Diesel Exhaust Particle Exposure Compromises Alveolar Macrophage Mitochondrial Bioenergetics



 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
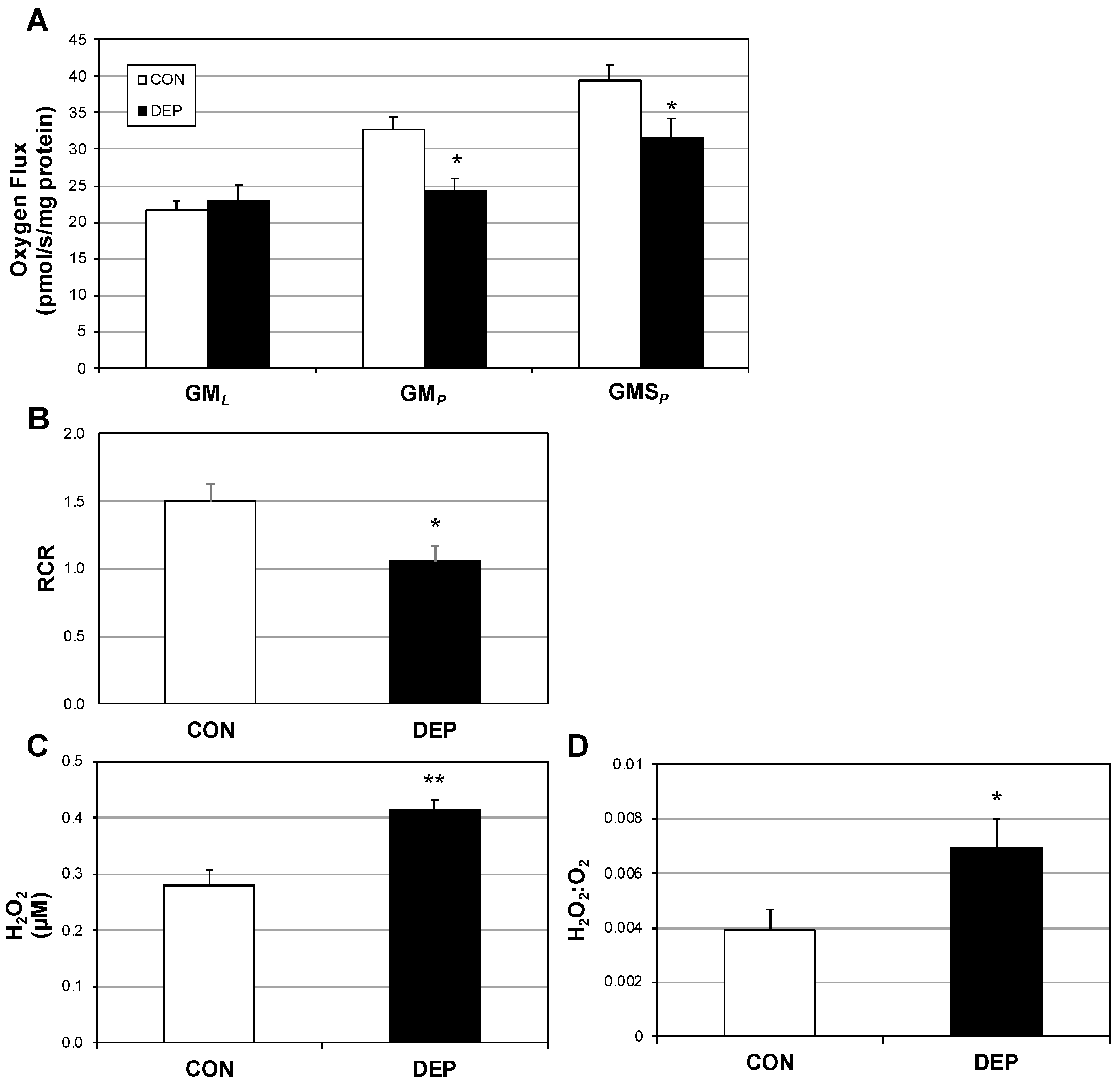
2.1. Lung Macrophage Mitochondrial Function Is Altered in Mice Following DEP Exposure
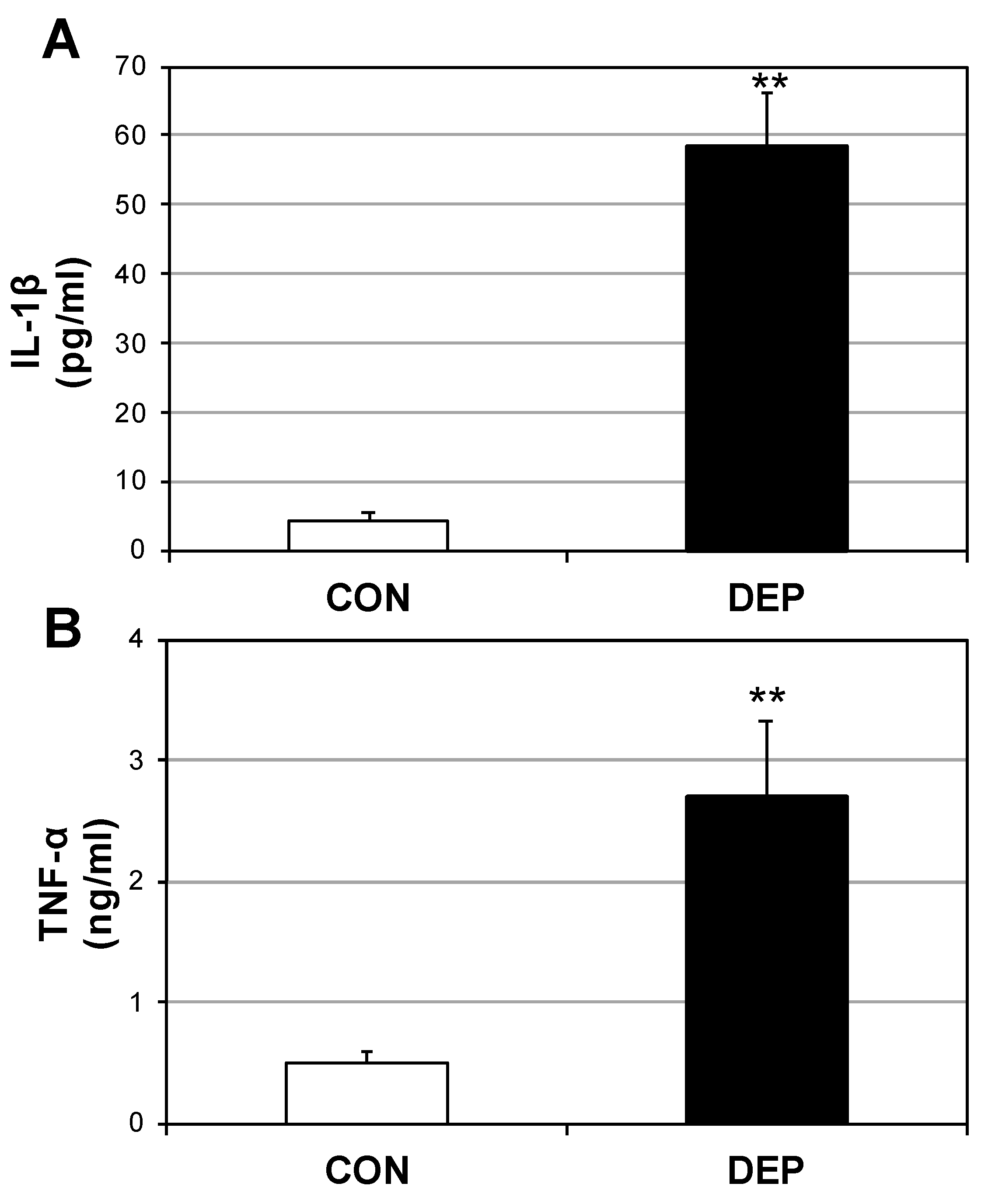
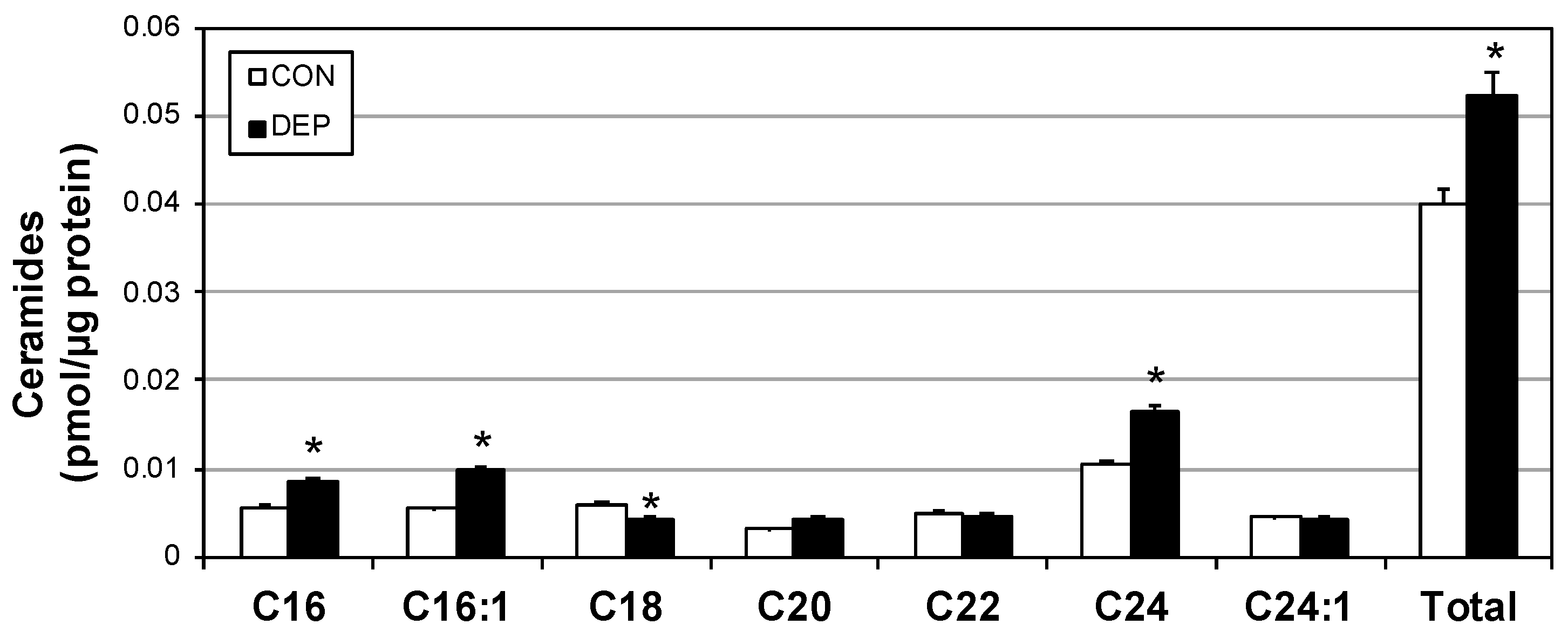
2.2. DEP Increases Plasma Cytokines and Ceramides
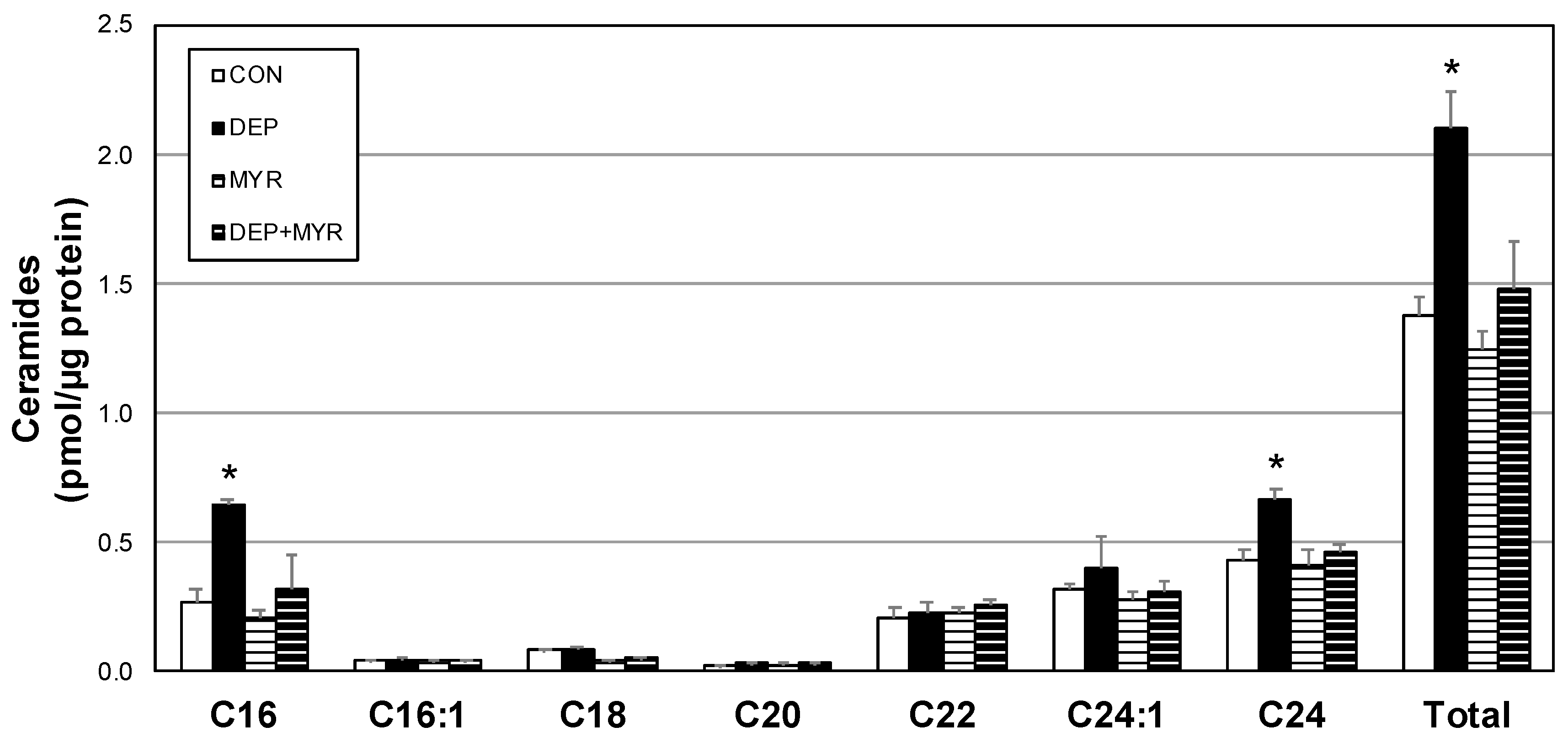
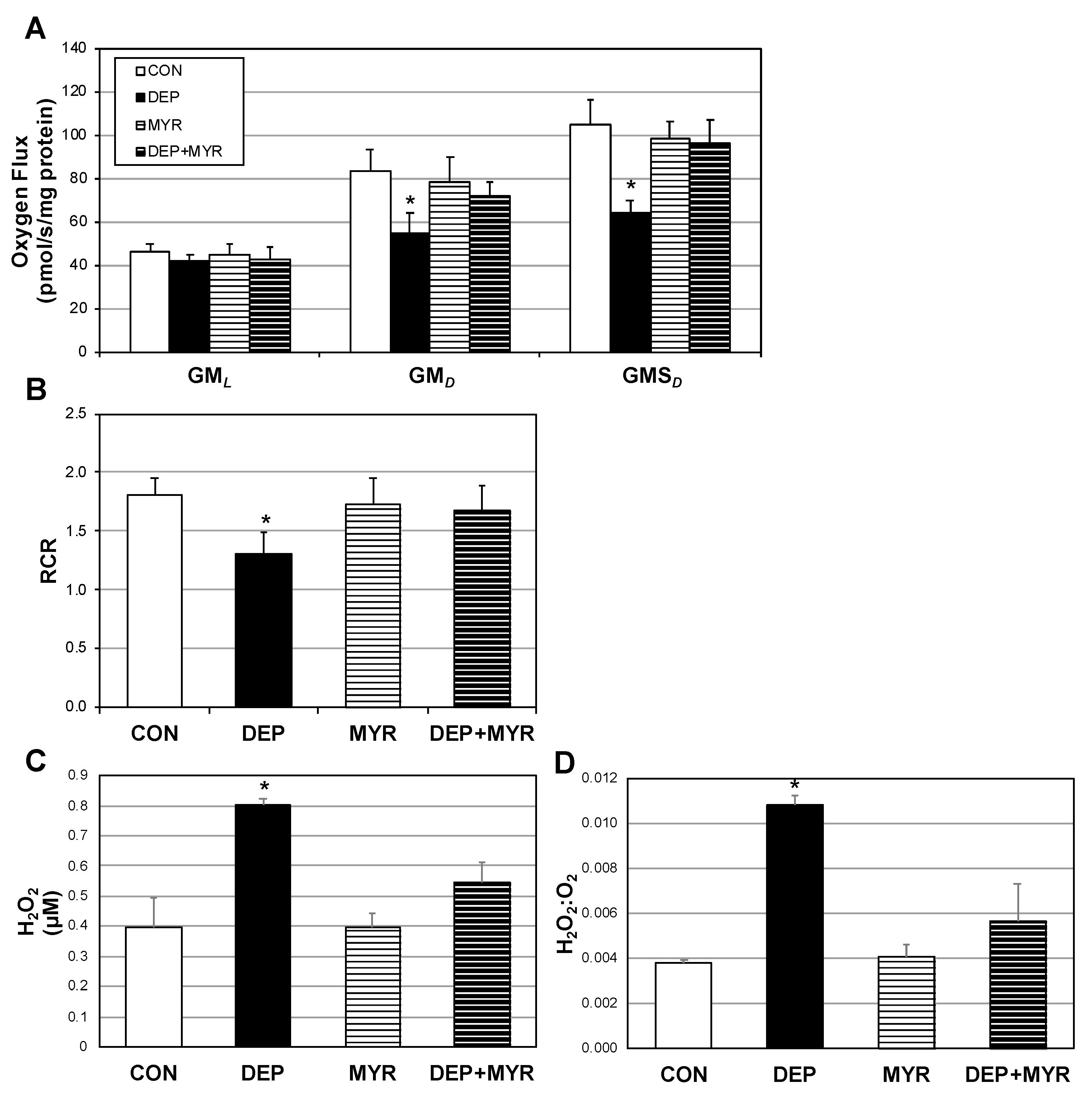
2.3. DEP Alters Primary Alveolar Macrophage Mitochondrial Function
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Diesel Exhaust Particles
4.3. Primary Alveolar Macrophage Culture
4.4. Mitochondrial Respiration
4.5. H2O2 Emission
4.6. Tissue Homogenization
4.7. Ceramide Analysis
4.8. ELISAs
4.9. Statistical Methods
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Fuentes, L.; Roszer, T.; Ricote, M. Inflammatory mediators and insulin resistance in obesity: Role of nuclear receptor signaling in macrophages. Mediat. Inflamm. 2010, 2010, 219583. [Google Scholar] [CrossRef]
- Tippetts, T.S.; Winden, D.R.; Swensen, A.C.; Nelson, M.B.; Thatcher, M.O.; Saito, R.R.; Condie, T.B.; Simmons, K.J.; Judd, A.M.; Reynolds, P.R.; et al. Cigarette smoke increases cardiomyocyte ceramide accumulation and inhibits mitochondrial respiration. BMC Cardiovasc. Disord. 2014, 14, 165. [Google Scholar] [CrossRef]
