The Pathogenic TSH β-Subunit Variant C105Vfs114X Causes a Modified Signaling Profile at TSHR

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
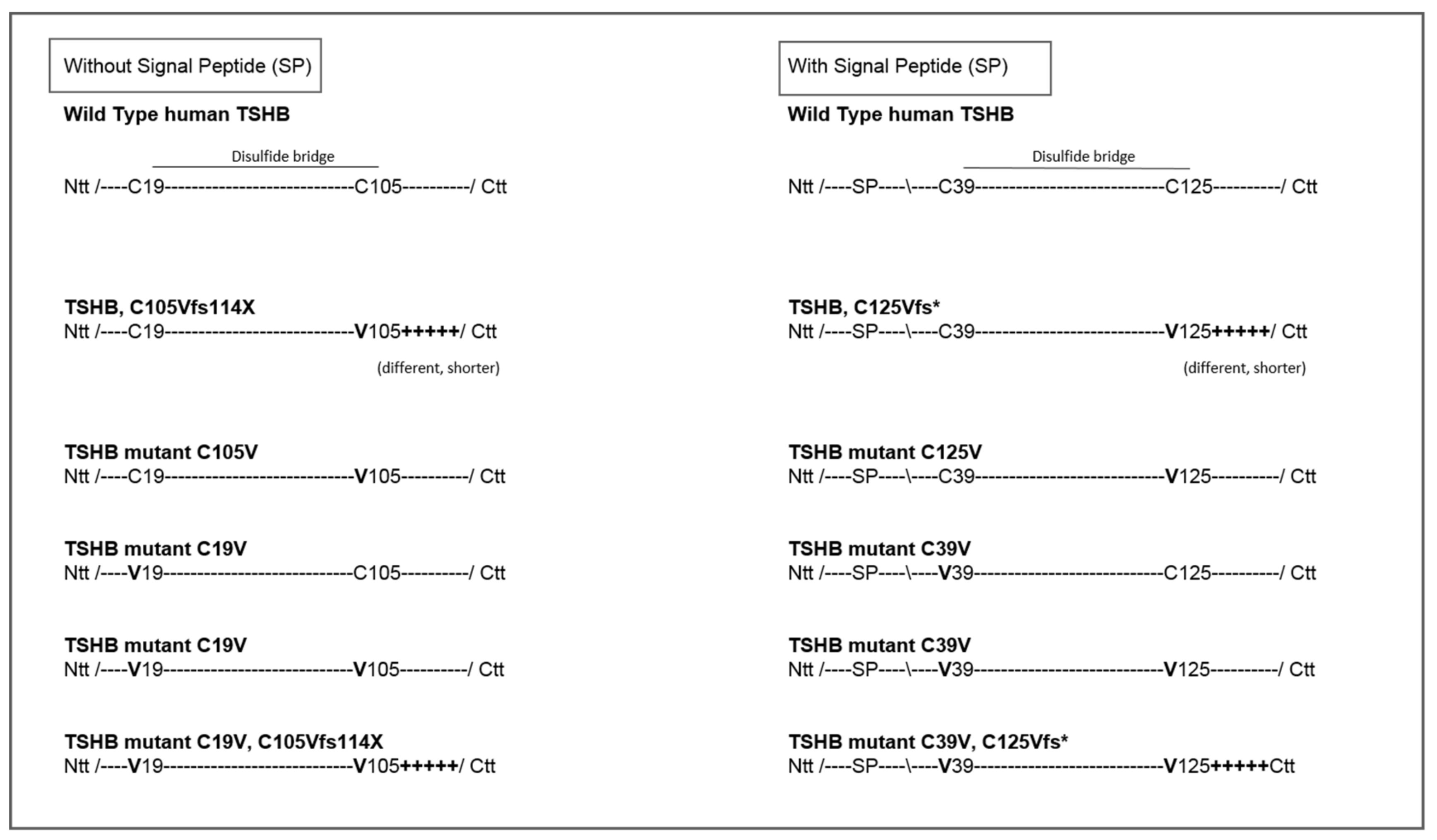
