RAS
3. EGFR
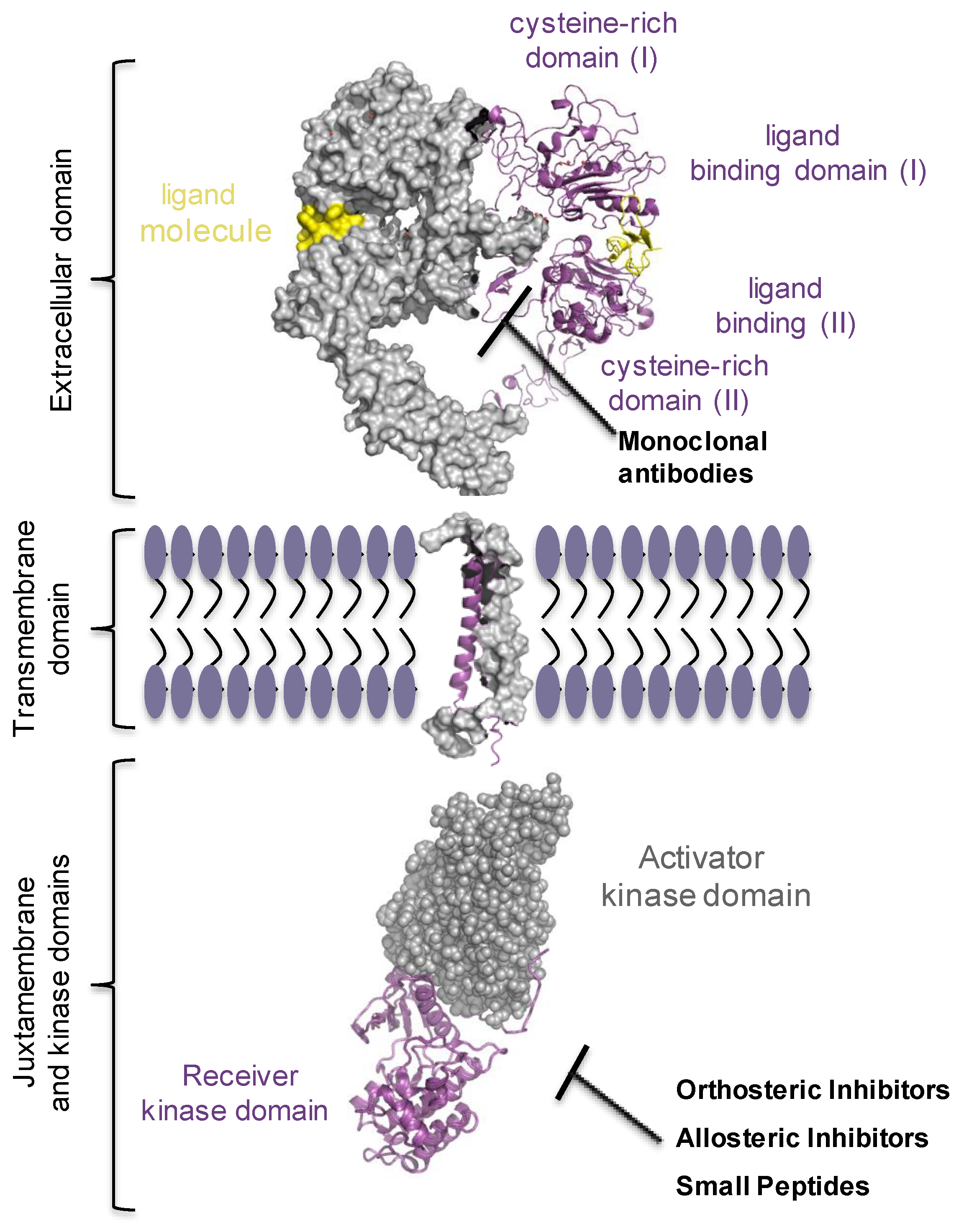
3.1. EGFR Biomarker
3.2. The Complexity of EGF Receptor Signalling in the Cells
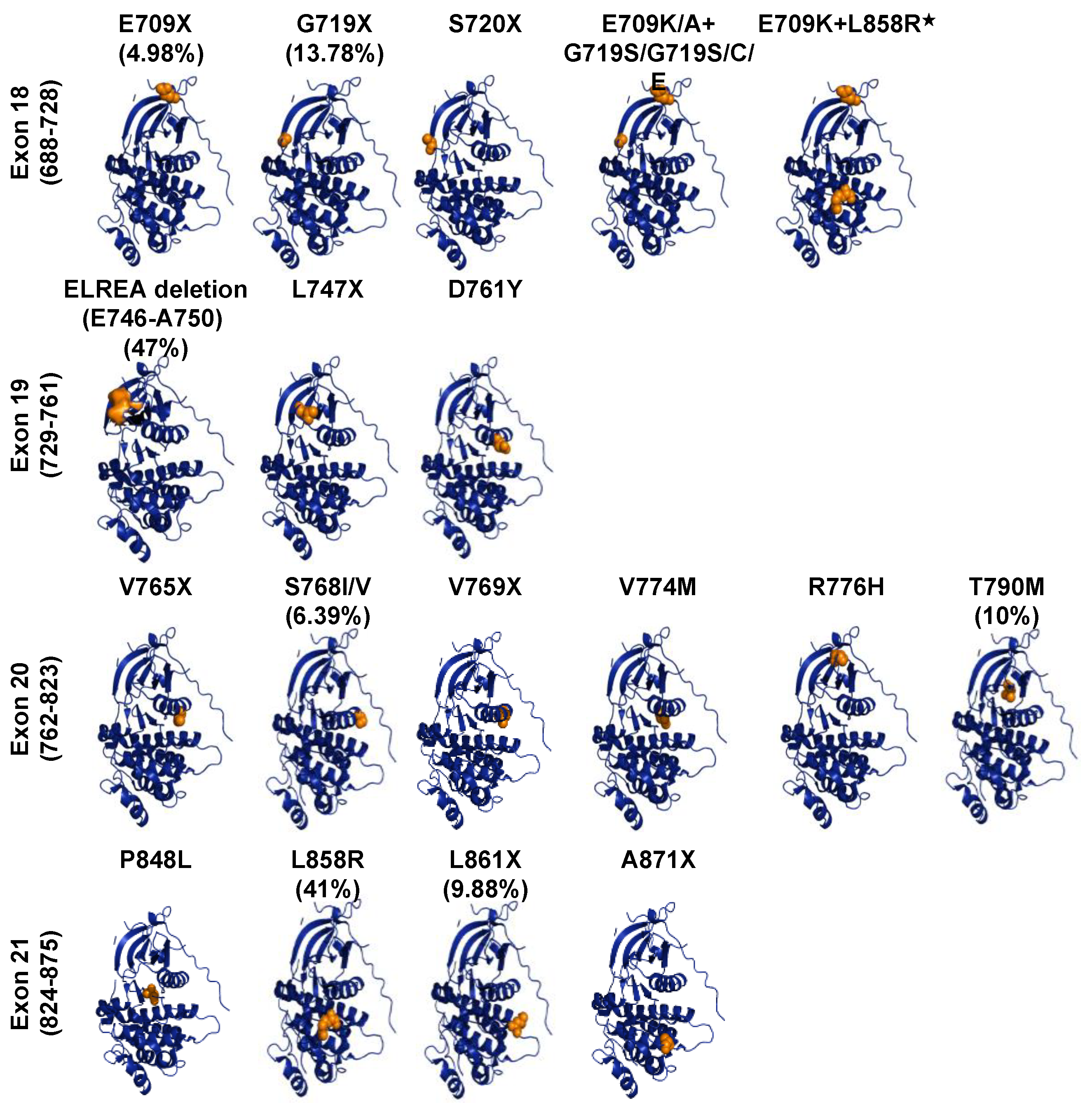
3.3. Oncogenic ErbB Variants in NSCLCs and Treatment Approaches
3.4. EGFR Targeting and Drug Resistant Mechanisms in NSCLC
3.5. EGFR and K-RAS Concomitant Mutations
4. Conclusions
Funding
Conflicts of Interest
Abbreviations
| K-RAS | Kirsten Rat Sarcoma |
| EGFR | Epidermal growth factor receptor |
| mTOR | mechanistic Target Of Rapamycin |
| ErbB2 | Epidermal Growth Factor Receptor 2 |
| VEGFR | Vascular Epidermal Growth Factor Receptor |
| PI3Ks | Phosphatidylinositol 3-kinase |
| ALK | Anaplastic Lymphoma Kinase |
| BRAF | v-Raf murine sarcoma viral oncogene homolog B |
| SCLC | Small Cell Lung Cancer |
| NSCLC | Non–small Cell Lung Cancer |
| WHO | World Health Organization |
| ctDNA | circulating tumour DNA |
| c-MET | Mesenchymal-epithelial transition factor |
| GDP | Guanosine diphosphate |
| GTP | Guanosine triphosphate |
| TBK1 | TANK-binding kinase 1 |
| MEK | mitogen/extracellular signal-related kinase |
| FDA | Food and Drug Administration |
| HSP90 | Heat shock protein 90 |
| GATA2 | GATA-binding factor 2 |
| BCL-XL | B-cell lymphoma extra-large |
| TKI | Tyrosine kinase inhibitor |
| TGFα | Transforming growth factor α |
| AR | Amphiregulin |
| EPG | Epigen |
| BTC | Betacellulin |
| HB-EGF | Heparin binding EGFR |
| EPR | Epiregulin |
| NRG | Neuregulins |
| N-RAS | Neuroblastoma RAS viral oncogene Homolog |
| H-RAS | Harvey RAS viral oncogene Homolog |
| PFS | Progression-free survival |
| MAPK | Mitogen-activated protein kinase |
| ICI | Immune checkpoint inhibitors |
| PD-1 | Programmed death protein 1 |
| PD-L1 | Programmed death ligand 1 |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dela Cruz, C.S.; Tanoue, L.T.; Matthay, R.A. Lung cancer: Epidemiology, etiology, and prevention. Clin. Chest Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-Small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Yoda, S.; Dagogo-Jack, I.; Hata, A.N. Targeting oncogenic drivers in lung cancer: Recent progress, current challenges and future opportunities. Pharmacol. Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Castellano, E.; Santos, E. Functional specificity of ras isoforms: So similar but so different. Genes Cancer 2011, 2, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011. [Google Scholar] [CrossRef]
- Ahrendt, S.A.; Anthony Decker, P.; Alawi, E.A.; Zhu, Y.R.; Sanchez-Cespedes, M.; Yang, S.C.; Haasler, G.B.; Kajdacsy-Balla, A.; Demeure, M.J.; Sidransky, D. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer 2001. [Google Scholar] [CrossRef]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017. [Google Scholar] [CrossRef]
- McCormick, F. KRAS as a therapeutic target. Clin. Cancer Res. 2015. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Marín-Ramos, N.I.; Ortega-Gutiérrez, S.; López-Rodríguez, M.L. Blocking ras inhibition as an antitumor strategy. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Cromm, P.M.; Spiegel, J.; Grossmann, T.N.; Waldmann, H. Direct modulation of small GTPase activity and function. Angew. Chem. Int. Ed. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F.; et al. Molecular epidemiology of EGFR and KRAS mutations in 3026 lung adenocarcinomas: Higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin. Cancer Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013. [Google Scholar] [CrossRef] [PubMed]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016. [Google Scholar] [CrossRef]
- Lito, P.; Solomon, M.; Li, L.S.; Hansen, R.; Rosen, N. Cancer therapeutics: Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016. [Google Scholar] [CrossRef] [PubMed]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 2018. [Google Scholar] [CrossRef]
- Misale, S.; Fatherree, J.P.; Cortez, E.; Li, C.; Bilton, S.; Timonina, D.; Myers, D.T.; Lee, D.; Gomez-Caraballo, M.; Greenberg, M.; et al. KRAS G12C NSCLC models are sensitive to direct targeting of KRAS in combination with PI3K inhibition. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Shimomura, I.; Yokoi, A.; Kohama, I.; Kumazaki, M.; Tada, Y.; Tatsumi, K.; Ochiya, T.; Yamamoto, Y. Drug library screen reveals benzimidazole derivatives as selective cytotoxic agents for KRAS-mutant lung cancer. Cancer Lett. 2019. [Google Scholar] [CrossRef]
