Claudin-2: Roles beyond Permeability Functions


Abstract
1. Introduction
Claudin-2 and the Claudin Family of Tight Junction Proteins
2. Properties of Claudin-2
2.1. Expression
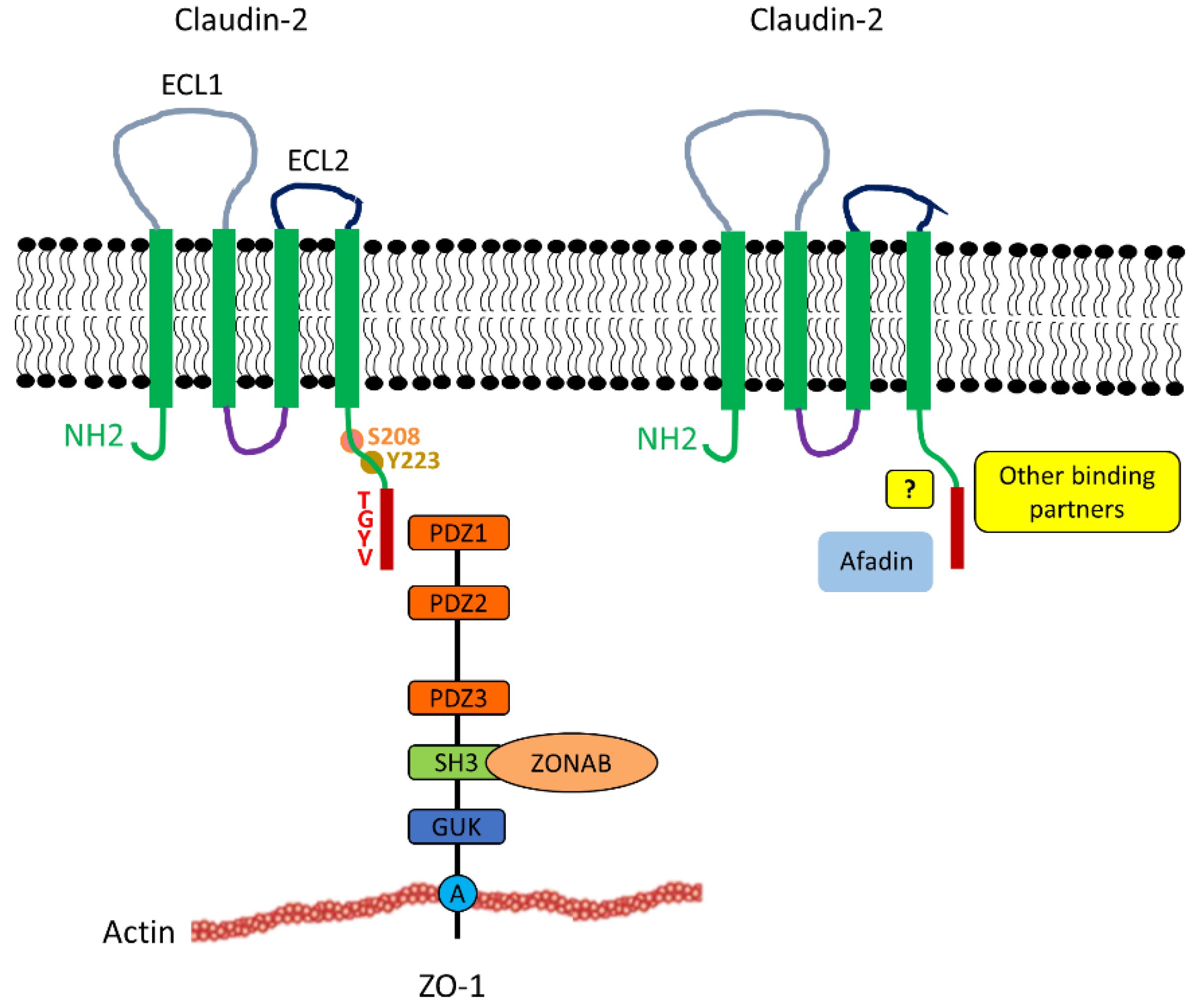
2.2. Structure and Interactions
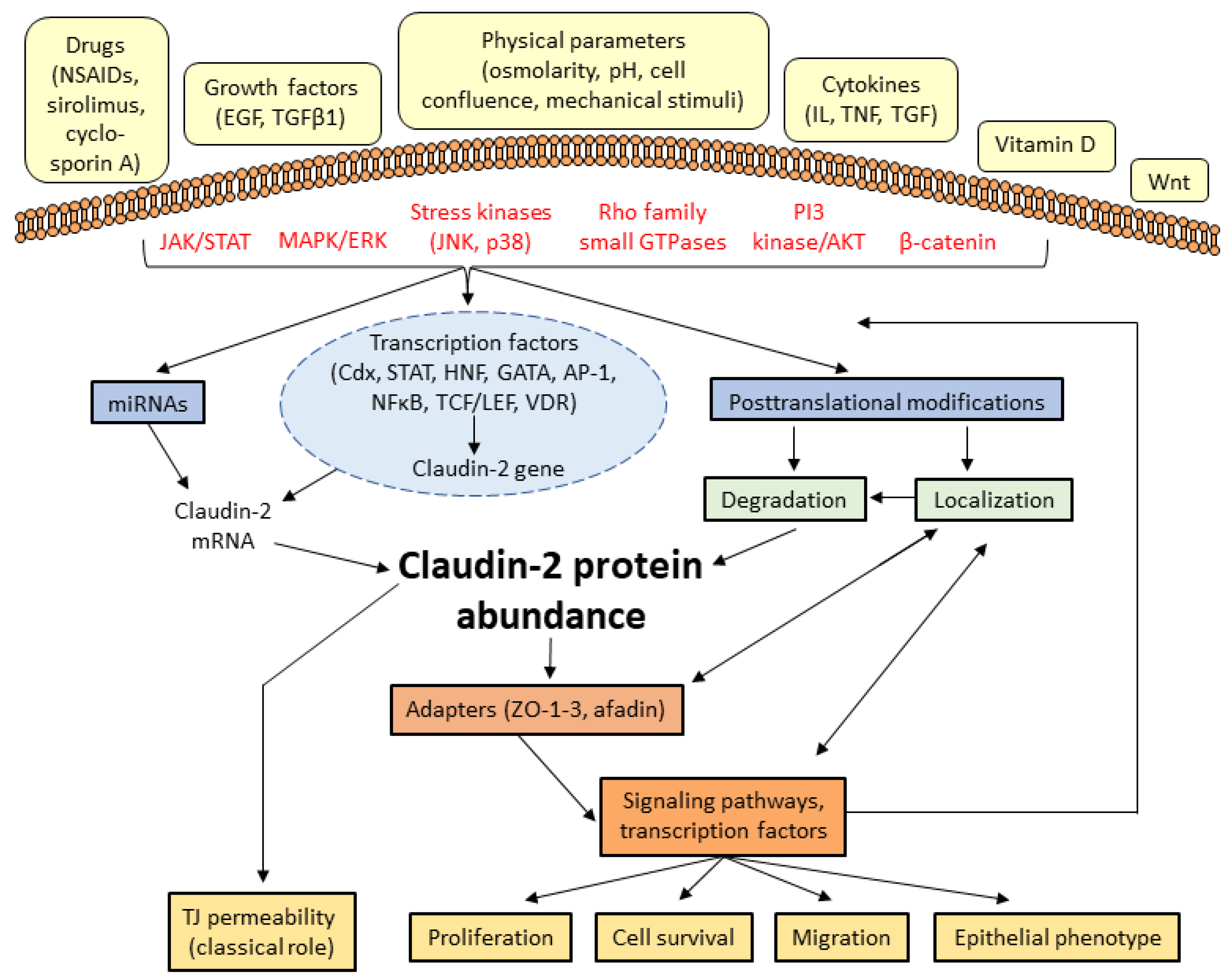
3. Regulation of Claudin-2
3.1. Context-Dependent Regulation of Claudin-2
3.2. Signal Transduction Pathways Regulating Claudin-2 Expression
3.3. Transcription Factors controlling claudin-2 expression
3.4. Claudin-2 Turnover, Trafficking, and Posttranslational Modifications
4. Functions of Claudin-2
4.1. Permeability Functions
4.2. Role in Proliferation
4.3. Migration
4.4. Signal-Modulating Effects of Claudin-2: Emerging Mechanisms Underlying Roles in Biological Processes
5. Claudin-2 in Diseases
5.1. Claudin-2 in Cancer and Metastasis Formation
5.2. Claudin-2 in Gut Inflammation
5.3. Claudin-2 in Kidney Disease
5.4. Development of Therapies Targeting Claudin-2
6. Open Questions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CagA | CagA: cytotoxin-associated gene |
| Cdx | Caudal-related homeobox |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial-mesenchymal transition |
| HNF | hepatocyte nuclear factor |
| IBD | inflammatory bowel disease |
| IL | Interleukin |
| MMP | matrix metalloprotease |
| TGFβ1 | transforming growth factor β1 |
| TNF | tumor necrosis factor |
| PDZ | postsynaptic density, disc-large, ZO-1 |
| TCF/LEF | T cell factor/lymphoid enhancer factor |
| ZONAB | Zonula occludens 1-associated nucleic acid-binding protein |
| TJ | tight junctions |
| ZO | zonula occludens |
References
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Anderson, J.M. Architecture of tight junctions and principles of molecular composition. Semin. Cell Dev. Biol. 2014, 36, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Weber, C.R.; Raleigh, D.R.; Yu, D.; Turner, J.R. Tight junction pore and leak pathways: A dynamic duo. Annu. Rev. Physiol. 2011, 73, 283–309. [Google Scholar] [CrossRef] [PubMed]
- Gunzel, D.; Fromm, M. Claudins and other tight junction proteins. Compr. Physiol. 2012, 2, 1819–1852. [Google Scholar] [PubMed]
- Tsukita, S.; Tanaka, H.; Tamura, A. The Claudins: From Tight Junctions to Biological Systems. Trends Biochem. Sci. 2019, 44, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Mineta, K.; Yamamoto, Y.; Yamazaki, Y.; Tanaka, H.; Tada, Y.; Saito, K.; Tamura, A.; Igarashi, M.; Endo, T.; Takeuchi, K.; et al. Predicted expansion of the claudin multigene family. FEBS Lett. 2011, 585, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Lal-Nag, M.; Morin, P.J. The claudins. Genome Biol. 2009, 10, 235. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Helftenbein, G.; Koslowski, M.; Sahin, U.; Tureci, O. Identification of new claudin family members by a novel PSI-BLAST based approach with enhanced specificity. Proteins 2006, 65, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M. Molecular basis of the core structure of tight junctions. Cold Spring Harb. Perspect. Biol. 2010, 2, a002907. [Google Scholar] [CrossRef] [PubMed]
- Krause, G.; Winkler, L.; Mueller, S.L.; Haseloff, R.F.; Piontek, J.; Blasig, I.E. Structure and function of claudins. Biochim. Biophys. Acta 2008, 1778, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Capaldo, C.T.; Nusrat, A. Claudin switching: Physiological plasticity of the Tight Junction. Semin. Cell Dev. Biol. 2015, 42, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Sasaki, H.; Fujimoto, K.; Tsukita, S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J. Cell Biol. 1998, 143, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, T.; Gu, X.; Golden, H.M.; Suh, E.; Rhoads, D.B.; Reinecker, H.C. Cloning of the human claudin-2 5’-flanking region revealed a TATA-less promoter with conserved binding sites in mouse and human for caudal-related homeodomain proteins and hepatocyte nuclear factor-1alpha. J. Biol. Chem 2002, 277, 21361–21370. [Google Scholar] [CrossRef] [PubMed]
- Gunzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.S.; Cheng, M.H.; Angelow, S.; Gunzel, D.; Kanzawa, S.A.; Schneeberger, E.E.; Fromm, M.; Coalson, R.D. Molecular basis for cation selectivity in claudin-2-based paracellular pores: Identification of an electrostatic interaction site. J. Gen. Physiol. 2009, 133, 111–127. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Furuse, K.; Sasaki, H.; Tsukita, S. Conversion of zonulae occludentes from tight to leaky strand type by introducing claudin-2 into Madin-Darby canine kidney I cells. J. Cell Biol. 2001, 153, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Amasheh, S.; Meiri, N.; Gitter, A.H.; Schoneberg, T.; Mankertz, J.; Schulzke, J.D.; Fromm, M. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J. Cell Sci. 2002, 115, 4969–4976. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Gomes, A.S.; Paul, D.L.; Goodenough, D.A. Study of claudin function by RNA interference. J. Biol. Chem. 2006, 281, 36117–36123. [Google Scholar] [CrossRef] [PubMed]
- Gupta, I.R.; Ryan, A.K. Claudins: Unlocking the code to tight junction function during embryogenesis and in disease. Clin. Genet. 2010, 77, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J. Emerging roles of claudins in human cancer. Int. J. Mol. Sci. 2013, 14, 18148–18180. [Google Scholar] [CrossRef] [PubMed]
- Hagen, S.J. Non-canonical functions of claudin proteins: Beyond the regulation of cell-cell adhesions. Tissue Barriers 2017, 5, e1327839. [Google Scholar] [CrossRef] [PubMed]
