Designing Microparticle-Impregnated Polyelectrolyte Composite: The Combination of ATRP, Fast Azidation, and Click Reaction Using a Single-Catalyst, Single-Pot Strategy


Abstract
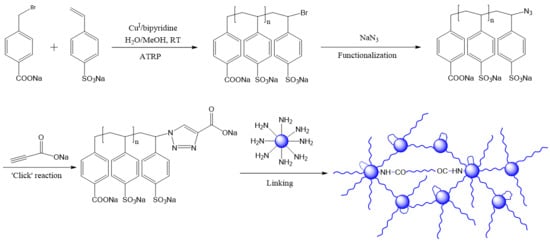
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Homopolymerization of NaSS via ATRP in Water/Methanol Mixed Solvent
2.2. Transformation of NaPSS–Br to NaPSS–N3
2.3. Transformation of NaPSS–N3 to NaPSS–COONa via ‘Click’ Reaction
2.4. Coupling of Polystyrene Microparticles via the Acid-End-Functionalized Polymer Chains and Kaiser Assay
2.5. Imaging of Microparticles Coupled through End-Functionalized Polyelectrolyte (NaCOO–NaPSS–COONa)
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Bromine-End-Functionalized Homopolymer NaPSS via ATRP
3.3. Transformation of Br-End of NaPSS (NaPSS–Br) to Azide (NaPSS-N3)
3.4. Transformation of NaPSS–N3 to Acid-End NaPSS (NaPSS–COONa)
3.5. Linking of Polystyrene Microparticles Using the NaOOC–NaPSS–COONa Chains
3.6. Kaiser Assay
3.7. Imaging
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oliveira, M.B.; Mano, J.F. Polymer-based microparticles in tissue engineering and regenerative medicine. Biotechnol. Prog. 2011, 27, 897–912. [Google Scholar] [CrossRef] [PubMed]
- Dorati, R.; DeTrizio, A.; Modena, T.; Conti, B.; Benazzo, F.; Gastaldi, G.; Genta, I. Biodegradable Scaffolds for Bone Regeneration Combined with Drug-Delivery Systems in Osteomyelitis Therapy. Pharmaceuticals 2017, 10, 96. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Primo, F.; Kumar, S.A.; Manciu, F.S.; Joddar, B. Fabrication of Surfactant-Dispersed HiPco Single-Walled Carbon Nanotube-Based Alginate Hydrogel Composites as Cellular Products. Int. J. Mol. Sci. 2019, 20, 4802. [Google Scholar] [CrossRef] [PubMed]
- Le-Masurier, S.P.; Duong, H.T.T.; Boyer, C.; Granville, A.M. Surface modification of polydopamine coated particles via glycopolymer brush synthesis for protein binding and FLIM testing. Polym. Chem. 2015, 6, 2504–2511. [Google Scholar] [CrossRef]
- Popping, B.; Deratani, A.; Sebille, B.; Desbois, N.; Lamarche, J.; Foissy, A. The effects of electrical charge on the adsorption of a weak cationic polyelectrolyte onto silica, silicon carbide and calcium fluoride. Colloids Surf. 1992, 64, 125–133. [Google Scholar] [CrossRef]
- Turnbull, G.; Clarke, J.; Picard, F.; Riches, P.; Jia, L.; Han, F.; Li, B.; Shu, W. 3D bioactive composite scaffolds for bone tissue engineering. Bioact. Mater. 2018, 3, 278–314. [Google Scholar] [CrossRef]
- Millesime, L.; Amiel, C.; Michel, F.; Chaufer, B. Surface Modification of Zirconium Oxide Particles with Charged Polymers. Langmuir 1996, 12, 3377–3382. [Google Scholar] [CrossRef]
- Nugroho, R.W.N.; Pettersson, T.; Odelius, K.; Hoglund, A.; Albertsson, A.-C. Force Interactions of Nonagglomerating Polylactide Particles Obtained Through Covalent Surface Grafting with Hydrophilic Polymers. Langmuir 2013, 29, 8873–8881. [Google Scholar] [CrossRef]
- Sangermano, M.; Perrot, A.; Gigot, A.; Rivolo, P.; Pirri, F.; Messori, M. Hydrophobic Scratch Resistant UV-Cured Epoxy Coating. Macromol. Mater. Eng. 2016, 301, 93–98. [Google Scholar] [CrossRef]