- Fakih, M.; O’Neil, B.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.; Govindan, R.; Rasmussen, E.; Morrow, P.K.H.; Ngang, J.; et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRAS G12C inhibitor, in advanced solid tumors. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- McCormick, F. K-Ras protein as a drug target. J. Mol. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Zhang, Y.; Bos, P.H.; Chambers, J.M.; Dupont, M.M.; Stockwell, B.R. K-rasg12d has a potential allosteric small molecule binding site. Biochemistry 2019. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, S.; Joy, S.T.; Arora, P.S.; Bar-Sagi, D. An orthosteric inhibitor of the RAS-SOS interaction. Enzymes 2013. [Google Scholar] [CrossRef]
- Patgiri, A.; Yadav, K.K.; Arora, P.S.; Bar-Sagi, D. An orthosteric inhibitor of the RAS-SOS interaction. Nat. Chem. Biol. 2011, 7, 585. [Google Scholar] [CrossRef]
- Hillig, R.C.; Sautier, B.; Schroeder, J.; Moosmayer, D.; Hilpmann, A.; Stegmann, C.M.; Werbeck, N.D.; Briem, H.; Boemer, U.; Weiske, J.; et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 Interaction. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008. [Google Scholar] [CrossRef]
- Barbie, D.A.; Spira, A.; Kelly, K.; Humeniuk, R.; Kawashima, J.; Kong, S.; Koczywas, M. Phase 1B study of momelotinib combined with trametinib in metastatic, kirsten rat sarcoma viral oncogene homolog-mutated non–small-cell lung cancer after platinum-based chemotherapy treatment failure. Clin. Lung Cancer 2018. [Google Scholar] [CrossRef]
- Tomasini, P.; Walia, P.; Labbe, C.; Jao, K.; Leighl, N.B. Targeting the KRAS pathway in non-small cell lung cancer. Oncologist 2016. [Google Scholar] [CrossRef]
- Ricciuti, B.; Brambilla, A.; Cortellini, A.; De Giglio, C.; Ficorella, A.; Sidoni, G.; Bellezza, L.; Crinò, V.; Ludovini, S.; Baglivo, G.; et al. Clinical outcomes to pemetrexed-based versus non-pemetrexed-based platinum doublets in patients with KRAS-mutant advanced non-squamous non-small cell lung cancer. Clin. Transl. Oncol. 2019, 1–9. [Google Scholar] [CrossRef]
- Athuluri-Divakar, S.K.; Vasquez-Del Carpio, R.; Dutta, K.; Baker, S.J.; Cosenza, S.C.; Basu, I.; Gupta, Y.K.; Reddy, M.V.R.; Ueno, L.; Hart, J.R.; et al. A small molecule RAS-mimetic disrupts RAS association with effector proteins to block signalling. Cell 2016. [Google Scholar] [CrossRef]
- Jeanson, A.; Tomasini, P.; Souquet-Bressand, M.; Brandone, N.; Boucekine, M.; Grangeon, M.; Chaleat, S.; Khobta, N.; Milia, J.; Mhanna, L.; et al. Efficacy of immune checkpoint inhibitors in KRAS-mutant non-small cell lung cancer (NSCLC). J. Thorac. Oncol. 2019, 14, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Ambrogio, C.; Köhler, J.; Zhou, Z.W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q.; et al. KRAS dimerization impacts MEK inhibitor sensitivity and oncogenic activity of mutant KRAS. Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Burgess, M.R.; Hwang, E.; Mroue, R.; Bielski, C.M.; Wandler, A.M.; Huang, B.J.; Firestone, A.J.; Young, A.; Lacap, J.A.; Crocker, L.; et al. KRAS allelic imbalance enhances fitness and modulates MAP kinase dependence in cancer. Cell 2017. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.A.; Giaccia, A.J. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat. Rev. Drug Discov. 2011. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J.; Hahn, W.C. Synthetic lethal vulnerabilities in kras-mutant cancers. Cold Spring Harb. Perspect. Med. 2018. [Google Scholar] [CrossRef]
- Kumar, M.S.; Hancock, D.C.; Molina-Arcas, M.; Steckel, M.; East, P.; Diefenbacher, M.; Armenteros-Monterroso, E.; Lassailly, F.; Matthews, N.; Nye, E.; et al. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell 2012. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Cheng, K.A.; Hata, A.N.; Faber, A.C.; Ebi, H.; Coffee, E.M.; Greninger, P.; Brown, R.D.; Godfrey, J.T.; Cohoon, T.J.; et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell 2013. [Google Scholar] [CrossRef]
- Puyol, M.; Martín, A.; Dubus, P.; Mulero, F.; Pizcueta, P.; Khan, G.; Guerra, C.; Santamaría, D.; Barbacid, M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 2010. [Google Scholar] [CrossRef]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non–small cell lung cancer, and other solid tumors. Cancer Discov. 2016. [Google Scholar] [CrossRef]
- Xue, Y.; Meehan, B.; Fu, Z.; Wang, X.Q.D.; Fiset, P.O.; Rieker, R.; Levins, C.; Kong, T.; Zhu, X.; Morin, G.; et al. SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nat. Commun. 2019, 10, 557. [Google Scholar] [CrossRef]