- Escaffit, F.; Boudreau, F.; Beaulieu, J.F. Differential expression of claudin-2 along the human intestine: Implication of GATA-4 in the maintenance of claudin-2 in differentiating cells. J. Cell Physiol. 2005, 203, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Aung, P.P.; Mitani, Y.; Sanada, Y.; Nakayama, H.; Matsusaki, K.; Yasui, W. Differential expression of claudin-2 in normal human tissues and gastrointestinal carcinomas. Virchows Arch. 2006, 448, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Markov, A.G.; Veshnyakova, A.; Fromm, M.; Amasheh, M.; Amasheh, S. Segmental expression of claudin proteins correlates with tight junction barrier properties in rat intestine. J. Comp. Physiol. B 2010, 180, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Lippoldt, A.; Liebner, S.; Andbjer, B.; Kalbacher, H.; Wolburg, H.; Haller, H.; Fuxe, K. Organization of choroid plexus epithelial and endothelial cell tight junctions and regulation of claudin-1, -2 and -5 expression by protein kinase C. Neuroreport 2000, 11, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Li, D.; Wang, X.; Zeng, Y.; Yan, Y.; Yang, L. Claudin-2 downregulation by KSHV infection is involved in the regulation of endothelial barrier function. J. Cutan Pathol. 2014, 41, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; Laoui, D.; Morias, Y.; Movahedi, K.; Raes, G.; De Baetselier, P.; Van Ginderachter, J.A. Claudin-1, claudin-2 and claudin-11 genes differentially associate with distinct types of anti-inflammatory macrophages in vitro and with parasite- and tumour-elicited macrophages in vivo. Scand J. Immunol. 2012, 75, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Van Itallie, C.; Rahner, C.; Anderson, J.M. Claudin extracellular domains determine paracellular charge selectivity and resistance but not tight junction fibril architecture. Am. J. Physiol. Cell Physiol. 2003, 284, C1346–C1354. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Van Itallie, C.M.; McCrea, H.J.; Rahner, C.; Anderson, J.M. Claudins create charge-selective channels in the paracellular pathway between epithelial cells. Am. J. Physiol. Cell Physiol. 2002, 283, C142–C147. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhuo, M.; Pei, L.; Rajagopal, M.; Yu, A.S. Comprehensive cysteine-scanning mutagenesis reveals Claudin-2 pore-lining residues with different intrapore locations. J. Biol. Chem 2014, 289, 6475–6484. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.R.; Liang, G.H.; Wang, Y.; Das, S.; Shen, L.; Yu, A.S.; Nelson, D.J.; Turner, J.R. Claudin-2-dependent paracellular channels are dynamically gated. Elife 2015, 4, e09906. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Angelow, S.; Linge, A.; Zhuo, M.; Yu, A.S. Claudin-2 pore function requires an intramolecular disulfide bond between two conserved extracellular cysteines. Am. J. Physiol. Cell Physiol. 2013, 305, C190–C196. [Google Scholar] [CrossRef] [PubMed]
- Tabaries, S.; McNulty, A.; Ouellet, V.; Annis, M.G.; Dessureault, M.; Vinette, M.; Hachem, Y.; Lavoie, B.; Omeroglu, A.; Simon, H.G.; et al. Afadin cooperates with Claudin-2 to promote breast cancer metastasis. Genes Dev. 2019, 33, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Nomme, J.; Antanasijevic, A.; Caffrey, M.; Van Itallie, C.M.; Anderson, J.M.; Fanning, A.S.; Lavie, A. Structural Basis of a Key Factor Regulating the Affinity between the Zonula Occludens First PDZ Domain and Claudins. J. Biol. Chem. 2015, 290, 16595–16606. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Mitic, L.L.; Anderson, J.M. Claudin-2 forms homodimers and is a component of a high molecular weight protein complex. J. Biol. Chem. 2011, 286, 3442–3450. [Google Scholar] [CrossRef] [PubMed]
- Tabaries, S.; Dupuy, F.; Dong, Z.; Monast, A.; Annis, M.G.; Spicer, J.; Ferri, L.E.; Omeroglu, A.; Basik, M.; Amir, E.; et al. Claudin-2 promotes breast cancer liver metastasis by facilitating tumor cell interactions with hepatocytes. Mol. Cell Biol. 2012, 32, 2979–2991. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.S.; Vedula, S.R.; Hunziker, W.; Lim, C.T. Kinetics of adhesion mediated by extracellular loops of claudin-2 as revealed by single-molecule force spectroscopy. J. Mol. Biol. 2008, 381, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Piontek, J.; Winkler, L.; Wolburg, H.; Muller, S.L.; Zuleger, N.; Piehl, C.; Wiesner, B.; Krause, G.; Blasig, I.E. Formation of tight junction: Determinants of homophilic interaction between classic claudins. FASEB J. 2008, 22, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Taga, S.; Watanabe, R.; Sato, T.; Shimobaba, S.; Sonoki, H.; Endo, S.; Matsunaga, T.; Sakai, H.; Yamaguchi, M.; et al. Clathrin-dependent endocytosis of claudin-2 by DFYSP peptide causes lysosomal damage in lung adenocarcinoma A549 cells. Biochim. Biophys. Acta 2015, 1848, 2326–2336. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Sasaki, H.; Tsukita, S. Manner of interaction of heterogeneous claudin species within and between tight junction strands. J. Cell Biol. 1999, 147, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Capaldo, C.T.; Farkas, A.E.; Hilgarth, R.S.; Krug, S.M.; Wolf, M.F.; Benedik, J.K.; Fromm, M.; Koval, M.; Parkos, C.; Nusrat, A. Proinflammatory cytokine-induced tight junction remodeling through dynamic self-assembly of claudins. Mol. Biol. Cell 2014, 25, 2710–2719. [Google Scholar] [CrossRef] [PubMed]
- Angelow, S.; Schneeberger, E.E.; Yu, A.S. Claudin-8 expression in renal epithelial cells augments the paracellular barrier by replacing endogenous claudin-2. J. Membr Biol. 2007, 215, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yeh, S.; Appleton, B.A.; Held, H.A.; Kausalya, P.J.; Phua, D.C.; Wong, W.L.; Lasky, L.A.; Wiesmann, C.; Hunziker, W.; et al. Convergent and divergent ligand specificity among PDZ domains of the LAP and zonula occludens (ZO) families. J. Biol. Chem. 2006, 281, 22299–22311. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Furuse, M.; Morita, K.; Kubota, K.; Saitou, M.; Tsukita, S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J. Cell Biol. 1999, 147, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Tietgens, A.J.; LoGrande, K.; Aponte, A.; Gucek, M.; Anderson, J.M. Phosphorylation of claudin-2 on serine 208 promotes membrane retention and reduces trafficking to lysosomes. J. Cell Sci. 2012, 125, 4902–4912. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, K.; Rai, T.; Kobayashi, K.; Sohara, E.; Suzuki, T.; Itoh, T.; Suda, S.; Hayama, A.; Sasaki, S.; Uchida, S. Disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins. Proc. Natl. Acad. Sci. USA 2004, 101, 4690–4694. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Matsuo, Y.; Matsunaga, T.; Endo, S.; Sakai, H.; Yamaguchi, M.; Yamazaki, Y.; Sugatani, J.; Ikari, A. Hypotonic Stress-Induced Down-Regulation of Claudin-1 and -2 Mediated by Dephosphorylation and Clathrin-Dependent Endocytosis in Renal Tubular Epithelial Cells. J. Biol. Chem. 2016, 291, 24787–24799. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Mitic, L.L.; Anderson, J.M. SUMOylation of claudin-2. Ann. N. Y. Acad. Sci. 2012, 1258, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Molina-Jijon, E.; Rodriguez-Munoz, R.; Namorado Mdel, C.; Pedraza-Chaverri, J.; Reyes, J.L. Oxidative stress induces claudin-2 nitration in experimental type 1 diabetic nephropathy. Free Radic. Biol. Med. 2014, 72, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, P.; Ahmad, R.; Chaturvedi, R.; Smith, J.J.; Midha, R.; Mittal, M.K.; Krishnan, M.; Chen, X.; Eschrich, S.; Yeatman, T.J.; et al. Claudin-2 expression increases tumorigenicity of colon cancer cells: Role of epidermal growth factor receptor activation. Oncogene 2011, 30, 3234–3247. [Google Scholar] [CrossRef] [PubMed]
- Amoozadeh, Y.; Anwer, S.; Dan, Q.; Venugopal, S.; Shi, Y.; Branchard, E.; Liedtke, E.; Ailenberg, M.; Rotstein, O.D.; Kapus, A.; et al. Cell confluence regulates claudin-2 expression: Possible role for ZO-1 and Rac. Am. J. Physiol. Cell Physiol. 2018, 314, C366–C378. [Google Scholar] [CrossRef] [PubMed]
- Amoozadeh, Y.; Dan, Q.; Xiao, J.; Waheed, F.; Szaszi, K. Tumor necrosis factor-alpha induces a biphasic change in claudin-2 expression in tubular epithelial cells: Role in barrier functions. Am. J. Physiol. Cell Physiol. 2015, 309, C38–C50. [Google Scholar] [CrossRef] [PubMed]
- Sonoki, H.; Sato, T.; Endo, S.; Matsunaga, T.; Yamaguchi, M.; Yamazaki, Y.; Sugatani, J.; Ikari, A. Quercetin Decreases Claudin-2 Expression Mediated by Up-Regulation of microRNA miR-16 in Lung Adenocarcinoma A549 Cells. Nutrients 2015, 7, 4578–4592. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yoshinaga, N.; Tanabe, S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J. Biol. Chem. 2011, 286, 31263–31271. [Google Scholar] [CrossRef] [PubMed]
- Mankertz, J.; Amasheh, M.; Krug, S.M.; Fromm, A.; Amasheh, S.; Hillenbrand, B.; Tavalali, S.; Fromm, M.; Schulzke, J.D. TNFalpha up-regulates claudin-2 expression in epithelial HT-29/B6 cells via phosphatidylinositol-3-kinase signaling. Cell Tissue Res. 2009, 336, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.B.; Harris, R.C. Epidermal growth factor receptor activation differentially regulates claudin expression and enhances transepithelial resistance in Madin-Darby canine kidney cells. J. Biol. Chem. 2004, 279, 3543–3552. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Takiguchi, A.; Atomi, K.; Sugatani, J. Epidermal growth factor increases clathrin-dependent endocytosis and degradation of claudin-2 protein in MDCK II cells. J. Cell Physiol. 2011, 226, 2448–2456. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.I.; Poulin, E.J.; Blask, E.; Bukhalid, R.; Whitehead, R.H.; Franklin, J.L.; Coffey, R.J. Myofibroblast keratinocyte growth factor reduces tight junctional integrity and increases claudin-2 levels in polarized Caco-2 cells. Growth Factors 2012, 30, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Sato, T.; Watanabe, R.; Yamazaki, Y.; Sugatani, J. Increase in claudin-2 expression by an EGFR/MEK/ERK/c-Fos pathway in lung adenocarcinoma A549 cells. Biochim. Biophys. Acta 2012, 1823, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Lipschutz, J.H.; Li, S.; Arisco, A.; Balkovetz, D.F. Extracellular signal-regulated kinases 1/2 control claudin-2 expression in Madin-Darby canine kidney strain I and II cells. J. Biol. Chem. 2005, 280, 3780–3788. [Google Scholar] [CrossRef] [PubMed]
- Martin-Martin, N.; Ryan, G.; McMorrow, T.; Ryan, M.P. Sirolimus and cyclosporine A alter barrier function in renal proximal tubular cells through stimulation of ERK1/2 signaling and claudin-1 expression. Am. J. Physiol. Renal Physiol. 2010, 298, F672–F682. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Kumar, B.; Pan, K.; Dhawan, P.; Singh, A.B. HDAC-4 regulates claudin-2 expression in EGFR-ERK1/2 dependent manner to regulate colonic epithelial cell differentiation. Oncotarget 2017, 8, 87718–87736. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hichino, A.; Okamoto, M.; Taga, S.; Akizuki, R.; Endo, S.; Matsunaga, T.; Ikari, A. Down-regulation of Claudin-2 Expression and Proliferation by Epigenetic Inhibitors in Human Lung Adenocarcinoma A549 Cells. J. Biol. Chem. 2017, 292, 2411–2421. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mumm, J.B.; Herbst, R.; Kolbeck, R.; Wang, Y. IL-22 Increases Permeability of Intestinal Epithelial Tight Junctions by Enhancing Claudin-2 Expression. J. Immunol. 2017, 199, 3316–3325. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Li, Y.; Zhang, J.; Chang, R.; Li, J.; Huo, L. IL-9 promotes the pathogenesis of ulcerative colitis through STAT3/SOCS3 signaling. Biosci. Rep. 2018, 38, 9476–9487. [Google Scholar] [CrossRef] [PubMed]
- Dan, Q.; Shi, Y.; Rabani, R.; Venugopal, S.; Xiao, J.; Anwer, S.; Ding, M.; Speight, P.; Pan, W.; Alexander, R.T.; et al. Claudin-2 suppresses GEF-H1, RHOA, and MRTF thereby impacting proliferation and profibrotic phenotype of tubular cells. J. Biol. Chem. 2019, 294, 15446–15465. [Google Scholar] [CrossRef] [PubMed]