- Ritzhaupt-Kleissl, E.; Boehm, J.; Hausselt, J.; Hanemann, T. Tuning the Polymer Refractive Index with Nanosized Organic Dopants. Mater. Sci. Eng. C 2006, 26, 1067–1071. [Google Scholar] [CrossRef]
- Ciprari, D.; Jacob, K.; Tannenbaum, R. Characterization of Polymer Nanocomposite Interphase and Its Impact on Mechanical Properties. Macromolecules 2006, 39, 6565–6573. [Google Scholar] [CrossRef]
- Huang, X.; Zheng, Y.; Jiang, P.; Yin, Y. Influence of Nanoparticle Surface Treatment on the Electrical Properties of Cycloaliphatic Epoxy Nanocomposites. IEEE Trans. Dielectr. Electr. Insul. 2017, 17, 635–643. [Google Scholar] [CrossRef]
- Kockmann, A.; Porsiel, J.C.; Saadat, R.; Garnweitner, G. Impact of nanoparticle surface modification on the mechanical properties of polystyrene-based nanocomposites. RSC Adv. 2018, 8, 11109–11118. [Google Scholar] [CrossRef]
- Akeroyd, N.; Klumperman, B. The combination of living radical polymerization and click chemistry for the synthesis of advanced macromolecular architectures. Eur. Polym. J. 2011, 47, 1207–1231. [Google Scholar] [CrossRef]
- De Graaf, A.J.; Mastrobattista, E.; van Nostrum, C.F.; Rijkers, D.T.; Hennink, W.E.; Vermonden, T. ATRP, subsequent azide substitution and ‘click’ chemistry: Three reactions using one catalyst in one pot. Chem. Commun. 2011, 47, 6972. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Tsarevsky, N.V. Macromolecular Engineering by Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2014, 136, 6513–6533. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Nakagawa, Y.; Gaynor, S.G. Synthesis of well-defined azido and amino end-functionalized polystyrene by atom transfer radical polymerization. Macromol. Rapid Commun. 1997, 18, 1057–1066. [Google Scholar] [CrossRef]
- Scriven, E.F.V.; Turnbull, K. Azides: Their Preparation and Synthetic Uses. Chem. Rev. 1998, 88, 297–368. [Google Scholar] [CrossRef]
- Coessens, V.; Matyjaszewski, K. End group transformation of polymers prepared by ATRP, substitution to azides. J. Macromol. Sci. Part A 1999, 36, 667–679. [Google Scholar] [CrossRef]
- Swetha, M.; Ramana, P.V.; Shirodkar, S.G. Simple and Efficient Method for the Synthesis of Azides in Water-THF Solvent System. Org. Prep. Proced. Int. 2011, 43, 348–353. [Google Scholar] [CrossRef]
- Bordwell, F.G.; Brannen, W.T. The Effect of the Carbonyl and Related Groups on the Reactivity of Halides in SN2 Reactions. J. Am. Chem. Soc. 1964, 86, 4645–4650. [Google Scholar] [CrossRef]
- Brase, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic Azides: An Exploding Diversity of a Unique Class of Compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef] [PubMed]
- Buxton, G.V.; Janovsky, I. Mechanism of the Oxidation of Iron(II) by Azide Radical. J. Chem. Soc. Faraday Trans. 1 1976, 72, 1884–1886. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Johnson, J.A.; Finn, M.G.; Koberstein, J.T.; Turro, N.J. Construction of Linear Polymers, Dendrimers, Networks, and Other Polymeric Architectures by Copper-Catalyzed Azide-Alkyne Cycloaddition “Click” Chemistry. Macromol. Rapid Commun. 2008, 29, 1052–1072. [Google Scholar] [CrossRef]
