Genome-Wide Correlation of 36 Agronomic Traits in the 287 Pepper (Capsicum) Accessions Obtained from the SLAF-seq-Based GWAS

Abstract
1. Introduction
2. Results
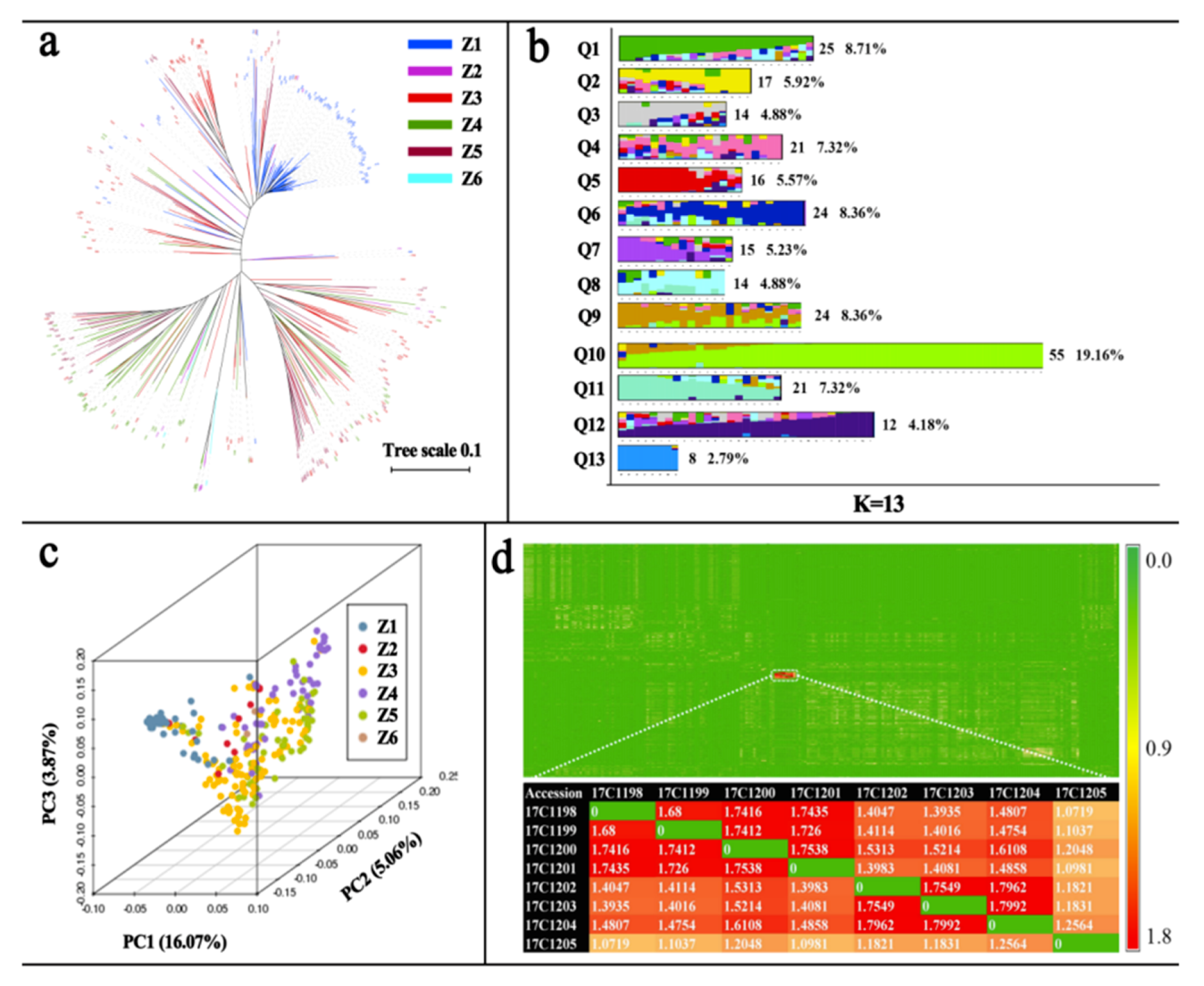
2.1. Genetic Diversity Analysis for the GWAS Population
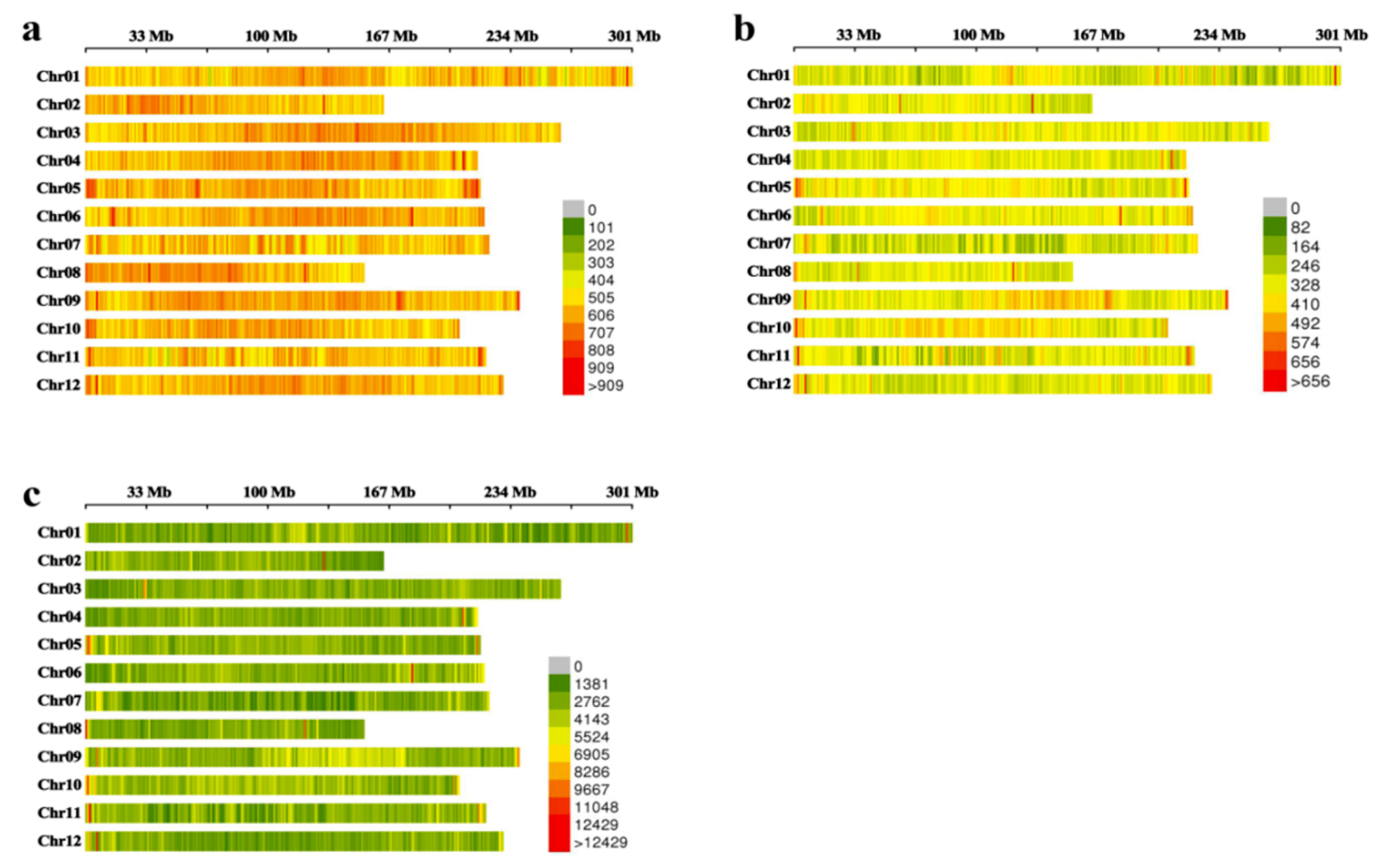
2.2. Identification and Distribution Analysis for Labels
2.3. Population Structure Analysis for the GWAS Population
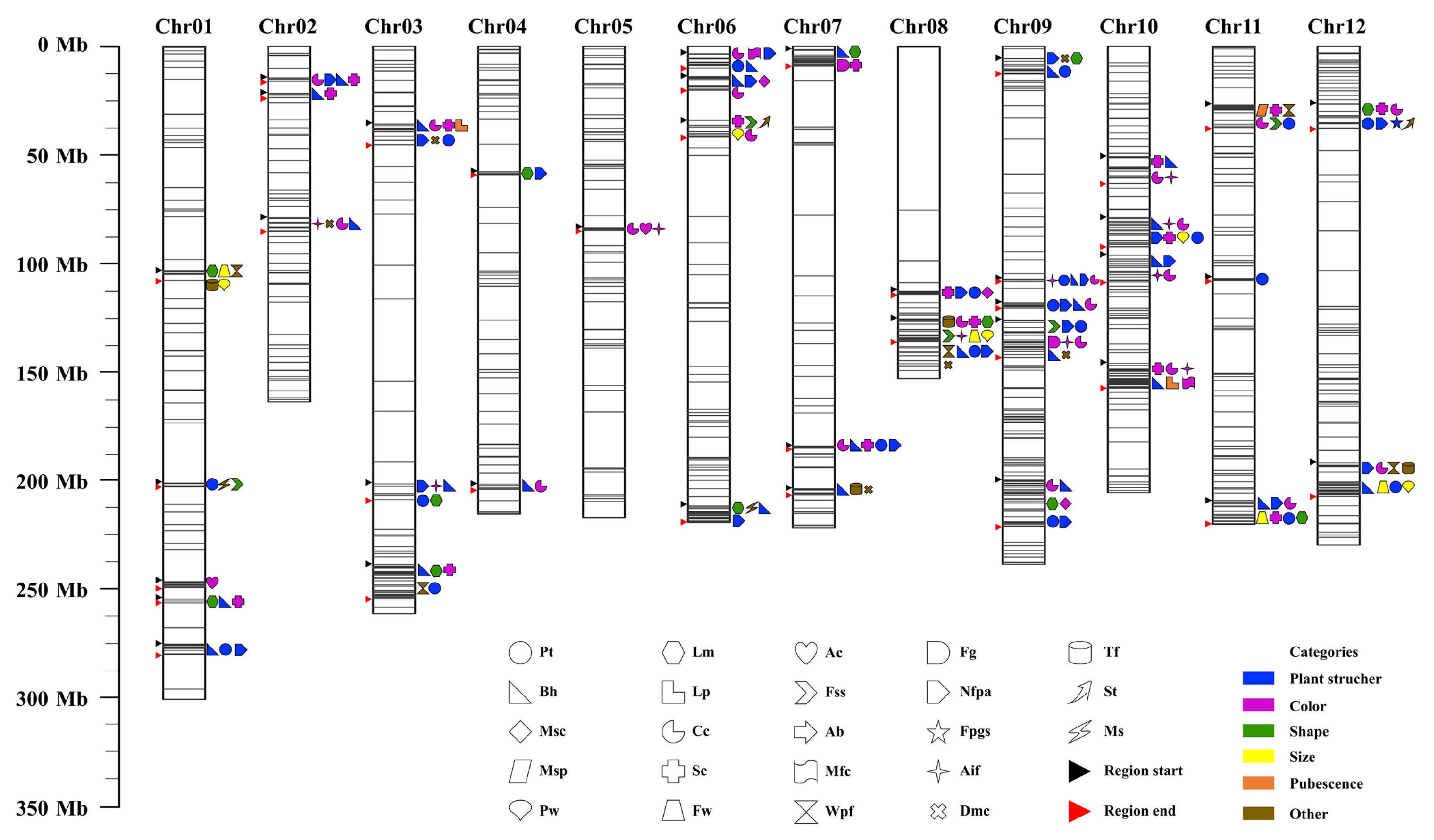
2.4. Large-Scale GWAS for 36 Agronomic Traits
2.4.1. The Three Stem-Related Traits
Plant Type
Branching Habit
Main Stem Pubescence
2.4.2. The Two Leaf-Related Traits
Leaf Margin
Leaf Pubescence
2.4.3. The Five Flower-Related Traits
Corolla Color
Style Color
Number of Flowers Per Axil
Flower Pedicel Growing State
Male Sterility
2.4.4. The Five Fruit-Related Traits
Anthocyanin on Immature Fruit
Mature Fruit Color
Spicy Type
Placenta Width
Fruit Width
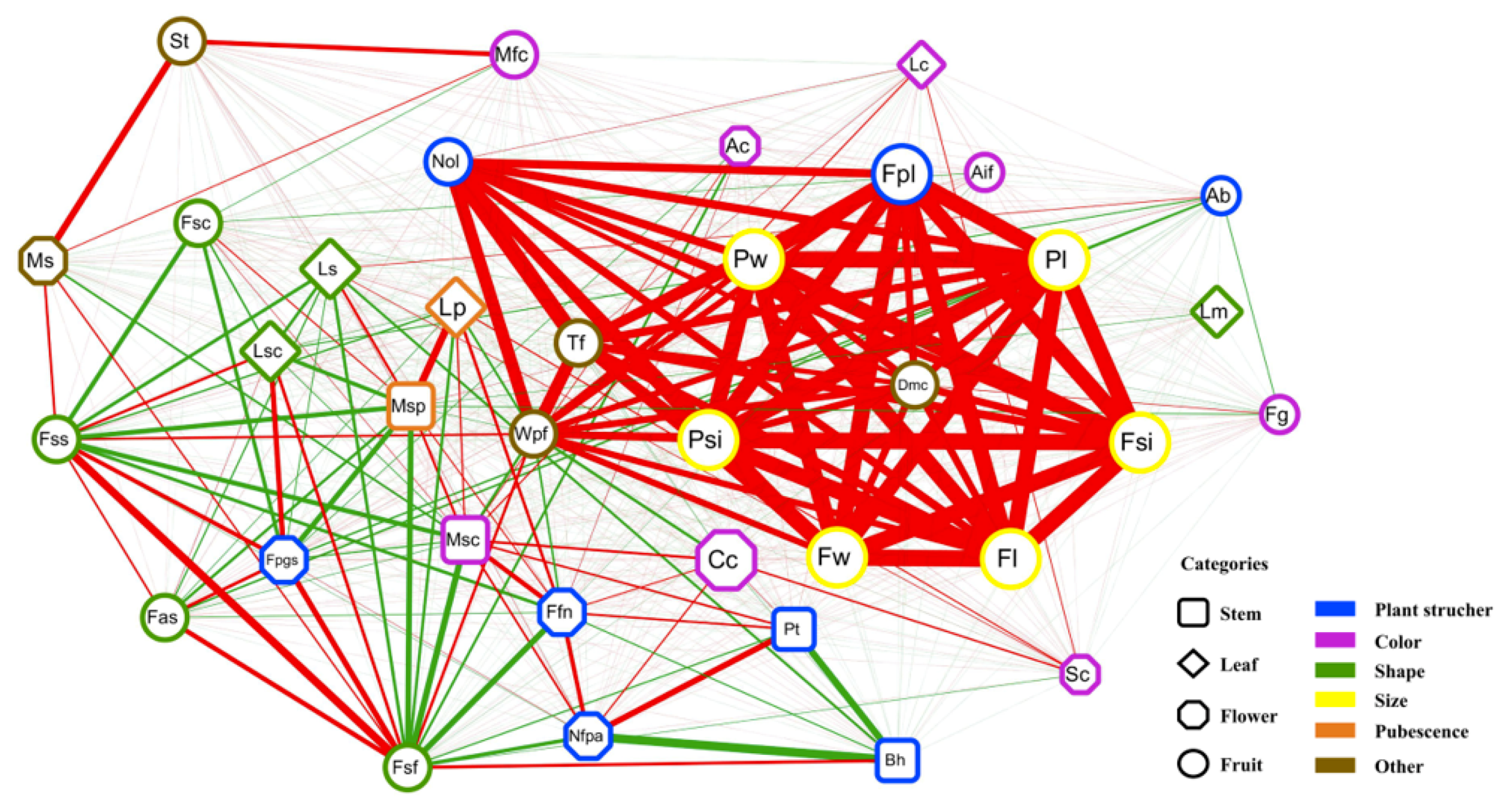
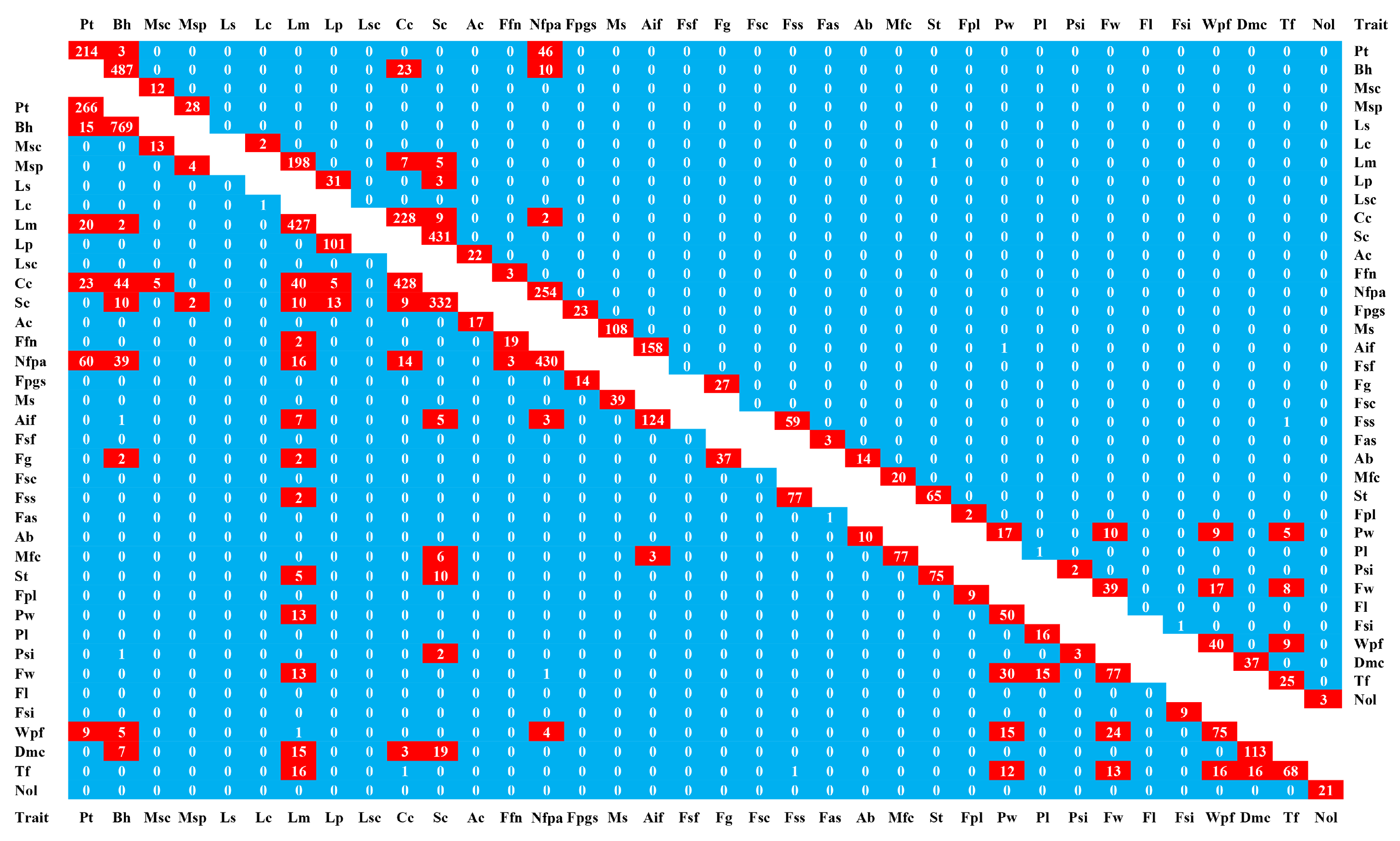
2.5. Global Exploration of Correlations Among the 36 Agronomic Traits
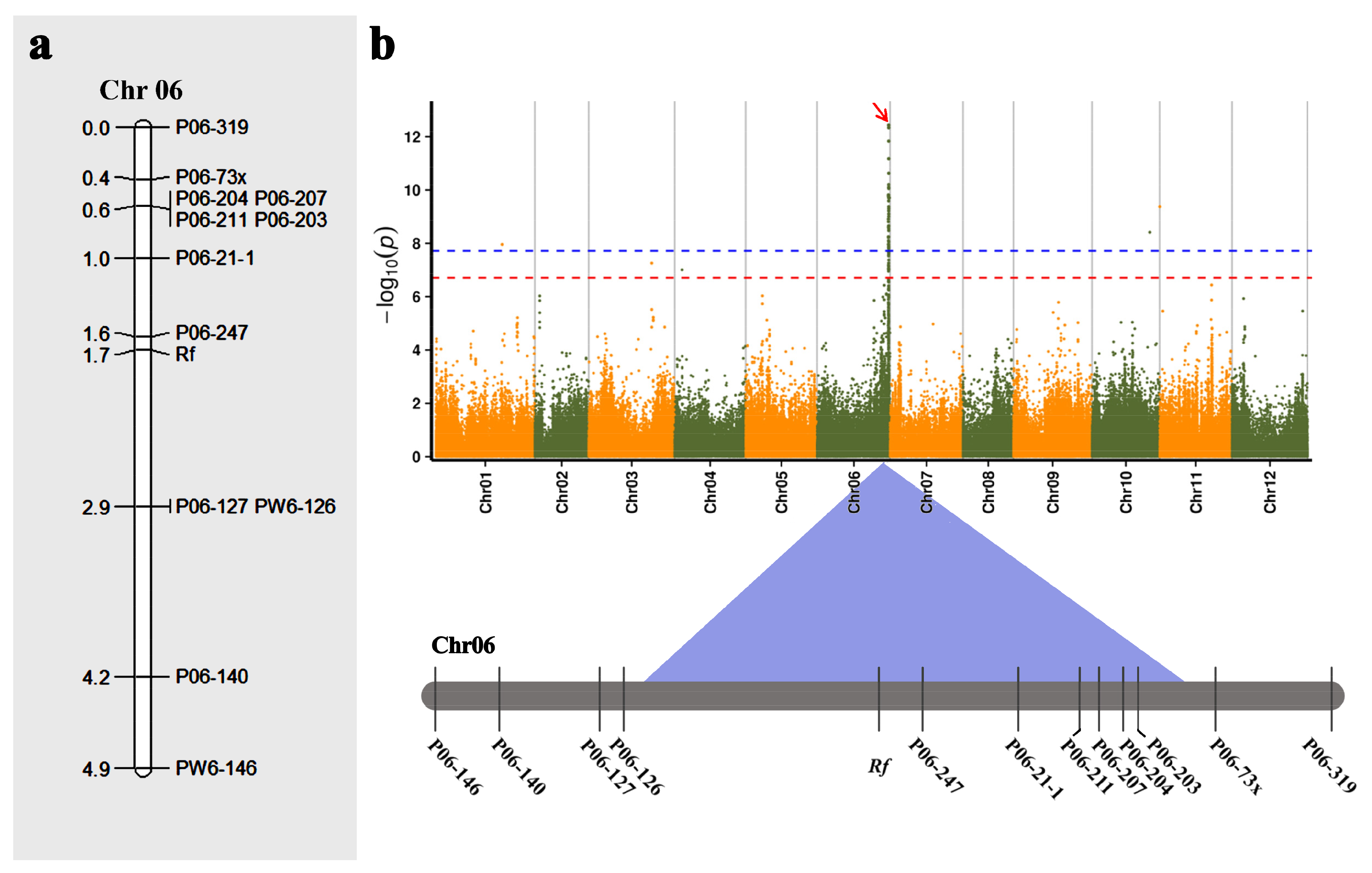
2.6. Verification of GWAS Results Based on Fine Mapping of Male-Sterility (Rf) Gene
3. Discussion
4. Materials and Methods
4.1. Global Exploration of Correlations among the 36 Agronomic Traits
4.2. Analysis of Phenotypic Data
4.3. Extraction of Genomic DNA
4.4. Design of Enzyme Digestion Scheme
4.5. Construction of SLAF Libraries and High-Throughput Sequencing
4.6. Identification and Distribution Analysis of Labels (SLAF Tags and SNPs)
4.7. Population Structure Analysis
4.8. Genome-Wide Association Analyses of 36 Agronomic Traits
4.9. Molecular Mapping of the Nuclear Fertility-Restoring Gene (Rf) for Cytoplasmic Male Sterility
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- International Plant Genetic Resources, I. Descriptors for Capsicum (Capsicum spp.); Bioversity International: Rome, Italy, 1995. [Google Scholar]
- Junior, E.; Silva, W.C.; de Carvalho, S.I.C.; Duarte, J.B. Identification of minimum descriptors for characterization of Capsicum spp. germplasm. Hortic. Bras. 2013, 31, 190–202. [Google Scholar]
- Li, X.; Zhang, B. (Eds.) Descriptors and Data Standard for Capisicum (Capisicum annum L.; Capisicum frutescens L.; Capisicum chinense, Capisicum bacctum, Capisicum pubescens), 1st ed.; China Agriculture Press, Ltd.: Beijing, China, 2006. (In Chinese) [Google Scholar]
- Danojevic, D.; Medic-Pap, S. Different multivariate analysis for fruit traits in sweet pepper breeding. Genet. Belgrade 2018, 50, 121–129. [Google Scholar] [CrossRef]
- Rao, V.V.R.; Jaisani, B.G.; Patel, G.J. Interrelationship and path coefficients of quantitative traits in Chilli. Indian J. Agric. Sci. 1974, 44, 462–465. [Google Scholar]
- Gill, H.S.; Asawa, B.M.; Thakur, P.C.; Thakur, T.C. Correlation, path-coefficient and multiple-regression analysis in sweetpepper. Indian J. Agric. Sci. 1977, 47, 408–410. [Google Scholar]
- Gupta, C.R.; Yadav, R.D.S. Genetic variability and path analysis in chili capsicum-annuum. Genet. Agrar. 1984, 38, 425–432. [Google Scholar]
- Espigares, T.; Peco, B. Mediterranean annual pasture dynamics—Impact of autumn drought. J. Ecol. 1995, 83, 135–142. [Google Scholar] [CrossRef]