- Tabaries, S.; Annis, M.G.; Hsu, B.E.; Tam, C.E.; Savage, P.; Park, M.; Siegel, P.M. Lyn modulates Claudin-2 expression and is a therapeutic target for breast cancer liver metastasis. Oncotarget 2015, 6, 9476–9487. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Hernandez, V.; Flores-Maldonado, C.; Rincon-Heredia, R.; Verdejo-Torres, O.; Bonilla-Delgado, J.; Meneses-Morales, I.; Gariglio, P.; Contreras, R.G. EGF regulates claudin-2 and -4 expression through Src and STAT3 in MDCK cells. J. Cell Physiol. 2015, 230, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Fujii, N.; Hahakabe, S.; Hayashi, H.; Yamaguchi, M.; Yamazaki, Y.; Endo, S.; Matsunaga, T.; Sugatani, J. Hyperosmolarity-Induced Down-Regulation of Claudin-2 Mediated by Decrease in PKCbeta-Dependent GATA-2 in MDCK Cells. J. Cell Physiol. 2015, 230, 2776–2787. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Dalgalan, D.; Mandell, E.K.; Parker, S.S.; Ghosh, S.; Wilson, J.M. PKCiota interacts with Rab14 and modulates epithelial barrier function through regulation of claudin-2 levels. Mol. Biol. Cell 2015, 26, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadi, R.; Ye, D.; Boivin, M.; Guo, S.; Hashimi, M.; Ereifej, L.; Ma, T.Y. Interleukin-6 modulation of intestinal epithelial tight junction permeability is mediated by JNK pathway activation of claudin-2 gene. PLoS ONE 2014, 9, e85345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.G.; Wu, S.; Xia, Y.; Sun, J. Salmonella infection upregulates the leaky protein claudin-2 in intestinal epithelial cells. PLoS ONE 2013, 8, e58606. [Google Scholar] [CrossRef] [PubMed]
- Mankertz, J.; Hillenbrand, B.; Tavalali, S.; Huber, O.; Fromm, M.; Schulzke, J.D. Functional crosstalk between Wnt signaling and Cdx-related transcriptional activation in the regulation of the claudin-2 promoter activity. Biochem. Biophys. Res. Commun. 2004, 314, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, L.; Citi, S. Cingulin regulates claudin-2 expression and cell proliferation through the small GTPase RhoA. Mol. Biol. Cell 2006, 17, 3569–3577. [Google Scholar] [CrossRef] [PubMed]
- Bruewer, M.; Hopkins, A.M.; Hobert, M.E.; Nusrat, A.; Madara, J.L. RhoA, Rac1, and Cdc42 exert distinct effects on epithelial barrier via selective structural and biochemical modulation of junctional proteins and F-actin. Am. J. Physiol. Cell Physiol. 2004, 287, C327–C335. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.B.; Shi, Q.; Li, G.; Zheng, J.H.; Lin, J.; Qiu, W. MicroRNA-488 inhibits progression of colorectal cancer via inhibition of the mitogen-activated protein kinase pathway by targeting claudin-2. Am. J. Physiol. Cell Physiol. 2019, 316, C33–C47. [Google Scholar] [CrossRef] [PubMed]
- Che, M.; Shi, T.; Feng, S.; Li, H.; Zhang, X.; Feng, N.; Lou, W.; Dou, J.; Tang, G.; Huang, C.; et al. The MicroRNA-199a/214 Cluster Targets E-Cadherin and Claudin-2 and Promotes High Glucose-Induced Peritoneal Fibrosis. J. Am. Soc. Nephrol. 2017, 28, 2459–2471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.G.; Wu, S.; Lu, R.; Zhou, D.; Zhou, J.; Carmeliet, G.; Petrof, E.; Claud, E.C.; Sun, J. Tight junction CLDN2 gene is a direct target of the vitamin D receptor. Sci. Rep. 2015, 5, 10642. [Google Scholar] [CrossRef] [PubMed]
- Lepage, D.; Belanger, E.; Jones, C.; Tremblay, S.; Allaire, J.M.; Bruneau, J.; Asselin, C.; Perreault, N.; Menendez, A.; Gendron, F.P.; et al. Gata4 is critical to maintain gut barrier function and mucosal integrity following epithelial injury. Sci. Rep. 2016, 6, 36776. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.J.; Suh, E.R.; Lynch, J.P. The role of Cdx proteins in intestinal development and cancer. Cancer Biol. Ther. 2004, 3, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Chen, H.X.; Wang, X.Y.; Deng, X.Y.; Xi, Y.X.; He, Q.; Peng, T.L.; Chen, J.; Chen, W.; Wong, B.C.; et al. pylori-encoded CagA disrupts tight junctions and induces invasiveness of AGS gastric carcinoma cells via Cdx2-dependent targeting of Claudin-2. Cell Immunol. 2013, 286, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Mutoh, H.; Sugano, K. Expression of Claudin-2 in intestinal metaplastic mucosa of Cdx2-transgenic mouse stomach. Scand J. Gastroenterol. 2010, 45, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, S.W.; Zhu, J.; Zuo, S.; Ma, Y.Y.; Chen, Z.Y.; Zhang, J.L.; Chen, G.W.; Liu, Y.C.; Wang, P.Y. Intestinal alkaline phosphatase inhibits the translocation of bacteria of gut-origin in mice with peritonitis: Mechanism of action. PLoS ONE 2015, 10, e0124835. [Google Scholar] [CrossRef] [PubMed]
- Peter, Y.; Comellas, A.; Levantini, E.; Ingenito, E.P.; Shapiro, S.D. Epidermal growth factor receptor and claudin-2 participate in A549 permeability and remodeling: Implications for non-small cell lung cancer tumor colonization. Mol. Carcinog. 2009, 48, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Burns, W.C.; Thomas, M.C. The molecular mediators of type 2 epithelial to mesenchymal transition (EMT) and their role in renal pathophysiology. Expert Rev. Mol. Med. 2010, 12, e17. [Google Scholar] [CrossRef] [PubMed]
- Carrozzino, F.; Soulie, P.; Huber, D.; Mensi, N.; Orci, L.; Cano, A.; Feraille, E.; Montesano, R. Inducible expression of Snail selectively increases paracellular ion permeability and differentially modulates tight junction proteins. Am. J. Physiol. Cell Physiol. 2005, 289, C1002–C1014. [Google Scholar] [CrossRef] [PubMed]
- Dukes, J.D.; Whitley, P.; Chalmers, A.D. The PIKfyve inhibitor YM201636 blocks the continuous recycling of the tight junction proteins claudin-1 and claudin-2 in MDCK cells. PLoS ONE 2012, 7, e28659. [Google Scholar] [CrossRef] [PubMed]
- Vasileva, E.; Citi, S. The role of microtubules in the regulation of epithelial junctions. Tissue Barriers 2018, 6, 1539596. [Google Scholar] [CrossRef] [PubMed]
- Shen, L. Tight junctions on the move: Molecular mechanisms for epithelial barrier regulation. Ann. N. Y. Acad. Sci. 2012, 1258, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Nusrat, A.; Turner, J.R.; Madara, J.L. Molecular physiology and pathophysiology of tight junctions. IV. Regulation of tight junctions by extracellular stimuli: Nutrients, cytokines, and immune cells. Am. J. Physiol. Gastrointest Liver Physiol. 2000, 279, G851–G857. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Colegio, O.R.; Anderson, J.M. The cytoplasmic tails of claudins can influence tight junction barrier properties through effects on protein stability. J. Membr. Biol. 2004, 199, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Fanning, A.S.; Bridges, A.; Anderson, J.M. ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol. Biol. Cell 2009, 20, 3930–3940. [Google Scholar] [CrossRef] [PubMed]