- Taylor, O.; Thatcher, M.; Carr, S.; Gibbs, J.; Trumbull, A.; Harrison, M.; Winden, D.; Pearson, M.; Tippetts, T.; Holland, W.; et al. High-Mobility Group Box 1 Disrupts Metabolic Function with Cigarette Smoke Exposure in a Ceramide-Dependent Manner. Int. J. Mol. Sci. 2017, 18, 1099. [Google Scholar] [CrossRef]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Terzano, C.; Di Stefano, F.; Conti, V.; Graziani, E.; Petroianni, A. Air pollution ultrafine particles: Toxicity beyond the lung. Eur. Rev. Med. Pharm. Sci. 2010, 14, 809–821. [Google Scholar]
- Barton, D.B.; Betteridge, B.C.; Earley, T.D.; Curtis, C.S.; Robinson, A.B.; Reynolds, P.R. Primary alveolar macrophages exposed to diesel particulate matter increase RAGE expression and activate RAGE signaling. Cell Tissue Res. 2014, 358, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Pope, C.A.; Thun, M.J.; Namboodiri, M.M.; Dockery, D.W.; Evans, J.S.; Speizer, F.E.; Heath, C.W. Particulate air pollution as a predictor of mortality in a prospective study of U.S. adults. Am. J. Respir. Crit. Care Med. 1995, 151, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Baeder, A.C.; Napa, K.; Richardson, S.T.; Taylor, O.J.; Andersen, S.G.; Wilcox, S.H.; Winden, D.R.; Reynolds, P.R.; Bikman, B.T. Oral Gingival Cell Cigarette Smoke Exposure Induces Muscle Cell Metabolic Disruption. Int. J. Dent. 2016, 2016, 2763160. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.R.; Wasley, K.M.; Allison, C.H. Diesel particulate matter induces receptor for advanced glycation end-products (RAGE) expression in pulmonary epithelial cells, and RAGE signaling influences NF-kappaB-mediated inflammation. Env. Health Perspect. 2011, 119, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, M.O.; Tippetts, T.S.; Nelson, M.B.; Swensen, A.C.; Winden, D.R.; Hansen, M.E.; Anderson, M.C.; Johnson, I.E.; Porter, J.P.; Reynolds, P.R.; et al. Ceramides mediate cigarette smoke-induced metabolic disruption in mice. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E919–E927. [Google Scholar] [CrossRef] [PubMed]
- McCoy, D.; Munro, A.; Stephan, C.; Grigg, J. Still failing to tackle air pollution. BMJ 2017, 358, j3802. [Google Scholar] [CrossRef] [PubMed]
- MacNee, W. Oxidative stress and lung inflammation in airways disease. Eur. J. Pharm. 2001, 429, 195–207. [Google Scholar] [CrossRef]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef]
- Teruel, T.; Hernandez, R.; Lorenzo, M. Ceramide mediates insulin resistance by tumor necrosis factor-α in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes 2001, 50, 2563–2571. [Google Scholar] [CrossRef]
- Bikman, B. Brigham Young University, Provo, UT. Unpublished Observation. 2019. [Google Scholar]
- Andersen, Z.J.; Raaschou-Nielsen, O.; Ketzel, M.; Jensen, S.S.; Hvidberg, M.; Loft, S.; Tjønneland, A.; Overvad, K.; Sørensen, M. Diabetes incidence and long-term exposure to air pollution: A cohort study. Diabetes Care 2012, 35, 92–98. [Google Scholar] [CrossRef]
- Brook, R.D.; Jerrett, M.; Brook, J.R.; Bard, R.L.; Finkelstein, M.M. The relationship between diabetes mellitus and traffic-related air pollution. J. Occup. Env. Med. 2008, 50, 32–38. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.H.; Neufer, P.D. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. 2012, 23, 142–153. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Invest. 2006, 116, 1793–1801. [Google Scholar] [CrossRef]
- Menke, A.; Casagrande, S.; Geiss, L.; Cowie, C.C. Prevalence of and Trends in Diabetes Among Adults in the United States 1988–2012. JAMA 2015, 314, 1021–1029. [Google Scholar] [CrossRef]
- Hu, C.; Jia, W. Diabetes in China: Epidemiology and Genetic Risk Factors and Their Clinical Utility in Personalized Medication. Diabetes 2018, 67, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Abuyassin, B.; Laher, I. Diabetes epidemic sweeping the Arab world. World J. Diabetes 2016, 7, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Lawal, A.O. Diesel Exhaust