- Golas, P.L.; Tsarevsky, N.V.; Sumerlin, B.S.; Matyjaszewski, K. Catalyst Performance in “Click” Coupling Reactions of Polymers Prepared by ATRP: Ligand and Metal Effects. Macromolecules 2006, 39, 6451–6457. [Google Scholar] [CrossRef]
- Iddon, P.D.; Robinson, K.L.; Armes, S.P. Polymerization of sodium 4-styrenesulfonate via atom transfer radical polymerization in protic media. Polymer 2004, 45, 759–768. [Google Scholar] [CrossRef]
- De, R.; Lee, H.; Das, B. Exploring the interactions in binary mixtures of polyelectrolytes: Influence of mixture composition, concentration, and temperature on counterion condensation. J. Mol. Liq. 2018, 251, 94–99. [Google Scholar] [CrossRef]
- De, R.; Ray, D.; Das, B. Influence of temperature, added electrolyte, and polymer molecular weight on the counterion-condensation phenomenon in aqueous solution of sodium polystyrenesulfonate: A scaling theory approach. RSC Adv. 2015, 5, 54890–54898. [Google Scholar] [CrossRef]
- Izunobi, J.U.; Higginbotham, C.L. Polymer Molecular Weight Analysis by1H NMR Spectroscopy. J. Chem. Educ. 2011, 88, 1098–1104. [Google Scholar] [CrossRef]
- Yameen, B.; Ali, M.; Alvarez, M.; Neumann, R.; Ensinger, W.; Knoll, W.; Azzaroni, O.A. Facile Route for the Preparation of Azide-Terminated Polymers. “Clicking” Polyelectrolyte Brushes on Planner Surfaces and Nanochannels. Polym. Chem. 2010, 1, 183–192. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Xia, J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K.; Shipp, D.A.; Wang, J.-L.; Grimaud, T.; Patten, T.E. Utilizing Halide Exchange To Improve Control of Atom Transfer Radical Polymerization. Macromolecules 1998, 31, 6836–6840. [Google Scholar] [CrossRef]
- Prakash, S.; Tuli, G.D.; Basu, S.K.; Madan, R.D. Advanced Inorganic Chemistry Volume 1; S Chand & Co: New Delhi, India, 2000; Volume 1, ISBN 9788121902632. [Google Scholar]
- De Bon, F.; Fantin, M.; Isse, A.A.; Gennaro, A. Electrochemically mediated ATRP in ionic liquids: Controlled polymerization of methyl acrylate in [BMIm][OTf]. Polym. Chem. 2018, 9, 646–655. [Google Scholar] [CrossRef]
- Evans, B.L. Structure and stability of inorganic azides. Proc. R. Soc. London. Ser. A Math. Phys. Sci. 1958, 246, 199–203. [Google Scholar]
- Evans, B.L.; Yoffe, A.D.; Gray, P. Physics and Chemistry of the Inorganic Azides. Chem. Rev. 1959, 59, 515–568. [Google Scholar] [CrossRef]
- Alfassi, Z.B.; Schuler, R.H. Reaction of azide radicals with aromatic compounds. Azide as a selective oxidant. J. Phys. Chem. 1985, 89, 3359–3363. [Google Scholar] [CrossRef]
- Chiang, M.; Wheeler, R. H-N3 and CH3-N3 Bond Dissociation Energies. Canad. J. Chem. 1968, 46, 3785–3788. [Google Scholar] [CrossRef]
- Worrell, B.T.; Malik, J.A.; Fokin, V.V. Direct evidence of a dinuclear copper intermediate in Cu(I)-catalyzed azide-alkyne cycloadditions. Science 2013, 340, 457–460. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; John Wiley & Sons: New York, NY, USA, 1984. [Google Scholar]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Capacchione, C.; Roma, A.D.; Bounerba, A.; Speranza, V.; Milione, S.; Grassi, A. Syndiotactic Polystyrene-block-Poly(methylmethacrylate) Copolymer via Click Chemistry. Macmol. Chem. Phys. 2013, 214, 1990–1997. [Google Scholar]