- Raya-Sandino, A.; Castillo-Kauil, A.; Dominguez-Calderon, A.; Alarcon, L.; Flores-Benitez, D.; Cuellar-Perez, F.; Lopez-Bayghen, B.; Chavez-Munguia, B.; Vazquez-Prado, J.; Gonzalez-Mariscal, L. Zonula occludens-2 regulates Rho proteins activity and the development of epithelial cytoarchitecture and barrier function. Biochim. Biophys. Acta 2017, 1864, 1714–1733. [Google Scholar] [CrossRef] [PubMed]
- Ares, G.; Buonpane, C.; Sincavage, J.; Yuan, C.; Wood, D.R.; Hunter, C.J. Caveolin 1 is Associated with Upregulated Claudin 2 in Necrotizing Enterocolitis. Sci. Rep. 2019, 9, 4982. [Google Scholar] [CrossRef] [PubMed]
- Itallie, C.M.; Anderson, J.M. Caveolin binds independently to claudin-2 and occludin. Ann. N. Y. Acad. Sci. 2012, 1257, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Marchiando, A.M.; Shen, L.; Graham, W.V.; Weber, C.R.; Schwarz, B.T.; Austin, J.R., 2nd; Raleigh, D.R.; Guan, Y.; Watson, A.J.; Montrose, M.H.; et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J. Cell Biol. 2010, 189, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, K.E.; Turner, J.R. Myosin light chain kinase: Pulling the strings of epithelial tight junction function. Ann. N. Y. Acad. Sci. 2012, 1258, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Nighot, P.K.; Hu, C.A.; Ma, T.Y. Autophagy enhancement of intestinal epithelial tight junction barrier function by targeting claudin-2 degradation. J. Biol. Chem. 2015, 290, 7234–7246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yan, J.; Xiao, Y.; Shen, Y.; Wang, J.; Ge, W.; Chen, Y. Inhibition of Autophagic Degradation Process Contributes to Claudin-2 Expression Increase and Epithelial Tight Junction Dysfunction in TNF-alpha Treated Cell Monolayers. Int. J. Mol. Sci. 2017, 18, 910–919. [Google Scholar]
- Huang, L.; Jiang, Y.; Sun, Z.; Gao, Z.; Wang, J.; Zhang, D. Autophagy Strengthens Intestinal Mucosal Barrier by Attenuating Oxidative Stress in Severe Acute Pancreatitis. Dig. Dis. Sci. 2018, 63, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Watanabe, R.; Sato, T.; Taga, S.; Shimobaba, S.; Yamaguchi, M.; Yamazaki, Y.; Endo, S.; Matsunaga, T.; Sugatani, J. Nuclear distribution of claudin-2 increases cell proliferation in human lung adenocarcinoma cells. Biochim. Biophys. Acta 2014, 1843, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; Milatz, S.; Krug, S.M.; Oelrich, B.; Schulzke, J.D.; Amasheh, S.; Gunzel, D.; Fromm, M. Claudin-2, a component of the tight junction, forms a paracellular water channel. J. Cell Sci. 2010, 123, 1913–1921. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; Gunzel, D.; Krug, S.M.; Schulzke, J.D.; Fromm, M.; Yu, A.S. Claudin-2-mediated cation and water transport share a common pore. Acta Physiol. (Oxf) 2017, 219, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; Gunzel, D.; Theune, D.; Czichos, C.; Schulzke, J.D.; Fromm, M. Water channels and barriers formed by claudins. Ann. N. Y. Acad. Sci. 2017, 1397, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Borovac, J.; Barker, R.S.; Rievaj, J.; Rasmussen, A.; Pan, W.; Wevrick, R.; Alexander, R.T. Claudin-4 forms a paracellular barrier, revealing the interdependence of claudin expression in the loose epithelial cell culture model opossum kidney cells. Am. J. Physiol. Cell Physiol. 2012, 303, C1278–C1291. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Tamura, A.; Suzuki, K.; Tsukita, S. Site-specific distribution of claudin-based paracellular channels with roles in biological fluid flow and metabolism. Ann. N. Y. Acad. Sci. 2017, 1405, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Muto, S.; Hata, M.; Taniguchi, J.; Tsuruoka, S.; Moriwaki, K.; Saitou, M.; Furuse, K.; Sasaki, H.; Fujimura, A.; Imai, M.; et al. Claudin-2-deficient mice are defective in the leaky and cation-selective paracellular permeability properties of renal proximal tubules. Proc. Natl. Acad. Sci. USA 2010, 107, 8011–8016. [Google Scholar] [CrossRef] [PubMed]
- Pei, L.; Solis, G.; Nguyen, M.T.; Kamat, N.; Magenheimer, L.; Zhuo, M.; Li, J.; Curry, J.; McDonough, A.A.; Fields, T.A.; et al. Paracellular epithelial sodium transport maximizes energy efficiency in the kidney. J. Clin. Investig. 2016, 126, 2509–2518. [Google Scholar] [CrossRef] [PubMed]
- Schnermann, J.; Huang, Y.; Mizel, D. Fluid reabsorption in proximal convoluted tubules of mice with gene deletions of claudin-2 and/or aquaporin1. Am. J. Physiol. Renal Physiol. 2013, 305, F1352–F1364. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Imasato, M.; Yamazaki, Y.; Tanaka, H.; Watanabe, M.; Eguchi, H.; Nagano, H.; Hikita, H.; Tatsumi, T.; Takehara, T.; et al. Claudin 2 deficiency reduces bile flow and increases susceptibility to cholesterol gallstone disease in mice. Gastroenterology 2014, 147, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Hayashi, H.; Imasato, M.; Yamazaki, Y.; Hagiwara, A.; Wada, M.; Noda, T.; Watanabe, M.; Suzuki, Y.; Tsukita, S. Loss of claudin-15, but not claudin-2, causes Na+ deficiency and glucose malabsorption in mouse small intestine. Gastroenterology 2011, 140, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Wada, M.; Tamura, A.; Takahashi, N.; Tsukita, S. Loss of claudins 2 and 15 from mice causes defects in paracellular Na+ flow and nutrient transport in gut and leads to death from malnutrition. Gastroenterology 2013, 144, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Sukka-Ganesh, B.; Mohammed, K.A.; Kaye, F.; Goldberg, E.P.; Nasreen, N. Ephrin-A1 inhibits NSCLC tumor growth via induction of Cdx-2 a tumor suppressor gene. BMC Cancer 2012, 12, 309. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Chaturvedi, R.; Olivares-Villagomez, D.; Habib, T.; Asim, M.; Shivesh, P.; Polk, D.B.; Wilson, K.T.; Washington, M.K.; Van Kaer, L.; et al. Targeted colonic claudin-2 expression renders resistance to epithelial injury, induces immune suppression, and protects from colitis. Mucosal Immunol. 2014, 7, 1340–1353. [Google Scholar] [CrossRef] [PubMed]
- Sonoki, H.; Tanimae, A.; Endo, S.; Matsunaga, T.; Furuta, T.; Ichihara, K.; Ikari, A. Kaempherol and Luteolin Decrease Claudin-2 Expression Mediated by Inhibition of STAT3 in Lung Adenocarcinoma A549 Cells. Nutrients 2017, 9, 597. [Google Scholar] [CrossRef] [PubMed]