- Hagley, E.A.C.; Bronskill, J.F.; Ford, E.J. Effect of the physical nature of leaf and fruit surfaces on oviposition by the codling moth, cydia-pomonella (lepidoptera, tortricidae). Can. Entomol. 1980, 112, 503–510. [Google Scholar] [CrossRef]
- Valverde, P.L.; Fornoni, J.; Nunez-Farfan, J. Defensive role of leaf trichomes in resistance to herbivorous insects in Datura stramonium. J. Evol. Biol. 2001, 14, 424–432. [Google Scholar] [CrossRef]
- Tewksbury, J.J.; Levey, D.J.; Huizinga, M.; Haak, D.C.; Traveset, A. Costs and benefits of capsaicin-mediated control of gut retention in dispersers of wild chilies. Ecology 2008, 89, 107–117. [Google Scholar] [CrossRef]
- Tewksbury, J.J.; Nabhan, G.P. Seed dispersal—Directed deterrence by capsaicin in chillies. Nature 2001, 412, 403–404. [Google Scholar] [CrossRef]
- Ben Chaim, A.; Paran, I.; Grube, R.C.; Jahn, M.; van Wijk, R.; Peleman, J. QTL mapping of fruit-related traits in pepper (Capsicum annuum). Theor. Appl. Genet. 2001, 102, 1016–1028. [Google Scholar] [CrossRef]
- Blum, E.; Mazourek, M.; O′Connell, M.; Curry, J.; Thorup, T.; Liu, K.D.; Jahn, M.; Paran, I. Molecular mapping of capsaicinoid biosynthesis genes and quantitative trait loci analysis for capsaicinoid content in Capsicum. Theor. Appl. Genet. 2003, 108, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.U.; Chaim, A.B.; Borovsky, Y.; Paran, I. Mapping of yield-related QTLs in pepper in an interspecific cross of Capsicum annuum and C-frutescens. Theor. Appl. Genet. 2003, 106, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Yarnes, S.C.; Ashrafi, H.; Reyes-Chin-Wo, S.; Hill, T.A.; Stoffel, K.M.; van Deynze, A. Identification of QTLs for capsaicinoids, fruit quality, and plant architecture-related traits in an interspecific Capsicum RIL population. Genome 2013, 56, 61–74. [Google Scholar] [CrossRef]
- Han, K.; Jeong, H.-J.; Yang, H.-B.; Kang, S.-M.; Kwon, J.-K.; Kim, S.; Choi, D.; Kang, B.-C. An ultra-high-density bin map facilitates high-throughput QTL mapping of horticultural traits in pepper (Capsicum annuum). DNA Res. 2016, 23, 81–91. [Google Scholar] [CrossRef]
- Li, X.; Wu, L.; Wang, J.; Sun, J.; Xia, X.; Geng, X.; Wang, X.; Xu, Z.; Xu, Q. Genome sequencing of rice subspecies and genetic analysis of recombinant lines reveals regional yield- and quality-associated loci. BMC Biol. 2018, 16, 102. [Google Scholar] [CrossRef]
- Peterson, P.A. Linkage of fruit shape and color genes in capsicum. Genetics 1959, 44, 407–419. [Google Scholar]
- Chaim, A.B.; Borovsky, Y.; de Jong, W.; Paran, I. Linkage of the A locus for the presence of anthocyanin and fs10.1, a major fruit-shape QTL in pepper. Theor. Appl. Genet. 2003, 106, 889–894. [Google Scholar] [CrossRef]
- Salvi, S.; Tuberosa, R. To clone or not to clone plant QTLs: Present and future challenges. Trends Plant Sci. 2005, 10, 297–304. [Google Scholar] [CrossRef]
- Yu, J.M.; Buckler, E.S. Genetic association mapping and genome organization of maize. Curr. Opin. Biotechnol. 2006, 17, 155–160. [Google Scholar] [CrossRef]
- Ngan Thi, P.; Sim, S. Genomic tools and their implications for vegetable breeding. Korean J. Hortic. Sci. Technol. 2017, 35, 149–164. [Google Scholar]