- Schlessinger, J. Ligand-Induced, receptor-mediated dimerization and activation of EGF receptor. Cell 2002. [Google Scholar] [CrossRef]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Zandi, R.; Larsen, A.B.; Andersen, P.; Stockhausen, M.T.; Poulsen, H.S. Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cell Signal. 2007. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, R.; Gee, J.M.; Harper, M. EGFR and cancer prognosis. Eur. J. Cancer 2001. [Google Scholar] [CrossRef]
- Suzuki, S.; Dobashi, Y.; Sakurai, H.; Nishikawa, K.; Hanawa, M.; Ooi, A. Protein overexpression and gene amplification of epidermal growth factor receptor in nonsmall cell lung carcinomas: An immunohistochemical and fluorescence in situ hybridization study. Cancer 2005. [Google Scholar] [CrossRef]
- Arcila, M.E.; Chaft, J.E.; Nafa, K.; Roy-Chowdhuri, S.; Lau, C.; Zaidinski, M.; Paik, P.K.; Zakowski, M.F.; Kris, M.G.; Ladanyi, M. Prevalence, clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin. Cancer Res. 2012. [Google Scholar] [CrossRef]
- Olayioye, M.A. New Embo members’ review: The erbb signalling network: receptor heterodimerization in development and cancer. EMBO J. 2000. [Google Scholar] [CrossRef]
- Hynes, N.E.; MacDonald, G. ErbB receptors and signalling pathways in cancer. Curr. Opin. Cell Biol. 2009. [Google Scholar] [CrossRef]
- Pawson, T. Regulation and targets of receptor tyrosine kinases. Eur. J. Cancer 2003. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005. [Google Scholar] [CrossRef]
- Roskoski, R.J. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharm. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.P.S.; Coelho, P.V.; Anazetti, M.; Simioni, P.U. Targeted therapies for the treatment of non-small-cell lung cancer: Monoclonal antibodies and biological inhibitors. Hum. Vaccin. Immunother. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Tam, K.Y. Combination strategies using EGFR-TKi in NSCLC therapy: Learning from the gap between pre-clinical results and clinical outcomes. Int. J. Biol. Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.-M.; Hu, Q.-H.; Yang, W.-T.; Wang, Y.; Tong, Y.-P.; Bao, W.-D. Targeting epidermal growth factor receptor in non-small-cell-lung cancer: Current state and future perspective. Anticancer Agents Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Serrano, A.; Gella, P.; Jiménez, E.; Zugazagoitia, J.; Paz-Ares Rodríguez, L. Targeting EGFR in lung cancer: Current standards and developments. Drugs 2018. [Google Scholar] [CrossRef]
- Lemmon, M.A. Ligand-Induced ErbB receptor dimerization. Exp. Cell Res. 2009. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signalling by receptor tyrosine kinases. Cell 2010. [Google Scholar] [CrossRef]
- Yarden, Y. The EGFR family and its ligands in human cancer. Signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 2001. [Google Scholar] [CrossRef]
- Maruyama, I.N. Activation of transmembrane cell-surface receptors via a common mechanism? The “rotation model”. BioEssays 2015. [Google Scholar] [CrossRef]
- Jones, S.; Rappoport, J.Z. Interdependent epidermal growth factor receptor signalling and trafficking. Int. J. Biochem. Cell Biol. 2014. [Google Scholar] [CrossRef]
- Macdonald-Obermann, J.L.; Pike, L.J. Different epidermal growth factor (EGF) receptor ligands show distinct kinetics and biased or partial agonism for homodimer and heterodimer formation. J. Biol. Chem. 2014. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Okada, S.; Ohshima, K.; Yamada, E.; Sato, M.; Uehara, Y.; Shimizu, H.; Pessin, J.E.; Mori, M. Differential activation of epidermal growth factor (EGF) receptor downstream signalling pathways by betacellulin and EGF. Endocrinology 2004. [Google Scholar] [CrossRef] [PubMed]
- Roepstorff, K.; Grandal, M.V.; Henriksen, L.; Knudsen, S.L.J.; Lerdrup, M.; Grøvdal, L.; Willumsen, B.M.; Van Deurs, B. Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 2009. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.C.; Chung, E.; Coffey, R.J. EGF receptor ligands. EGF Recept. Fam. Biol. Mech. Role Cancer 2003. [Google Scholar] [CrossRef]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.J.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. EGF Recept. Family Biol. Mech. Role Cancer 2003. [Google Scholar] [CrossRef]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell. Biol. 2011. [Google Scholar] [CrossRef]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990. [Google Scholar] [CrossRef]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A structural perspective on the regulation of the epidermal growth factor receptor. Annu. Rev. Biochem. 2015. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR family: Not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2014. [Google Scholar] [CrossRef]