Particles and the Induction of Macrophage Activation and Dysfunction. Inflammation 2018, 41, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Shiraiwa, M.; Selzle, K.; Poschl, U. Hazardous components and health effects of atmospheric aerosol particles: Reactive oxygen species, soot, polycyclic aromatic compounds and allergenic proteins. Free Radic. Res. 2012, 46, 927–939. [Google Scholar] [CrossRef]
- Ji, J.; Upadhyay, S.; Xiong, X.; Malmlöf, M.; Sandström, T.; Gerde, P.; Palmberg, L. Multi-cellular human bronchial models exposed to diesel exhaust particles: Assessment of inflammation, oxidative stress and macrophage polarization. Part. Fibre Toxicol. 2018, 15, 19. [Google Scholar] [CrossRef]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Nelson, M.B.; Swensen, A.C.; Winden, D.R.; Bodine, J.S.; Bikman, B.T.; Reynolds, P.R. Cardiomyocyte mitochondrial respiration is reduced by receptor for advanced glycation end-product signaling in a ceramide-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H63–H69. [Google Scholar] [CrossRef]
- Hodson, A.E.; Tippetts, T.S.; Bikman, B.T. Insulin treatment increases myocardial ceramide accumulation and disrupts cardiometabolic function. Cardiovasc. Diabetol. 2015, 14, 153. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Holland, W.L.; Wilson, L.; Tanner, J.M.; Kearns, D.; Cahoon, J.M.; Pettey, D.; Losee, J.; Duncan, B.; Gale, D.; et al. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes 2012, 61, 1848–1859. [Google Scholar] [CrossRef]