- Ingvarsson, P.K.; Street, N.R. Association genetics of complex traits in plants. New Phytol. 2011, 189, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, H.D.; Bajaj, D.; Narnoliya, L.; Das, S.; Kumar, V.; Gowda, C.L.L.; Sharma, S.; Tyagi, A.K.; Parida, S.K. Genome-Wide Scans for Delineation of Candiate Genes Regulating Seed-Protein Content in Chickpea. Front. Plant Sci. 2016, 7, 302. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.A.R.; Willcox, M.; Burgueno, J.; Romay, C.; Swarts, K.; Trachsel, S.; Preciado, E.; Terron, A.; Delgado, H.V.; Vidal, V.; et al. A study of allelic diversity underlying flowering-time adaptation in maize landraces. Nat. Genet. 2017, 49, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Sonah, H.; O′Donoughue, L.; Cober, E.; Rajcan, I.; Belzile, F. Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol. J. 2015, 13, 211–221. [Google Scholar] [CrossRef]
- Sauvage, C.; Segura, V.; Bauchet, G.; Stevens, R.; Thi, D.P.; Nikoloski, Z.; Fernie, A.R.; Causse, M. Genome-Wide Association in Tomato Reveals 44 Candidate Loci for Fruit Metabolic Traits. Plant Physiol. 2014, 165, 1120–1132. [Google Scholar] [CrossRef]
- Zhao, J.; Artemyeva, A.; del Carpio, D.P.; Basnet, R.K.; Zhang, N.; Gao, J.; Li, F.; Bucher, J.; Wang, X.; Visser, R.G.F.; et al. Design of a Brassica rapa core collection for association mapping studies. Genome 2010, 53, 884–898. [Google Scholar] [CrossRef]
- Zhang, P.; Zhu, Y.; Wang, L.; Chen, L.; Zhou, S. Mining candidate genes associated with powdery mildew resistance in cucumber via super-BSA by specific length amplified fragment (SLAF) sequencing. BMC Genom. 2015, 16, 1058. [Google Scholar] [CrossRef]
- Sakiroglu, M.; Brummer, E.C. Identification of loci controlling forage yield and nutritive value in diploid alfalfa using GBS-GWAS. Theor. Appl. Genet. 2017, 130, 261–268. [Google Scholar] [CrossRef]
- Nimmakayala, P.; Abburi, V.L.; Abburi, L.; Alaparthi, S.B.; Cantrell, R.; Park, M.; Choi, D.; Hankins, G.; Malkaram, S.; Reddy, U.K. Linkage disequilibrium and population-structure analysis among Capsicum annuum L. cultivars for use in association mapping. Mol. Genet. Genom. 2014, 289, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Nimmakayala, P.; Abburi, V.L.; Saminathan, T.; Alaparthi, S.B.; Almeida, A.; Davenport, B.; Nadimi, M.; Davidson, J.; Tonapi, K.; Yadav, L.; et al. Genome-wide Diversity and Association Mapping for Capsaicinoids and Fruit Weight in Capsicum annuum L. Sci. Rep. 2016, 6, 38081. [Google Scholar] [CrossRef] [PubMed]
- Nimmakayala, P.; Abburi, V.L.; Saminathan, T.; Almeida, A.; Davenport, B.; Davidson, J.; Reddy, C.V.C.M.; Hankins, G.; Ebert, A.; Choi, D.; et al. Genome-Wide Divergence and Linkage Disequilibrium Analyses for Capsicum baccatum Revealed by Genome-Anchored Single Nucleotide Polymorphisms. Front. Plant Sci. Sci. 2016, 7, 1646. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.-K.; Manivannan, A.; Karna, S.; Jun, T.-H.; Yang, E.-Y.; Choi, S.; Kim, J.-H.; Kim, D.-S.; Lee, E.-S. Whole Genome Resequencing of Capsicum baccatum and Capsicum annuum to Discover Single Nucleotide Polymorphism Related to Powdery Mildew Resistance. Sci. Rep. 2018, 8, 5188. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Gore, M.; Buckler, E.S.; Yu, J. Status and Prospects of Association Mapping in Plants. Plant Genome 2008, 1, 5–20. [Google Scholar] [CrossRef]
- Brachi, B.; Faure, N.; Horton, M.; Flahauw, E.; Vazquez, A.; Nordborg, M.; Bergelson, J.; Cuguen, J.; Roux, F. Linkage and Association Mapping of Arabidopsis thaliana Flowering Time in Nature. PLoS Genet. 2010, 6, e1000940. [Google Scholar] [CrossRef] [PubMed]