- Buchert, M.; Papin, M.; Bonnans, C.; Darido, C.; Raye, W.S.; Garambois, V.; Pelegrin, A.; Bourgaux, J.F.; Pannequin, J.; Joubert, D.; et al. Symplekin promotes tumorigenicity by up-regulating claudin-2 expression. Proc. Natl. Acad. Sci. USA 2010, 107, 2628–2633. [Google Scholar] [CrossRef] [PubMed]
- Maruhashi, R.; Akizuki, R.; Sato, T.; Matsunaga, T.; Endo, S.; Yamaguchi, M.; Yamazaki, Y.; Sakai, H.; Ikari, A. Elevation of sensitivity to anticancer agents of human lung adenocarcinoma A549 cells by knockdown of claudin-2 expression in monolayer and spheroid culture models. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Sato, T.; Takiguchi, A.; Atomi, K.; Yamazaki, Y.; Sugatani, J. Claudin-2 knockdown decreases matrix metalloproteinase-9 activity and cell migration via suppression of nuclear Sp1 in A549 cells. Life Sci. 2011, 88, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Behrens, A.; Sibilia, M.; Wagner, E.F. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet. 1999, 21, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Paquet-Fifield, S.; Koh, S.L.; Cheng, L.; Beyit, L.M.; Shembrey, C.; Molck, C.; Behrenbruch, C.; Papin, M.; Gironella, M.; Guelfi, S.; et al. Tight Junction Protein Claudin-2 Promotes Self-Renewal of Human Colorectal Cancer Stem-like Cells. Cancer Res. 2018, 78, 2925–2938. [Google Scholar] [CrossRef] [PubMed]
- Larre, I.; Castillo, A.; Flores-Maldonado, C.; Contreras, R.G.; Galvan, I.; Munoz-Estrada, J.; Cereijido, M. Ouabain modulates ciliogenesis in epithelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 20591–20596. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, I.; Dynlacht, B.D. Cilium assembly and disassembly. Nat. Cell Biol. 2016, 18, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Mima, S.; Takehara, M.; Takada, H.; Nishimura, T.; Hoshino, T.; Mizushima, T. NSAIDs suppress the expression of claudin-2 to promote invasion activity of cancer cells. Carcinogenesis 2008, 29, 1994–2000. [Google Scholar] [CrossRef] [PubMed]
- Dreymueller, D.; Theodorou, K.; Donners, M.; Ludwig, A. Fine Tuning Cell Migration by a Disintegrin and Metalloproteinases. Mediators Inflamm. 2017, 2017, 9621724. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Takiguchi, A.; Atomi, K.; Sato, T.; Sugatani, J. Decrease in claudin-2 expression enhances cell migration in renal epithelial Madin-Darby canine kidney cells. J. Cell Physiol. 2011, 226, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- Takehara, M.; Nishimura, T.; Mima, S.; Hoshino, T.; Mizushima, T. Effect of claudin expression on paracellular permeability, migration and invasion of colonic cancer cells. Biol. Pharm Bull. 2009, 32, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Xu, X.; Niu, Q.; Zhang, X.; Wei, Y.; Wang, Z.; Zhang, W.; Yan, J.; Ru, Y.; Fu, Z.; et al. Spi-B-Mediated Silencing of Claudin-2 Promotes Early Dissemination of Lung Cancer Cells from Primary Tumors. Cancer Res. 2017, 77, 4809–4822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, H.; Li, Q.; Li, T. CLDN2 inhibits the metastasis of osteosarcoma cells via down-regulating the afadin/ERK signaling pathway. Cancer Cell Int. 2018, 18, 160. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.; Casanova, J. A common framework for EMT and collective cell migration. Development 2016, 143, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Che, J.; Yue, D.; Zhang, B.; Zhang, H.; Huo, Y.; Gao, L.; Zhen, H.; Yang, Y.; Cao, B. Claudin-3 Inhibits Lung Squamous Cell Carcinoma Cell Epithelial-mesenchymal Transition and Invasion via Suppression of the Wnt/beta-catenin Signaling Pathway. Int. J. Med. Sci. 2018, 15, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.; Yoon, C.H.; Kim, R.K.; Lim, E.J.; Oh, Y.S.; Hwang, S.G.; An, S.; Yoon, G.; Gye, M.C.; Yi, J.M.; et al. Claudin-1 induces epithelial-mesenchymal transition through activation of the c-Abl-ERK signaling pathway in human liver cells. Oncogene 2013, 32, 4873–4882. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Yoshida, M.; Nishiumi, S.; Furuse, M.; Azuma, T. Claudin-2 Regulates Colorectal Inflammation via Myosin Light Chain Kinase-Dependent Signaling. Dig. Dis. Sci. 2013, 58, 1546–1559. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, L.; Paschoud, S.; Pulimeno, P.; Foglia, A.; Citi, S. The cytoplasmic plaque of tight junctions: A scaffolding and signalling center. Biochim. Biophys. Acta 2008, 1778, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Matter, K. Tight junctions as regulators of tissue remodelling. Curr. Opin. Cell Biol. 2016, 42, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Herve, J.C.; Derangeon, M.; Sarrouilhe, D.; Bourmeyster, N. Influence of the scaffolding protein Zonula Occludens (ZOs) on membrane channels. Biochim. Biophys. Acta 2014, 1838, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Matsui, T.; Tamura, A.; Uji, M.; Tsukita, S. The association of microtubules with tight junctions is promoted by cingulin phosphorylation by AMPK. J. Cell Biol. 2013, 203, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Torisawa, T.; Oiwa, K.; Tsukita, S. AMPK-dependent phosphorylation of cingulin reversibly regulates its binding to actin filaments and microtubules. Sci. Rep. 2018, 8, 15550. [Google Scholar] [CrossRef] [PubMed]
- Pollack, V.; Sarkozi, R.; Banki, Z.; Feifel, E.; Wehn, S.; Gstraunthaler, G.; Stoiber, H.; Mayer, G.; Montesano, R.; Strutz, F.; et al. Oncostatin M-induced effects on EMT in human proximal tubular cells: Differential role of ERK signaling. Am. J. Physiol. Renal Physiol. 2007, 293, F1714–F1726. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, T.; Sakaguchi, T.; Gu, X.; Reinecker, H.C. Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology 2000, 118, 1001–1011. [Google Scholar] [CrossRef]
- Kinugasa, T.; Huo, Q.; Higashi, D.; Shibaguchi, H.; Kuroki, M.; Tanaka, T.; Futami, K.; Yamashita, Y.; Hachimine, K.; Maekawa, S.; et al. Selective up-regulation of claudin-1 and claudin-2 in colorectal cancer. Anticancer Res. 2007, 27, 3729–3734. [Google Scholar] [CrossRef]