- Rodionov, V.O.; Presolski, S.I.; Diaz, D.D.; Fokin, V.V.; Finn, M.G. Ligand-Accelerated Cu-Catalyzed Azide−Alkyne Cycloaddition: A Mechanistic Report. J. Am. Chem. Soc. 2007, 129, 12705–12712. [Google Scholar] [CrossRef] [PubMed]
- Chernykh, A.; Agag, T.; Ishida, H. Synthesis of linear polymers containing benzoxazine moieties in the main chain with high molecular design versatility via click reaction. Polymer 2009, 50, 382–390. [Google Scholar] [CrossRef]
- Lewis, W.G.; Magallon, F.G.; Fokin, V.V.; Finn, M.G. Discovery and Characterization of Catalyst for Azide-Alkyne Cycloaddition by Fluorescence Quenching. J. Am. Chem. Soc. 2014, 123, 9152–9153. [Google Scholar] [CrossRef]
- Gu, S.; Xia, H.; Du, J.; Yang, L.; Cai, Y.; Zhou, Y.; Huang, J. Surface modification of polysulfones via one-pot ATRP and click chemistry: Zwitterionic graft complex and their hemocompatibility. Fibers Polym. 2016, 17, 161–165. [Google Scholar] [CrossRef]
- Singh, M.S.; Chowdhury, S.; Koley, S. Advances of azide-alkyne cycloaddition-click chemistry over the recent decade. Tetrahedron 2016, 72, 5257–5283. [Google Scholar] [CrossRef]
- D’Estea, M.; Eglina, E.; Alini, M.A. Systematic Analysis of DMTMM vs. EDC/NHS for Ligation of Amines to Hyaluronan in Water. Carbohyd. Polym. 2014, 108, 239–246. [Google Scholar] [CrossRef]
- Pelet, J.M.; Putnam, D. An In-Depth Analysis of Polymer-Analogous Conjugation using DMTMM. Bioconjug. Chem. 2011, 22, 329–337. [Google Scholar] [CrossRef]
- Poli, E.; Chaleix, V.; Damià, C.; Hjezi, Z.; Champion, E.; Sol, V. Efficient quantification of primary amine functions grafted onto apatite ceramics by using two UV-Vis spectrophotometric methods. Anal. Methods 2014, 6, 9622–9627. [Google Scholar] [CrossRef]
- De, R.; Das, B. Coiling/uncoiling behaviour of sodium polystyrenesulfonate in 2-ethoxyethanol–water mixed solvent media as probed using viscometry. Polym. Int. 2014, 63, 1959–1964. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De, R.; Jung, M.; Lee, H. Designing Microparticle-Impregnated Polyelectrolyte Composite: The Combination of ATRP, Fast Azidation, and Click Reaction Using a Single-Catalyst, Single-Pot Strategy. Int. J. Mol. Sci. 2019, 20, 5582. https://doi.org/10.3390/ijms20225582
De R, Jung M, Lee H. Designing Microparticle-Impregnated Polyelectrolyte Composite: The Combination of ATRP, Fast Azidation, and Click Reaction Using a Single-Catalyst, Single-Pot Strategy. International Journal of Molecular Sciences. 2019; 20(22):5582. https://doi.org/10.3390/ijms20225582
Chicago/Turabian StyleDe, Ranjit, Minhyuk Jung, and Hohjai Lee. 2019. "Designing Microparticle-Impregnated Polyelectrolyte Composite: The Combination of ATRP, Fast Azidation, and Click Reaction Using a Single-Catalyst, Single-Pot Strategy" International Journal of Molecular Sciences 20, no. 22: 5582. https://doi.org/10.3390/ijms20225582
APA StyleDe, R., Jung, M., & Lee, H. (2019). Designing Microparticle-Impregnated Polyelectrolyte Composite: The Combination of ATRP, Fast Azidation, and Click Reaction Using a Single-Catalyst, Single-Pot Strategy. International Journal of Molecular Sciences, 20(22), 5582. https://doi.org/10.3390/ijms20225582