- Crowell, S.; Korniliev, P.; Falcao, A.; Ismail, A.; Gregorio, G.; Mezey, J.; McCouch, S. Genome-wide association and high-resolution phenotyping link Oryza sativa panicle traits to numerous trait-specific QTL clusters. Nat. Commun. 2016, 7, 10527. [Google Scholar] [CrossRef]
- Sallam, A.; Arbaoui, M.; El-Esawi, M.; Abshire, N.; Martsch, R. Identification and Verification of QTL Associated with Frost Tolerance Using Linkage Mapping and GWAS in Winter Faba Bean. Front. Plant Sci. 2016, 7, 1098. [Google Scholar] [CrossRef]
- Tian, F.; Bradbury, P.J.; Brown, P.J.; Hung, H.; Sun, Q.; Flint-Garcia, S.; Rocheford, T.R.; McMullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 2011, 43, 159. [Google Scholar] [CrossRef]
- Han, K.; Lee, H.-Y.; Ro, N.-Y.; Hur, O.-S.; Lee, J.-H.; Kwon, J.-K.; Kang, B.-C. QTL mapping and GWAS reveal candidate genes controlling capsaicinoid content in Capsicum. Plant Biotechnol. J. 2018, 16, 1546–1558. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, G.; Chen, B.; Du, H.; Zhang, F.; Zhang, H.; Wang, Q.; Geng, S. Candidate genes for first flower node identified in pepper using combined SLAF-seq and BSA. PLoS ONE 2018, 13, e0194071. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chao, J.; Cheng, X.; Wang, R.; Sun, B.; Wang, H.; Luo, S.; Xu, X.; Wu, T.; Li, Y. Mapping of a Novel Race Specific Resistance Gene to Phytophthora Root Rot of Pepper (Capsicum annuum) Using Bulked Segregant Analysis Combined with Specific Length Amplified Fragment Sequencing Strategy. PLoS ONE 2016, 11, e0151401. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Wang, S.; Liu, J.; Pan, B.; Diao, W.; Ge, W.; Gao, C.; Snyder, J.C. Rapid identification of QTLs underlying resistance to Cucumber mosaic virus in pepper (Capsicum frutescens). Theor. Appl. Genet. 2017, 130, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Liang, Z.; Yan, T.; Xu, Y.; Xuan, L.; Tang, J.; Zhou, G.; Lohwasser, U.; Hua, S.; Wang, H.; et al. Whole-genome resequencing of a worldwide collection of Rapeseed accessions reveals the genetic basis of ecotype divergence. Mol. Plant 2019, 12, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Wei, L.; Li, X.; Wang, Y.; Wu, J.; Liu, M.; Zhang, C.; Chen, Z.; Xiao, Z.; Jian, H.; et al. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 2019, 10, 1154. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Han, J.-H.; Kwon, J.-K.; Park, M.; Kim, B.-D.; Choi, D. Fine mapping of pepper trichome locus 1 controlling trichome formation in Capsicum annuum L. CM334. Theor. Appl. Genet. 2010, 120, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-R.; Kim, K.-T.; Kim, H.-J.; Han, J.-H.; Kim, J.-H.; Yeom, S.-I.; Kim, H.J.; Kang, W.-H.; Jinxia, S.; Park, S.-W.; et al. QTL analysis of fruit length using rRAMP, WRKY, and AFLP markers in chili pepper. Hortic. Environ. Biotechnol. 2011, 52, 602–613. [Google Scholar] [CrossRef]
- Lee, J.; Park, S.J.; Hong, S.C.; Han, J.-H.; Choi, D.; Yoon, J.B. QTL mapping for capsaicin and dihydrocapsaicin content in a population of Capsicum annuum ″NB1′xCapsicum chinense″ Bhut Jolokia′. Plant Breed. 2016, 135, 376–383. [Google Scholar] [CrossRef]