- Moldvay, J.; Jackel, M.; Paska, C.; Soltesz, I.; Schaff, Z.; Kiss, A. Distinct claudin expression profile in histologic subtypes of lung cancer. Lung Cancer 2007, 57, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Xin, S.; Huixin, C.; Benchang, S.; Aiping, B.; Jinhui, W.; Xiaoyan, L.; Yu, W.B.; Minhu, C. Expression of Cdx2 and claudin-2 in the multistage tissue of gastric carcinogenesis. Oncology 2007, 73, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Zhang, X.; Liu, Z.; Liu, Q.; Wang, L.; Lu, Y.; Liu, Y.; Wang, M.; Yang, M.; Jin, X.; et al. The distinct expression patterns of claudin-2, -6, and -11 between human gastric neoplasms and adjacent non-neoplastic tissues. Diagn. Pathol. 2013, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- Virman, J.; Soini, Y.; Kujala, P.; Luukkaala, T.; Salminen, T.; Sunela, K.; Kellokumpu-Lehtinen, P.L. Claudins as prognostic factors for renal cell cancer. Anticancer Res. 2014, 34, 4181–4187. [Google Scholar] [PubMed]
- Song, X.; Li, X.; Tang, Y.; Chen, H.; Wong, B.; Wang, J.; Chen, M. Expression of claudin-2 in the multistage process of gastric carcinogenesis. Histol. Histopathol. 2008, 23, 673–682. [Google Scholar] [PubMed]
- Jung, H.; Jun, K.H.; Jung, J.H.; Chin, H.M.; Park, W.B. The expression of claudin-1, claudin-2, claudin-3, and claudin-4 in gastric cancer tissue. J. Surg. Res. 2011, 167, e185–e191. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Zhang, R.G.; Duan, G.C. Pathogenic mechanisms of the oncoprotein CagA in H. pylori-induced gastric cancer (Review). Oncol. Rep. 2016, 36, 3087–3094. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Haase, G.; Ben-Ze’ev, A. Wnt signaling in cancer stem cells and colon cancer metastasis. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.R.; Nalle, S.C.; Tretiakova, M.; Rubin, D.T.; Turner, J.R. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab. Investig. 2008, 88, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Mezheyeuski, A.; Strell, C.; Hrynchyk, I.; Guren, T.K.; Dragomir, A.; Doroshenko, T.; Pashkova, O.; Gorgun, J.; Ruksha, K.; Pfeiffer, P.; et al. Treatment-related survival associations of claudin-2 expression in fibroblasts of colorectal cancer. Virchows Arch. 2018, 472, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Soini, Y. Claudins 2, 3, 4, and 5 in Paget’s disease and breast carcinoma. Hum. Pathol. 2004, 35, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Huh, J.H.; Lee, S.; Kang, H.; Kim, G.I.; An, H.J. Down-regulation of claudin-2 in breast carcinomas is associated with advanced disease. Histopathology 2008, 53, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Tabaries, S.; Dong, Z.; Annis, M.G.; Omeroglu, A.; Pepin, F.; Ouellet, V.; Russo, C.; Hassanain, M.; Metrakos, P.; Diaz, Z.; et al. Claudin-2 is selectively enriched in and promotes the formation of breast cancer liver metastases through engagement of integrin complexes. Oncogene 2011, 30, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Kimbung, S.; Kovacs, A.; Bendahl, P.O.; Malmstrom, P.; Ferno, M.; Hatschek, T.; Hedenfalk, I. Claudin-2 is an independent negative prognostic factor in breast cancer and specifically predicts early liver recurrences. Mol. Oncol. 2014, 8, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Barmeyer, C.; Fromm, M.; Schulzke, J.D. Active and passive involvement of claudins in the pathophysiology of intestinal inflammatory diseases. Pflugers Arch. 2017, 469, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Landy, J.; Ronde, E.; English, N.; Clark, S.K.; Hart, A.L.; Knight, S.C.; Ciclitira, P.J.; Al-Hassi, H.O. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J. Gastroenterol. 2016, 22, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Amasheh, M.; Fromm, A.; Krug, S.M.; Amasheh, S.; Andres, S.; Zeitz, M.; Fromm, M.; Schulzke, J.D. TNFalpha-induced and berberine-antagonized tight junction barrier impairment via tyrosine kinase, Akt and NFkappaB signaling. J. Cell Sci. 2010, 123, 4145–4155. [Google Scholar] [CrossRef] [PubMed]
- Zeissig, S.; Burgel, N.; Gunzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Luettig, J.; Rosenthal, R.; Barmeyer, C.; Schulzke, J.D. Claudin-2 as a mediator of leaky gut barrier during intestinal inflammation. Tissue Barriers 2015, 3, e977176. [Google Scholar] [CrossRef] [PubMed]
- Aghdassi, A.A.; Weiss, F.U.; Mayerle, J.; Lerch, M.M.; Simon, P. Genetic susceptibility factors for alcohol-induced chronic pancreatitis. Pancreatology 2015, 15, S23–S31. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Hou, J. Claudins in barrier and transport function-the kidney. Pflugers Arch. 2017, 469, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Fromm, M.; Piontek, J.; Rosenthal, R.; Gunzel, D.; Krug, S.M. Tight junctions of the proximal tubule and their channel proteins. Pflugers Arch. 2017, 469, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Szaszi, K.; Amoozadeh, Y. New insights into functions, regulation, and pathological roles of tight junctions in kidney tubular epithelium. Int. Rev. Cell Mol. Biol. 2014, 308, 205–271. [Google Scholar] [PubMed]
- Balkovetz, D.F.; Chumley, P.; Amlal, H. Downregulation of claudin-2 expression in renal epithelial cells by metabolic acidosis. Am. J. Physiol. Renal Physiol. 2009, 297, F604–F611. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.E.; DiGeronimo, R.J.; Arthur, D.E.; King, J.M. Remodeling of the tight junction during recovery from exposure to hydrogen peroxide in kidney epithelial cells. Free Radic Biol. Med. 2009, 47, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.B.; Sugimoto, K.; Dhawan, P.; Harris, R.C. Juxtacrine activation of EGFR regulates claudin expression and increases transepithelial resistance. Am. J. Physiol. Cell Physiol. 2007, 293, C1660–C1668. [Google Scholar] [CrossRef] [PubMed]
- Maggiorani, D.; Dissard, R.; Belloy, M.; Saulnier-Blache, J.S.; Casemayou, A.; Ducasse, L.; Gres, S.; Belliere, J.; Caubet, C.; Bascands, J.L.; et al. Shear Stress-Induced Alteration of Epithelial Organization in Human Renal Tubular Cells. PLoS ONE 2015, 10, e0131416. [Google Scholar] [CrossRef] [PubMed]