- Bessman, N.J.; Freed, D.M.; Lemmon, M.A. Putting together structures of epidermal growth factor receptors. Curr. Opin. Struct. Biol. 2014. [Google Scholar] [CrossRef]
- Bae, J.H.; Schlessinger, J. Asymmetric tyrosine kinase arrangements in activation or autophosphorylation of receptor tyrosine kinases. Mol. Cells. 2010. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.M. Structure-Based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008. [Google Scholar] [CrossRef] [PubMed]
- Huse, M.; Kuriyan, J. The conformational plasticity of protein kinases. Cell 2002. [Google Scholar] [CrossRef]
- Kawamoto, T.; Sato, J.D.; Le, A.; Polikoff, J.; Sato, G.H.; Mendelsohn, J. Growth stimulation of A431 cells by epidermal growth factor: Identification of high-affinity receptors for epidermal growth factor by an anti-receptor monoclonal antibody. Proc. Natl. Acad. Sci. USA 1983. [Google Scholar] [CrossRef] [PubMed]
- Greulich, H.; Chen, T.-H.; Feng, W.; Janne, P.A.; Alvarez, J.V.; Zappaterra, M.; Bulmer, S.E.; Frank, D.A.; Hahn, W.C.; Sellers, W.R.; et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005, 2, e313. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Sharpless, N.E.; Wong, K.K. EGFR targeted therapy: View from biological standpoint. Cell Cycle. 2006. [Google Scholar] [CrossRef][Green Version]
- Politi, K.; Zakowski, M.F.; Fan, P.D.; Schonfeld, E.A.; Pao, W.; Varmus, H.E. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006. [Google Scholar] [CrossRef]
- Riese, D.J.; Gallo, R.M.; Settleman, J. Mutational activation of ErbB family receptor tyrosine kinases: Insights into mechanisms of signal transduction and tumorigenesis. BioEssays 2007. [Google Scholar] [CrossRef]
- Shigematsu, H.; Gazdar, A.F. Somatic mutations of epidermal growth factor receptor signalling pathway in lung cancers. Int. J. Cancer 2006. [Google Scholar] [CrossRef]
- Chan, S.K.; Gullick, W.J.; Hill, M.E. Mutations of the epidermal growth factor receptor in non-small cell lung cancer—Search and destroy. Eur. J. Cancer 2006. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung, cancer: Correlation with clinical response to gefitinib therapy. Science 2004. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Onozato, R.; Yatabe, Y.; Mitsudomi, T. EGFR T790M mutation: A double role in lung cancer cell survival? J. Thorac. Oncol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Park, E.; Yun, C.H.; Sng, N.J.; Lucena-Araujo, A.R.; Yeo, W.L.; Huberman, M.S.; Cohen, D.W.; Nakayama, S.; Ishioka, K.; et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci. Transl. Med. 2013. [Google Scholar] [CrossRef]
- Yun, C.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007. [Google Scholar] [CrossRef]
- Eck, M.J.; Yun, C.H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochimica Biophysica Acta Proteins Proteomics 2010. [Google Scholar] [CrossRef]
- Massarelli, E.; Johnson, F.M.; Erickson, H.S.; Wistuba, I.I.; Papadimitrakopoulou, V. Uncommon epidermal growth factor receptor mutations in non-small cell lung cancer and their mechanisms of EGFR tyrosine kinase inhibitors sensitivity and resistance. Lung Cancer 2013. [Google Scholar] [CrossRef]
- Sgambato, A.; Casaluce, F.; Maione, P.; Rossi, A.; Rossi, E.; Napolitano, A.; Palazzolo, G.; Bareschino, M.A.; Schettino, C.; Sacco, P.C.; et al. The role of EGFR tyrosine kinase inhibitors in the first-line treatment of advanced non small cell lung cancer patients harboring EGFR mutation. Curr. Med. Chem. 2012, 19, 3337–3352. [Google Scholar] [CrossRef]
- Shawver, L.K.; Slamon, D.; Ullrich, A. Smart drugs: Tyrosine Kinase Inhibitors in Cancer Therapy. Cancer Cell. 2002, 1, 117–123. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.Y.; Chiu, C.H.; Li, L.H.; Hsiao, C.Y.; Tzen, C.Y.; Chang, K.T.; Chen, Y.M.; Perng, R.P.; Tsai, S.F.; Tsai, C.M. Mutation in the tyrosine kinase domain of epidermal growth factor receptor is a predictive and prognostic factor for gefitinib treatment in patients with non-small cell lung cancer. Clin. Cancer Res. 2005. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Besse, B.; Lynch, T.J.; Miller, V.A.; Wong, K.K.; Gitlitz, B.; Eaton, K.; Zacharchuk, C.; Freyman, A.; Powell, C.; et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: Results of a phase II trial in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Molina, M.A.; Costa, C.; Simonetti, S.; Gimenez-Capitan, A.; Bertran-Alamillo, J.; Mayo, C.; Moran, T.; Mendez, P.; Cardenal, F.; et al. Pretreatment EGFR T790M mutation and BRCA1 MRNA expression in erlotinib-treated advanced non-small-cell lung cancer patients with EGFR mutations. Clin. Cancer Res. 2011. [Google Scholar] [CrossRef]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002. [Google Scholar] [CrossRef]