- Dwivedi, N.; Kumar, R.; Paliwal, R.; Kumar, U.; Kumar, S.; Singh, M.; Singh, R.K. QTL mapping for important horticultural traits in pepper (Capsicum annuum L.). J. Plant Biochem. Biotechnol. 2015, 24, 154–160. [Google Scholar] [CrossRef]
- Siddique, M.I.; Lee, H.-Y.; Ro, N.-Y.; Han, K.; Venkatesh, J.; Solomon, A.M.; Patil, A.S.; Changkwian, A.; Kwon, J.-K.; Kang, B.-C. Identifying candidate genes for Phytophthora capsici resistance in pepper (Capsicum annuum) via genotyping-by-sequencing-based QTL mapping and genome-wide association study. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Liu, H.; Wu, L.; Warburton, M.; Yan, J. Genome-wide Association Studies in Maize: Praise and Stargaze. Mol. Plant 2017, 10, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yan, W.; Agrama, H.; Jia, L.; Shen, X.; Jackson, A.; Moldenhauer, K.; Yeater, K.; McClung, A.; Wu, D. Mapping QTLs for improving grain yield using the USDA rice mini-core collection. Planta 2011, 234, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Huang, L.; Ren, X.; Chen, Y.; Zhou, X.; Xia, Y.; Huang, J.; Lei, Y.; Yan, L.; Wan, L.; et al. Diversity characterization and association analysis of agronomic traits in a Chinese peanut (Arachis hypogaea L.) mini-core collection. J. Integr. Plant Biol. 2014, 56, 159–169. [Google Scholar] [CrossRef]
- Xie, D.; Dai, Z.; Yang, Z.; Tang, Q.; Sun, J.; Yang, X.; Song, X.; Lu, Y.; Zhao, D.; Zhang, L.; et al. Genomic variations and association study of agronomic traits in flax. BMC Genom. 2018, 19, 512. [Google Scholar] [CrossRef]
- Wang, N. QTL Analysis and Effect of Different Cultivation Conditions on Restoration of Cytoplasmic Male Sterility in Capsicum. D; Chinese Academy of Agricultural Sciences: Beijing, China, 2015. (In Chinese) [Google Scholar]
- Chen, J. Development and Application of Genome-Wide SSR and SNP Markers in Pepper (Capsicum spp.), D; SouthChina Agricultural University: Guangzhou, China, 2016. (In Chinese) [Google Scholar]
- Chunthawodtiporn, J.; Hill, T.; Stoffel, K.; van Deynze, A. Quantitative Trait Loci Controlling Fruit Size and Other Horticultural Traits in Bell Pepper (Capsicum annuum). Plant Genome 2018, 11. [Google Scholar] [CrossRef]
- Thorup, T.A.; Tanyolac, B.; Livingstone, K.D.; Popovsky, S.; Paran, I.; Jahn, M. Candidate gene analysis of organ pigmentation loci in the Solanaceae. Proc. Natl. Acad. Sci. USA 2000, 97, 11192–11197. [Google Scholar] [CrossRef]
- Efrati, A.; Eyal, Y.; Paran, I. Molecular mapping of the chlorophyll retainer (cl) mutation in pepper (Capsicum spp.) and screening for candidate genes using tomato ESTs homologous to structural genes of the chlorophyll catabolism pathway. Genome 2005, 48, 347–351. [Google Scholar] [CrossRef]
- Lightbourn, G.J.; Griesbach, R.J.; Novotny, J.A.; Clevidence, B.A.; Rao, D.D.; Stommel, J.R. Effects of anthocyanin and carotenoid combinations on foliage and immature fruit color of Capsicum annuum L. J. Hered. 2008, 99, 105–111. [Google Scholar] [CrossRef]
- De Jong, W.S.; Eannetta, N.T.; de Jong, D.M.; Bodis, M. Candidate gene analysis of anthocyanin pigmentation loci in the Solanaceae. Theor. Appl. Genet. 2004, 108, 423–432. [Google Scholar] [CrossRef]
- Rinaldi, R.; van Deynze, A.; Portis, E.; Rotino, G.L.; Toppino, L.; Hill, T.; Ashrafi, H.; Barchi, L.; Lanteri, S. New Insights on Eggplant/Tomato/Pepper Synteny and Identification of Eggplant and Pepper Orthologous QTL. Front. Plant Sci. 2016, 7, 1031. [Google Scholar] [CrossRef] [PubMed]