- Lewis, J.B.; Bodine, J.S.; Gassman, J.R.; Muñoz, S.A.; Milner, D.C.; Dunaway, T.M.; Egbert, K.M.; Monson, T.D.; Broberg, D.S.; Arroyo, J.A.; et al. Transgenic up-regulation of Claudin-6 decreases fine diesel particulate matter (DPM)-induced pulmonary inflammation. Env. Sci. Pollut. Res. Int. 2018, 25, 18179–18188. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.E.; Tippetts, T.S.; Brassfield, E.S.; Tucker, B.J.; Ockey, A.; Swensen, A.C.; Anthonymuthu, T.S.; Washburn, T.D.; Kane, D.A.; Prince, J.T.; et al. Mitochondrial fission mediates ceramide-induced metabolic disruption in skeletal muscle. Biochem. J. 2013, 456, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Dallon, B.W.; Parker, B.A.; Hodson, A.E.; Tippetts, T.S.; Harrison, M.E.; Appiah, M.M.A.; Witt, J.E.; Gibbs, J.L.; Gray, H.M.; Sant, T.M.; et al. Insulin selectively reduces mitochondrial uncoupling in brown adipose tissue in mice. Biochem. J. 2018, 475, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.E.; Simmons, K.J.; Tippetts, T.S.; Thatcher, M.O.; Saito, R.R.; Hubbard, S.T.; Trumbull, A.M.; Parker, B.A.; Taylor, O.J.; Bikman, B.T. Lipopolysaccharide Disrupts Mitochondrial Physiology in Skeletal Muscle via Disparate Effects on Sphingolipid Metabolism. Shock 2015, 44, 585–592. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibbs, J.L.; Dallon, B.W.; Lewis, J.B.; Walton, C.M.; Arroyo, J.A.; Reynolds, P.R.; Bikman, B.T. Diesel Exhaust Particle Exposure Compromises Alveolar Macrophage Mitochondrial Bioenergetics. Int. J. Mol. Sci. 2019, 20, 5598. https://doi.org/10.3390/ijms20225598
Gibbs JL, Dallon BW, Lewis JB, Walton CM, Arroyo JA, Reynolds PR, Bikman BT. Diesel Exhaust Particle Exposure Compromises Alveolar Macrophage Mitochondrial Bioenergetics. International Journal of Molecular Sciences. 2019; 20(22):5598. https://doi.org/10.3390/ijms20225598
Chicago/Turabian StyleGibbs, Jonathan L., Blake W. Dallon, Joshua B. Lewis, Chase M. Walton, Juan A. Arroyo, Paul R. Reynolds, and Benjamin T. Bikman. 2019. "Diesel Exhaust Particle Exposure Compromises Alveolar Macrophage Mitochondrial Bioenergetics" International Journal of Molecular Sciences 20, no. 22: 5598. https://doi.org/10.3390/ijms20225598
APA StyleGibbs, J. L., Dallon, B. W., Lewis, J. B., Walton, C. M., Arroyo, J. A., Reynolds, P. R., & Bikman, B. T. (2019). Diesel Exhaust Particle Exposure Compromises Alveolar Macrophage Mitochondrial Bioenergetics. International Journal of Molecular Sciences, 20(22), 5598. https://doi.org/10.3390/ijms20225598