- Mulloy, R.; Ferrand, A.; Kim, Y.; Sordella, R.; Bell, D.W.; Haber, D.A.; Anderson, K.S.; Settleman, J. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007. [Google Scholar] [CrossRef]
- Wu, J.Y.; Wu, S.G.; Yang, C.H.; Chang, Y.L.; Chang, Y.C.; Hsu, Y.C.; Shih, J.Y.; Yang, P.C. Comparison of gefitinib and erlotinib in advanced NSCLC and the effect of EGFR mutations. Lung Cancer 2011. [Google Scholar] [CrossRef]
- Carey, K.D.; Garton, A.J.; Romero, M.S.; Kahler, J.; Thomson, S.; Ross, S.; Park, F.; Haley, J.D.; Gibson, N.; Sliwkowski, M.X. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006. [Google Scholar] [CrossRef]
- Vyse, S.; Huang, P.H. Targeting EGFR exon 20 insertion mutations in non-small cell lung cancer. Signal Transduct. Target. Ther. 2019. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Gale, C.M.; Lifshits, E.; Gonzales, A.J.; Shimamura, T.; Zhao, F.; Vincent, P.W.; Naumov, G.N.; Bradner, J.E.; et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Sequist, L.V.; Arcila, M.E.; Moran, T.; Chmielecki, J.; Lin, Y.-L.; Pan, Y.; Wang, L.; de Stanchina, E.; Shien, K.; et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc. Natl. Acad. Sci. USA 2012. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008. [Google Scholar] [CrossRef] [PubMed]
- Yuza, Y.; Glatt, K.A.; Jiang, J.; Greulich, H.; Minami, Y.; Woo, M.S.; Shimamura, T.; Shapiro, G.; Lee, J.C.; Ji, H.; et al. Allele-Dependent variation in the relative cellular potency of distinct EGFR inhibitors. Cancer Biol. 2007. [Google Scholar] [CrossRef][Green Version]
- Ning, J.; Wu, Q.; Liu, Z.; Wang, J.; Lin, X. Mapping inhibitor response to the in-frame deletions, insertions and duplications of epidermal growth factor receptor (EGFR) in non-small cell lung cancer. J. Recept. Signal Transduct. 2016. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Lo, P.C.; Nishino, M.; Dahlberg, S.E.; Lindeman, N.I.; Butaney, M.; Jackman, D.M.; Johnson, B.E.; Jänne, P.A. Natural history and molecular characteristics of lung cancers harboring EGFR exon 20 insertions. J. Thorac. Oncol. 2013. [Google Scholar] [CrossRef]
- Qin, B.-D.; Jiao, X.-D.; Yuan, L.-Y.; Liu, K.; Wang, Z.; Qin, W.-X.; Zang, Y.-S. The effectiveness of afatinib and osimertinib in a chinese patient with advanced lung adenocarcinoma harboring a rare triple EGFR mutation (R670W/H835L/L833V): A case report and literature review. OncoTargets Ther. 2018, 4739–4745. [Google Scholar] [CrossRef]
- Friedman, R. Drug resistance in cancer: Molecular evolution and compensatory proliferation. Oncotarget 2016. [Google Scholar] [CrossRef]
- Sini, C.; Tuzi, A.; Rossi, G.; Russo, A.; Pezzuto, A. Acquired resistance in oncogene-addicted non-small-cell lung cancer. Future Oncol. 2018, 14, 29–40. [Google Scholar] [CrossRef]
- Tomasello, C.; Baldessari, C.; Napolitano, M.; Orsi, G.; Grizzi, G.; Bertolini, F.; Barbieri, F.; Cascinu, S. Resistance to EGFR inhibitors in non-small cell lung cancer: Clinical management and future perspectives. Crit. Rev. Oncol. Hematol. 2018, 123, 149–161. [Google Scholar] [CrossRef]
- Kohsaka, S.; Petronczki, M.; Solca, F.; Maemondo, M. Tumor clonality and resistance mechanisms in EGFR mutation-positive non-small-cell lung cancer: Implications for therapeutic sequencing. Future Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Wang, T.Y.; Riely, G.J.; Miller, V.A.; Pan, Q.; Ladanyi, M.; Zakowski, M.F.; Heelan, R.T.; Kris, M.G.; Varmus, H.E. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Inukai, M.; Toyooka, S.; Ito, S.; Asano, H.; Ichihara, S.; Soh, J.; Suehisa, H.; Ouchida, M.; Aoe, K.; Aoe, M.; et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res. 2006. [Google Scholar] [CrossRef]
- Su, K.Y.; Chen, H.Y.; Li, K.C.; Kuo, M.L.; Yang, J.C.H.; Chan, W.K.; Ho, B.C.; Chang, G.C.; Shih, J.Y.; Yu, S.L.; et al. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J. Clin. Oncol. 2012. [Google Scholar] [CrossRef]
- Maheswaran, S.; Sequist, L.V.; Nagrath, S.; Ulkus, L.; Brannigan, B.; Collura, C.V.; Inserra, E.; Diederichs, S.; Iafrate, A.J.; Bell, D.W.; et al. Detection of mutations in EGFR in circulating lung-cancer cells. N. Engl. J. Med. 2008. [Google Scholar] [CrossRef]