- Borovsky, Y.; Paran, I. Characterization of fs10.1, a major QTL controlling fruit elongation in Capsicum. Theor. Appl. Genet. 2011, 123, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Ben Chaim, A.; Borovsky, Y.; Rao, G.U.; Tanyolac, B.; Paran, I. fs3.1, a major fruit shape QTL conserved in Capsicum. Genome 2003, 46, 1–9. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.R.; Rego, E.R.d.; Pessoa, A.M.D.S.; Rego, M.M.D. Correlation network analysis between phenotypic and genotypic traits of chili pepper. Pesqui. Agropecu. Bras. 2016, 51, 372–377. [Google Scholar] [CrossRef][Green Version]
- Olawuyi, O.J.; Jonathan, S.G.; Babatunde, F.E.; Babalola, B.J.; Yaya, O.O.S.; Agbolade, J.O.; Aina, D.A.; Egun, C.J. Accession * treatment interaction, variability and correlation studies of pepper (Capsicum spp.) under the influence of arbuscular mycorrhiza fungus (Glomus clarum) and cow dung. Am. J. Plant Sci. 2014, 5, 683–690. [Google Scholar] [CrossRef][Green Version]
- Epskamp, S.; Cramer, A.O.J.; Waldorp, L.J.; Schmittmann, V.D.; Borsboom, D. qgraph: Network Visualizations of Relationships in Psychometric Data. J. Stat. Softw. 2012, 48, 1–18. [Google Scholar] [CrossRef]
- Fulton, T.M.; Chunwongse, J.; Tanksley, S.D. Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol. Biol. Report. 1995, 13, 207–209. [Google Scholar] [CrossRef]
- Lee, J.-H.; An, J.-T.; Siddique, M.I.; Han, K.; Choi, S.; Kwon, J.-K.; Kang, B.-C. Identification and molecular genetic mapping of Chili veinal mottle virus (ChiVMV) resistance genes in pepper (Capsicum annuum). Mol. Breed. 2017, 37, 121. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An Efficient Method of Large-Scale De Novo SNP Discovery and Genotyping Using High-Throughput Sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; P. Genome Project Data. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6, Molecular Evolutionary Genetics Analysis Version 6. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Hardy, O.J.; Vekemans, X. SPAGEDi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Lippert, C.; Listgarten, J.; Liu, Y.; Kadie, C.M.; Davidson, R.I.; Heckerman, D. FaST linear mixed models for genome-wide association studies. Nat. Methods 2011, 8, 833–835. [Google Scholar] [CrossRef]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348. [Google Scholar] [CrossRef]
- Wang, P.; Lu, Q.; Ai, Y.; Wang, Y.; Li, T.; Wu, L.; Liu, J.; Cheng, Q.; Sun, L.; Shen, H. Candidate Gene Selection for Cytoplasmic Male Sterility in Pepper (Capsicum annuum L.) through Whole Mitochondrial Genome Sequencing. Int. J. Mol. Sci. 2019, 20, 578. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plant Organ | Trait | Model | Chromosome | Number of SNPs (p < 1.707 × 10−8) in Peak Region | Trait-Associated Peak Region | ||
|---|---|---|---|---|---|---|---|
| Start | End | Size (Mbp) | |||||
| Stem | Pt | FaST-LMM | Chr01 | 9 | 278425275 | 280417460 | 1.992185 |
| Chr04 | 2 | 23932585 | 23972683 | 0.040098 | |||
| Chr05 | 5 | 17744799 | 19221034 | 1.476235 | |||
| Chr06 | 10 | 5675663 | 5759294 | 0.083631 | |||
| Chr09 | 10 | 12510495 | 19373800 | 6.863305 | |||
| Chr09 | 14 | 118192571 | 119757960 | 1.565389 | |||