- Gamero-Estevez, E.; Andonian, S.; Jean-Claude, B.; Gupta, I.; Ryan, A.K. Temporal Effects of Quercetin on Tight Junction Barrier Properties and Claudin Expression and Localization in MDCK II Cells. Int. J. Mol. Sci. 2019, 20, 4889. [Google Scholar] [CrossRef] [PubMed]
- Martin-Martin, N.; Dan, Q.; Amoozadeh, Y.; Waheed, F.; McMorrow, T.; Ryan, M.P.; Szaszi, K. RhoA and Rho kinase mediate cyclosporine A and sirolimus-induced barrier tightening in renal proximal tubular cells. Int. J. Biochem. Cell Biol. 2012, 44, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.; Molina-Jijon, E.; Medina-Campos, O.N.; Rodriguez-Munoz, R.; Reyes, J.L.; Loredo, M.L.; Tapia, E.; Sanchez-Lozada, L.G.; Barrera-Oviedo, D.; Pedraza-Chaverri, J. Renal tight junction proteins are decreased in cisplatin-induced nephrotoxicity in rats. Toxicol. Mech. Methods 2014, 24, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Bialik, J.F.; Ding, M.; Speight, P.; Dan, Q.; Miranda, M.Z.; Di Ciano-Oliveira, C.; Kofler, M.M.; Rotstein, O.D.; Pedersen, S.F.; Szaszi, K.; et al. Profibrotic epithelial phenotype: A central role for MRTF and TAZ. Sci. Rep. 2019, 9, 4323. [Google Scholar] [CrossRef] [PubMed]
- Takigawa, M.; Iida, M.; Nagase, S.; Suzuki, H.; Watari, A.; Tada, M.; Okada, Y.; Doi, T.; Fukasawa, M.; Yagi, K.; et al. Creation of a Claudin-2 Binder and Its Tight Junction-Modulating Activity in a Human Intestinal Model. J. Pharmacol. Exp. Ther. 2017, 363, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Hata, T.; Tada, M.; Iida, M.; Watari, A.; Okada, Y.; Doi, T.; Kuniyasu, H.; Yagi, K.; Kondoh, M. Safety evaluation of a human chimeric monoclonal antibody that recognizes the extracellular loop domain of claudin-2. Eur. J. Pharm. Sci. 2018, 117, 161–167. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cell Line or Transgenic Mouse | Change in Claudin-2 Expression | Downstream Functional Effect | Signaling Components Downstream from Claudin-2 | Ref. |
|---|---|---|---|---|
| Villin-claudin-2 transgenic mice | Claudin-2 overexpression | Increased colonocyte proliferation | PI-3K/Bcl-2 pathway | [116] |
| Caco2 human colon cancer cell | Claudin-2 silencing | Reduced EGF-induced proliferation | [51] | |
| SW480 and HCT116 colon cancer cell lines | Claudin-2 overexpression | Increased proliferation and anchorage-independent growth | ||
| LLC-PK1 porcine kidney tubular cells | Claudin-2 silencing | Reduced proliferation | GEF-H1-mediated RhoA activation, increase in p27kip1 | [67] |
| Pro-fibrotic epithelial shift | RhoA-mediated MRTF activation | |||
| A549 human lung adenocarcinoma cells | Claudin-2 downregulation/ knockdown | Reduced G1/S transition | Cyclin D1 and E1, ZONAB | [103] |
| Increased sensitivity to anti-cancer agents; increased intracellular drug accumulation and reduced efflux | Decrease in phosho-c-Jun and nuclear Sp1; reduced expression of multidrug resistance-associated protein/ABCC2 | [119] | ||
| Reduced migration | Decreased Sp1, reduced MMP-9 expression, activity | [120] | ||
| A549 human lung adenocarcinoma cells | Flavonoid- or epigenetic inhibitor-induced claudin-2 downregulation; | Reduced proliferation; partial rescue of phenotype by claudin-2 reexpression | [64,117] | |
| HT-29 colorectal cancer cell line | Symplekin silencing-induced claudin-2 downregulation; | Reduced anchorage-dependent growth and Zonab nuclear localization; rescue of phenotype by claudin-2 reexpression | [118] | |
| A549 human lung adenocarcinoma cells | Endocytosis and lysosomal degradation of claudin-2 induced by a peptide mimic (DFYSP) of a conserved ECL2 region | Claudin-2 accumulation in the lysosomes, cellular injury and necrotic cell death | Cathepsin B release from lysosomes | [40] |
| MDCK canine tubular cells | Inducible knockdown of claudin-2 | Enhanced migration in a wound-healing assay | Increased MMP-9 mRNA and activity | [120] |
| Human colon cancer stem-like cells (patient-derived CCP1 cells) | Claudin-2 overexpression | Self-renewal of cancer stem cells; enhanced tumor initiation, progression, and metastasis | YAP and miRNAs (especially miR-222-3p) | [122] |
| Caco2 human colon cancer cell | Claudin-2 overexpression | Enhanced migration | Effect independent from MMP-2 and 9 | [128] |
| AGS stomach carcinoma cells | Claudin-2 overexpression | Enhanced migration | Likely via increased MMP-1, -2 and 9 expression | [125] |
| T-84 colonic adenocarcinoma, AGS and KATO-III stomach carcinoma cells; A549 lung adenocarcinoma cell lines | Claudin-2 silencing | Reduced migration | [125] | |
| Claudin-2 overexpression | Augmented migration | |||
| Non-steroidal anti-inflammatory drugs (NSAIDs)-induced claudin-2 downregulation | Reduced migration; claudin-2 reexpression counteracted the effect | |||
| U2OS osteosarcoma cell line | Claudin-2 overexpression | Reduced migration and invasion | Afadin-mediated ERK inhibition | [130] |
| Fetal osteoblast cell line hFOB.1.19 | Claudin-2 silencing | Augmented migration and invasion | ERK activation, afadin reduction | |
| Claudin-2 KO mice | Claudin-2 KO | Augmented TNFα-induced colorectal inflammation | NFκB, myosin light chain kinase | [135] |
| Claudin-2 KO mice | Claudin-2 KO | Augmented energy demand of transport processes; increased ischemia-reperfusion kidney injury | [110] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venugopal, S.; Anwer, S.; Szászi, K. Claudin-2: Roles beyond Permeability Functions. Int. J. Mol. Sci. 2019, 20, 5655. https://doi.org/10.3390/ijms20225655
Venugopal S, Anwer S, Szászi K. Claudin-2: Roles beyond Permeability Functions. International Journal of Molecular Sciences. 2019; 20(22):5655. https://doi.org/10.3390/ijms20225655
Chicago/Turabian StyleVenugopal, Shruthi, Shaista Anwer, and Katalin Szászi. 2019. "Claudin-2: Roles beyond Permeability Functions" International Journal of Molecular Sciences 20, no. 22: 5655. https://doi.org/10.3390/ijms20225655
APA StyleVenugopal, S., Anwer, S., & Szászi, K. (2019). Claudin-2: Roles beyond Permeability Functions. International Journal of Molecular Sciences, 20(22), 5655. https://doi.org/10.3390/ijms20225655