- Wu, S.G.; Shih, J.Y. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol. Cancer 2018. [Google Scholar] [CrossRef]
- Del Re, M.; Crucitta, S.; Gianfilippo, G.; Passaro, A.; Petrini, I.; Restante, G.; Michelucci, A.; Fogli, S.; de Marinis, F.; Porta, C.; et al. Understanding the mechanisms of resistance in EGFR-positive NSCLC: From tissue to liquid biopsy to guide treatment strategy. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Saarenheimo, J.; Eigeliene, N.; Andersen, H.; Tiirola, M.; Jekunen, A. The value of liquid biopsies for guiding therapy decisions in non-small cell lung cancer. Front. Oncol. 2019. [Google Scholar] [CrossRef]
- Balak, M.N.; Gong, Y.; Riely, G.J.; Somwar, R.; Li, A.R.; Zakowski, M.F.; Chiang, A.; Yang, G.; Ouerfelli, O.; Kris, M.G.; et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin. Cancer Res. 2006. [Google Scholar] [CrossRef]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res. 2008. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Jänne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008. [Google Scholar] [CrossRef]
- Kosaka, T.; Yatabe, Y.; Endoh, H.; Yoshida, K.; Hida, T.; Tsuboi, M.; Tada, H.; Kuwano, H.; Mitsudomi, T. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin. Cancer Res. 2006. [Google Scholar] [CrossRef]
- Bell, D.W.; Gore, I.; Okimoto, R.A.; Godin-Heymann, N.; Sordella, R.; Mulloy, R.; Sharma, S.V.; Brannigan, B.W.; Mohapatra, G.; Settleman, J.; et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat. Genet. 2005. [Google Scholar] [CrossRef]
- Jorge, S.E.D.C.; Kobayashi, S.S.; Costa, D.B. Epidermal growth factor receptor (EGFR) mutations in lung cancer: Preclinical and clinical data. Braz. J. Med. Biol. Res. 2014. [Google Scholar] [CrossRef]
- Costa, D.B.; Halmos, B.; Kumar, A.; Schumer, S.T.; Huberman, M.S.; Boggon, T.J.; Tenen, D.G.; Kobayashi, S. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007. [Google Scholar] [CrossRef]
- Bean, J.; Riely, G.J.; Balak, M.; Marks, J.L.; Ladanyi, M.; Miller, V.A.; Pao, W. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin. Cancer Res. 2008. [Google Scholar] [CrossRef]
- Konecny, G.E.; Pegram, M.D.; Venkatesan, N.; Finn, R.; Yang, G.; Rahmeh, M.; Untch, M.; Rusnak, D.W.; Spehar, G.; Mullin, R.J.; et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006. [Google Scholar] [CrossRef]
- Wecker, H.; Waller, C.F. Afatinib. Recent Results Cancer Res. 2018, 211, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.W.; Bridges, A.J.; Denny, W.A.; Doherty, A.; Greis, K.D.; Hicks, J.L.; Hook, K.E.; Keller, P.R.; Leopold, W.R.; Loo, J.A.; et al. Specific, irreversible inactivation of the epidermal growth factor receptor and ErbB2, by a new class of tyrosine kinase inhibitor. Proc. Natl. Acad. Sci. USA 2002. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Riely, G.J. Second-Generation epidermal growth factor receptor tyrosine kinase inhibitors in lung cancers. J. Natl. Compr. Cancer Netw. 2013. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.A.; Kuzmic, P.; Solowiej, J.; Bergqvist, S.; Bolanos, B.; Almaden, C.; Nagata, A.; Ryan, K.; Feng, J.; Dalvie, D.; et al. Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, S.-H.; Ahn, J.S.; Ahn, M.-J.; Park, K.; Sun, J.-M. Efficacy and safety of afatinib for egfr-mutant non-small cell lung cancer, compared with gefitinib or erlotinib. Cancer Res. Treat. 2019, 51, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Yang, J.C.H.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.M.; Boyer, M.; et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zhou, C.; Hu, C.P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): An open-label, randomised phase 3 trial. Lancet Oncol. 2014. [Google Scholar] [CrossRef]
- Dhillon, S. Neratinib in early-stage breast cancer: A profile of its use in the EU. Clin. Drug Investig. 2019. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 2017. [Google Scholar] [CrossRef]
- Kosaka, T.; Tanizaki, J.; Paranal, R.M.; Endoh, H.; Lydon, C.; Capelletti, M.; Repellin, C.E.; Choi, J.; Ogino, A.; Calles, A.; et al. Response heterogeneity of EGFR and HER2 exon 20 insertions to covalent EGFR and HER2 inhibitors. Cancer Res. 2017. [Google Scholar] [CrossRef]
- Yasuda, H.; Kobayashi, S.; Costa, D.B. EGFR exon 20 insertion mutations in non-small-cell lung cancer: Preclinical data and clinical implications. Lancet Oncol. 2012. [Google Scholar] [CrossRef]