| Chr09 | 10 | 171029237 | 173134444 | 2.105207 | |||
| Chr11 | 3 | 37287718 | 40113909 | 2.826191 | |||
| Chr11 | 17 | 107259335 | 107501665 | 0.24233 | |||
| Bh | EMMAX | Chr01 | 59 | 275555035 | 277575138 | 2.020103 | |
| Chr02 | 6 | 83643192 | 85422044 | 1.778852 | |||
| Chr02 | 21 | 104238462 | 104524382 | 0.28592 | |||
| Chr03 | 13 | 253213778 | 254639900 | 1.426122 | |||
| Chr06 | 14 | 13971060 | 18672334 | 4.701274 | |||
| Chr07 | 9 | 5373860 | 7707856 | 2.333996 | |||
| Chr07 | 35 | 184612786 | 184825374 | 0.212588 | |||
| Chr09 | 10 | 191839433 | 200002779 | 8.163346 | |||
| Msc | FaST-LMM | Chr06 | 2 | 20231877 | 20231907 | 0.00003 | |
| Chr08 | 6 | 114388151 | 114431559 | 0.043408 | |||
| Chr09 | 2 | 214330476 | 214330504 | 0.000028 | |||
| Msp | EMMAX | Chr11 | 28 | 27131483 | 28706358 | 1.574875 | |
| Leaf | Ls | GLM | - | - | - | - | - |
| Lc | GLM | - | - | - | - | - | |
| Lm | GLM | Chr01 | 8 | 158420891 | 158729605 | 0.308714 | |
| Chr04 | 7 | 58610574 | 59222198 | 0.611624 | |||
| Chr06 | 7 | 212371837 | 212424874 | 0.053037 | |||
| Chr08 | 7 | 131427437 | 134733324 | 3.305887 | |||
| Chr12 | 4 | 12354265 | 19260025 | 6.90576 | |||
| Lp | EMMAX | Chr10 | 9 | 162170912 | 162221229 | 0.050317 | |
| Chr10 | 4 | 197927729 | 200867123 | 2.939394 | |||
| Chr12 | 3 | 22681366 | 22692847 | 0.011481 | |||
| Lsc | GLM | - | - | - | - | - | |
| Flower | Cc | EMMAX | Chr02 | 4 | 68060203 | 68060282 | 0.000079 |
| Chr04 | 6 | 189062369 | 192962061 | 3.899692 | |||
| Chr06 | 8 | 41630661 | 41857768 | 0.227107 | |||
| Chr07 | 2 | 214559213 | 214585881 | 0.026668 | |||
| Chr07 | 6 | 220688885 | 220941545 | 0.25266 | |||
| Chr08 | 8 | 135096972 | 135952487 | 0.855515 | |||
| Chr09 | 5 | 201471658 | 202362543 | 0.890885 | |||
| Chr11 | 5 | 28177723 | 28191389 | 0.013666 | |||
| Chr11 | 6 | 214289893 | 216304968 | 2.015075 | |||
| Sc | GLM | Chr01 | 3 | 211183454 | 211183884 | 0.00043 | |
| Chr10 | 24 | 155035157 | 155097491 | 0.062334 | |||
| Chr11 | 114 | 27555236 | 29305711 | 1.750475 | |||
| Ac | FaST-LMM | Chr01 | 17 | 246812579 | 249477491 | 2.664912 | |
| Chr07 | 2 | 130948617 | 130973590 | 0.024973 | |||
| Ffn | FaST-LMM | - | - | - | - | - | |
| Nfpa | EMMAX | Chr01 | 3 | 220458255 | 220462790 | 0.004535 | |
| Chr01 | 7 | 280054497 | 280417460 | 0.362963 | |||
| Chr04 | 3 | 57766474 | 58558409 | 0.791935 | |||
| Chr05 | 2 | 206654389 | 206654579 | 0.00019 | |||
| Chr06 | 10 | 13971060 | 19735646 | 5.764586 | |||
| Chr06 | 39 | 219098248 | 219470017 | 0.371769 | |||
| Chr07 | 3 | 185249295 | 185271463 | 0.022168 | |||
| Chr09 | 22 | 10463866 | 12820022 | 2.356156 | |||
| Chr09 | 12 | 118192571 | 119641888 | 1.449317 | |||
| Chr10 | 7 | 29037048 | 33183732 | 4.146684 | |||
| Chr11 | 5 | 209423789 | 210867712 | 1.443923 | |||
| Chr12 | 20 | 192018036 | 193789860 | 1.771824 | |||
| Fpgs | EMMAX | Chr12 | 23 | 32930746 | 38073141 | 5.142395 | |
| Ms | FaST-LMM | Chr06 | 101 | 214443037 | 215536517 | 1.09348 | |
| Fruit | Aif | EMMAX | Chr08 | 4 | 130745390 | 131501395 | 0.756005 |
| Chr09 | 6 | 104816188 | 104816287 | 0.000099 | |||
| Chr09 | 5 | 167226310 | 167226390 | 0.00008 | |||
| Chr10 | 86 | 148782468 | 155625398 | 6.84293 | |||
| Fsf | EMMAX | - | - | - | - | - | |
| Fg | FaST-LMM | Chr07 | 8 | 6280167 | 6685034 | 0.404867 | |
| Chr09 | 3 | 39263 | 1201526 | 1.162263 | |||
| Chr12 | 8 | 180112718 | 186338088 | 6.22537 | |||
| Fsc | EMMAX | - | - | - | - | - | |
| Fss | FaST-LMM | Chr01 | 3 | 202642462 | 203121398 | 0.478936 | |
| Chr04 | 2 | 15454448 | 15454730 | 0.000282 | |||
| Chr09 | 4 | 94667918 | 94693832 | 0.025914 | |||
| Chr09 | 8 | 126292337 | 127360615 | 1.068278 | |||
| Chr11 | 9 | 30762829 | 49631336 | 18.868507 | |||
| Fas | FaST-LMM | - | - | - | - | - | |
| Ab | FaST-LMM | - | - | - | - | - | |
| Mfc | EMMAX | Chr06 | 17 | 3758563 | 10060744 | 6.302181 | |
| St | FaST-LMM | Chr04 | 2 | 3047655 | 3280037 | 0.232382 | |
| Chr05 | 2 | 54372525 | 54372671 | 0.000146 | |||
| Chr06 | 3 | 40301536 | 40301894 | 0.000358 | |||
| Chr10 | 3 | 201908626 | 203356884 | 1.448258 | |||
| Chr12 | 3 | 6307012 | 12354265 | 6.047253 | |||
| Chr12 | 45 | 35222952 | 35714999 | 0.492047 | |||
| Fpl | FaST-LMM | - | - | - | - | - | |
| Pw | FaST-LMM | Chr08 | 4 | 133185108 | 133433188 | 0.24808 | |
| Chr12 | 203552366 | 206591342 | 3.038976 | ||||
| Pl | FaST-LMM | - | - | - | - | - | |
| Psi | FaST-LMM | - | - | - | - | - | |
| Fw | FaST-LMM | Chr01 | 4 | 103860245 | 105083614 | 1.223369 | |
| Chr08 | 22 | 133045048 | 133584887 | 0.539839 | |||
| Chr12 | 9 | 201671262 | 206591342 | 4.92008 | |||
| Fl | FaST-LMM | - | - | - | - | - | |
| Fsi | EMMAX | - | - | - | - | - | |
| Wpf | FaST-LMM | Chr01 | 7 | 103860245 | 105083614 | 1.223369 | |
| Chr05 | 2 | 168537584 | 168540175 | 0.002591 | |||
| Chr08 | 4 | 133396929 | 133433188 | 0.036259 | |||
| Chr12 | 14 | 200798023 | 211814051 | 11.016028 | |||
| Dmc | FaST-LMM | Chr02 | 7 | 69956806 | 79414868 | 9.458062 | |
| Chr11 | 5 | 85204434 | 85204791 | 0.000357 | |||
| Tf | FaST-LMM | Chr01 | 3 | 103860245 | 105083388 | 1.223143 | |
| Chr08 | 3 | 126042291 | 133396929 | 7.354638 | |||
| Chr11 | 5 | 128979925 | 128979983 | 0.000058 | |||
| Chr12 | 8 | 200798023 | 207472045 | 6.674022 | |||
| Nol | FaST-LMM | - | - | - | - | - | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, L.; Wang, P.; Wang, Y.; Cheng, Q.; Lu, Q.; Liu, J.; Li, T.; Ai, Y.; Yang, W.; Sun, L.; et al. Genome-Wide Correlation of 36 Agronomic Traits in the 287 Pepper (Capsicum) Accessions Obtained from the SLAF-seq-Based GWAS. Int. J. Mol. Sci. 2019, 20, 5675. https://doi.org/10.3390/ijms20225675
Wu L, Wang P, Wang Y, Cheng Q, Lu Q, Liu J, Li T, Ai Y, Yang W, Sun L, et al. Genome-Wide Correlation of 36 Agronomic Traits in the 287 Pepper (Capsicum) Accessions Obtained from the SLAF-seq-Based GWAS. International Journal of Molecular Sciences. 2019; 20(22):5675. https://doi.org/10.3390/ijms20225675
Chicago/Turabian StyleWu, Lang, Peng Wang, Yihao Wang, Qing Cheng, Qiaohua Lu, Jinqiu Liu, Ting Li, Yixin Ai, Wencai Yang, Liang Sun, and et al. 2019. "Genome-Wide Correlation of 36 Agronomic Traits in the 287 Pepper (Capsicum) Accessions Obtained from the SLAF-seq-Based GWAS" International Journal of Molecular Sciences 20, no. 22: 5675. https://doi.org/10.3390/ijms20225675
APA StyleWu, L., Wang, P., Wang, Y., Cheng, Q., Lu, Q., Liu, J., Li, T., Ai, Y., Yang, W., Sun, L., & Shen, H. (2019). Genome-Wide Correlation of 36 Agronomic Traits in the 287 Pepper (Capsicum) Accessions Obtained from the SLAF-seq-Based GWAS. International Journal of Molecular Sciences, 20(22), 5675. https://doi.org/10.3390/ijms20225675