- Burstein, H.J.; Sun, Y.; Dirix, L.Y.; Jiang, Z.; Paridaens, R.; Tan, A.R.; Awada, A.; Ranade, A.; Jiao, S.; Schwartz, G.; et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J. Clin. Oncol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Minkovsky, N.; Berezov, A. BIBW-2992, a dual receptor tyrosine kinase inhibitor for the treatment of solid tumors. Curr. Opin. Investig. Drugs 2008, 9, 1336–1346. [Google Scholar] [PubMed]
- Janjigian, Y.Y.; Azzoli, C.G.; Krug, L.M.; Pereira, L.K.; Rizvi, N.A.; Pietanza, M.C.; Kris, M.G.; Ginsberg, M.S.; Pao, W.; Miller, V.A.; et al. Phase I/II trial of cetuximab and erlotinib in patients with lung adenocarcinoma and acquired resistance to erlotinib. Clin. Cancer Res. 2011. [Google Scholar] [CrossRef]
- Walter, A.O.; Sjin, R.T.T.; Haringsma, H.J.; Sun, J.; Ohashi, K.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Wang, Z.; Zhu, Z.; et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M mediated resistance in NSCLC. Cancer Discov. 2013. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.S.; Kumarakulasinghe, N.B.; Huang, Y.Q.; Ang, Y.L.E.; Choo, J.R.E.; Goh, B.C.; Soo, R.A. Third generation EGFR TKIs: Current data and future directions. Mol. Cancer 2018. [Google Scholar] [CrossRef]
- Sukrithan, V.; Deng, L.; Barbaro, A.; Cheng, H. Emerging drugs for EGFR-mutated non-small cell lung cancer. Expert Opin. Emerg. Drugs 2019, 24, 5–16. [Google Scholar] [CrossRef]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014. [Google Scholar] [CrossRef]
- Liao, B.C.; Lin, C.C.; Lee, J.H.; Yang, J.C.H. Optimal management of EGFR-mutant non-small cell lung cancer with disease progression on first-line tyrosine kinase inhibitor therapy. Lung Cancer. 2017. [Google Scholar] [CrossRef]
- Hirano, T.; Yasuda, H.; Tani, T.; Hamamoto, J.; Oashi, A.; Ishioka, K.; Arai, D.; Nukaga, S.; Miyawaki, M.; Kawada, I.; et al. In vitro modeling to determine mutation specificity of EGFR tyrosine kinase inhibitors against clinically relevant EGFR mutants in non-small-cell lung cancer. Oncotarget 2015. [Google Scholar] [CrossRef]
- Robichaux, J.P.; Elamin, Y.Y.; Tan, Z.; Carter, B.W.; Zhang, S.; Liu, S.; Li, S.; Chen, T.; Poteete, A.; Estrada-Bernal, A.; et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Osimertinib as first-line therapy in advanced NSCLC: A profile of its use. Drugs Perspect. 2018. [Google Scholar] [CrossRef] [PubMed]
- Senthil Rajappa, M.V.K. Integrating osimertinib in clinical practice for non-small cell lung cancer treatment. Adv. Ther. 2019, 36, 1279–1290. [Google Scholar]
- Holleman, M.S.; van Tinteren, H.; Groen, H.J.M.; Al, M.J.; Uyl-de Groot, C.A. First-Line tyrosine kinase inhibitors in EGFR mutation-positive non-small-cell lung cancer: A network meta-analysis. OncoTargets Ther. 2019. [Google Scholar] [CrossRef]
- Attili, I.; Karachaliou, N.; Conte, P.; Bonanno, L.; Rosell, R. Therapeutic approaches for T790M mutation positive non-small-cell lung cancer. Expert Rev. Anticancer 2018, 18, 1021–1030. [Google Scholar] [CrossRef]
- Lelais, G.; Epple, R.; Marsilje, T.H.; Long, Y.O.; McNeill, M.; Chen, B.; Lu, W.; Anumolu, J.; Badiger, S.; Bursulaya, B.; et al. Discovery of (R,E)-N-(7-Chloro-1-(1-[4-(Dimethylamino)but-2-Enoyl]Azepan-3-Yl)-1H-Benzo[d]Imid Azol-2-Yl)-2-Methylisonicotinamide (EGF816), a novel, potent, and WT sparing covalent inhibitor of oncogenic (L858R, Ex19del) and resistant (T790M) EGFR mutants. J. Med. Chem. 2016, 59, 6671–6689. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.-H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef]
- Singh, J.; Evans, E.; Hagel, M.; Labinski, M.; Dubrovskiy, A.; Nacht, M.; Petter, R.C.; Prasad, A.; Sheets, M.; St. Martin, T.; et al. Superiority of a novel EGFR targeted covalent inhibitor over its reversible counterpart in overcoming drug resistance. Medchemcomm 2012. [Google Scholar] [CrossRef]
- Liang, S.I.; van Lengerich, B.; Eichel, K.; Cha, M.; Patterson, D.M.; Yoon, T.Y.; von Zastrow, M.; Jura, N.; Gartner, Z.J. Phosphorylated EGFR dimers are not sufficient to activate ras. Cell Rep. 2018. [Google Scholar] [CrossRef]
- Yang, R.Y.C.; Yang, K.S.; Pike, L.J.; Marshall, G.R. Targeting the dimerization of epidermal growth factor receptors with small-molecule inhibitors. Chem. Biol. Drug Des. 2010. [Google Scholar] [CrossRef]
- Wang, S.; Song, Y.; Liu, D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S Resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef] [PubMed]
- To, C.; Jang, J.; Chen, T.; Park, E.; Mushajiang, M.; De Clercq, D.J.H.; Xu, M.; Wang, S.; Cameron, M.D.; Heppner, D.E.; et al. Single and dual targeting of mutant EGFR with an allosteric inhibitor. Cancer Discov. 2019. [Google Scholar] [CrossRef] [PubMed]
- Fulton, M.D.; Hanold, L.E.; Ruan, Z.; Patel, S.; Beedle, A.M.; Kannan, N.; Kennedy, E.J. Conformationally constrained peptides target the allosteric kinase dimer interface and inhibit EGFR activation. Bioorganic Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fogli, S.; Polini, B.; Del Re, M.; Petrini, I.; Passaro, A.; Crucitta, S.; Rofi, E.; Danesi, R. EGFR-TKIs in non-small-cell lung cancer: Focus on clinical pharmacology and mechanisms of resistance. Pharmacogenomics 2018, 19, 727–740. [Google Scholar] [CrossRef]
- Schoeberl, B.; Faber, A.C.; Li, D.; Liang, M.C.; Crosby, K.; Onsum, M.; Burenkova, O.; Pace, E.; Walton, Z.; Nie, L.; et al. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res. 2010. [Google Scholar] [CrossRef]
- La Monica, S.; Minari, R.; Cretella, D.; Flammini, L.; Fumarola, C.; Bonelli, M.; Cavazzoni, A.; Digiacomo, G.; Galetti, M.; Madeddu, D.; et al. Third generation EGFR inhibitor osimertinib combined with pemetrexed or cisplatin exerts long-lasting anti-tumor effect in EGFR-mutated pre-clinical models of NSCLC. J. Exp. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 amplification: A potential mechanism of acquired resistance to egfr inhibition in EGFR-mutant lung cancers that lack the second-site EGFR T790M mutation. Cancer Discov. 2012. [Google Scholar] [CrossRef]
- Zhou, B.B.S.; Peyton, M.; He, B.; Liu, C.; Girard, L.; Caudler, E.; Lo, Y.; Baribaud, F.; Mikami, I.; Reguart, N.; et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell 2006. [Google Scholar] [CrossRef]
- Ercan, D.; Xu, C.; Yanagita, M.; Monast, C.S.; Pratilas, C.A.; Montero, J.; Butaney, M.; Shimamura, T.; Sholl, L.; Ivanova, E.V.; et al. Reactivation of ERK signalling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V. Development of PI3K/AKT/MTOR pathway inhibitors and their application in personalized therapy for non-small-cell lung cancer. J. Thorac. Oncol. 2012. [Google Scholar] [CrossRef]
- Yochum, Z.A.; Cades, J.; Wang, H.; Chatterjee, S.; Simons, B.W.; O’Brien, J.P.; Khetarpal, S.K.; Lemtiri-Chlieh, G.; Myers, K.V.; Huang, E.H.B.; et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene 2019. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, T.; Lowell, A.M.; Engelman, J.A.; Shapiro, G.I. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res. 2005. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, C.; Mack, P.C.; Scagliotti, G.V.; Baas, P.; Barlesi, F.; Bivona, T.G.; Herbst, R.S.; Mok, T.S.; Peled, N.; Pirker, R.; et al. Liquid biopsy for advanced non-small cell lung cancer (NSCLC): A statement paper from the IASLC. J. Thorac. Oncol. 2018, 9, 1248–1268. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, J.N.; Santoni-Rugiu, E.; Grauslund, M.; Melchior, L.; Sørensen, J.B. Concomitant driver mutations in advanced EGFR-mutated non-small-cell lung cancer and their impact on erlotinib treatment. Oncotarget 2018, 9, 26195–26208. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Zhao, C.; Li, J.; Su, C.; Chen, X.; Ren, S.; Li, X.; Zhou, C. Clinical features and therapeutic options in non-small cell lung cancer patients with concomitant mutations of EGFR, ALK, ROS1, KRAS or BRAF. Cancer Med. 2019, 8, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Rachiglio, A.M.; Fenizia, F.; Piccirillo, M.C.; Galetta, D.; Crinò, L.; Vincenzi, B.; Barletta, E.; Pinto, C.; Ferraù, F.; Lambiase, M.; et al. The presence of concomitant mutations affects the activity of EGFR tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer (NSCLC) patients. Cancers 2019, 11, 341. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aran, V.; Omerovic, J. Current Approaches in NSCLC Targeting K-RAS and EGFR. Int. J. Mol. Sci. 2019, 20, 5701. https://doi.org/10.3390/ijms20225701
Aran V, Omerovic J. Current Approaches in NSCLC Targeting K-RAS and EGFR. International Journal of Molecular Sciences. 2019; 20(22):5701. https://doi.org/10.3390/ijms20225701
Chicago/Turabian StyleAran, Veronica, and Jasminka Omerovic. 2019. "Current Approaches in NSCLC Targeting K-RAS and EGFR" International Journal of Molecular Sciences 20, no. 22: 5701. https://doi.org/10.3390/ijms20225701
APA StyleAran, V., & Omerovic, J. (2019). Current Approaches in NSCLC Targeting K-RAS and EGFR. International Journal of Molecular Sciences, 20(22), 5701. https://doi.org/10.3390/ijms20225701