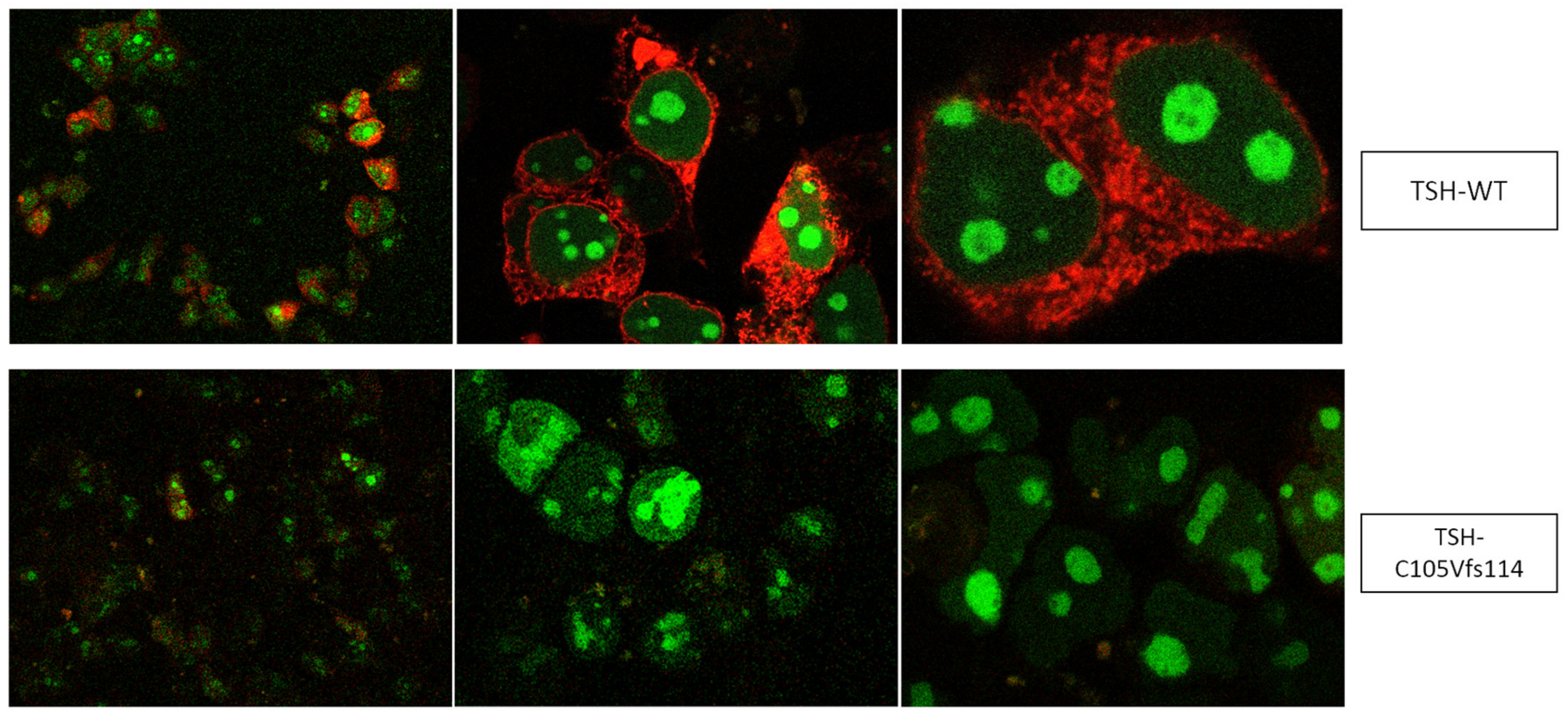
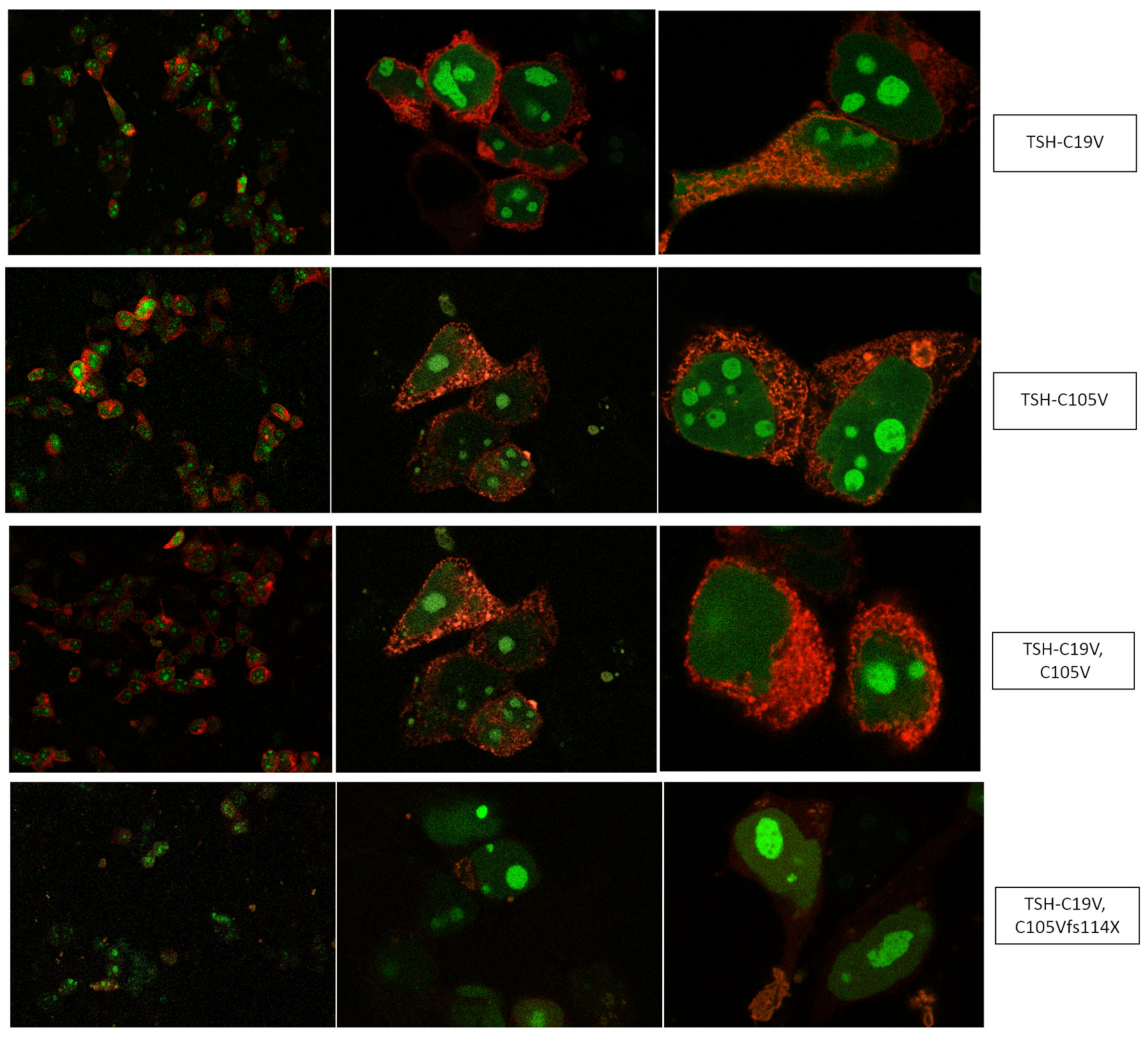
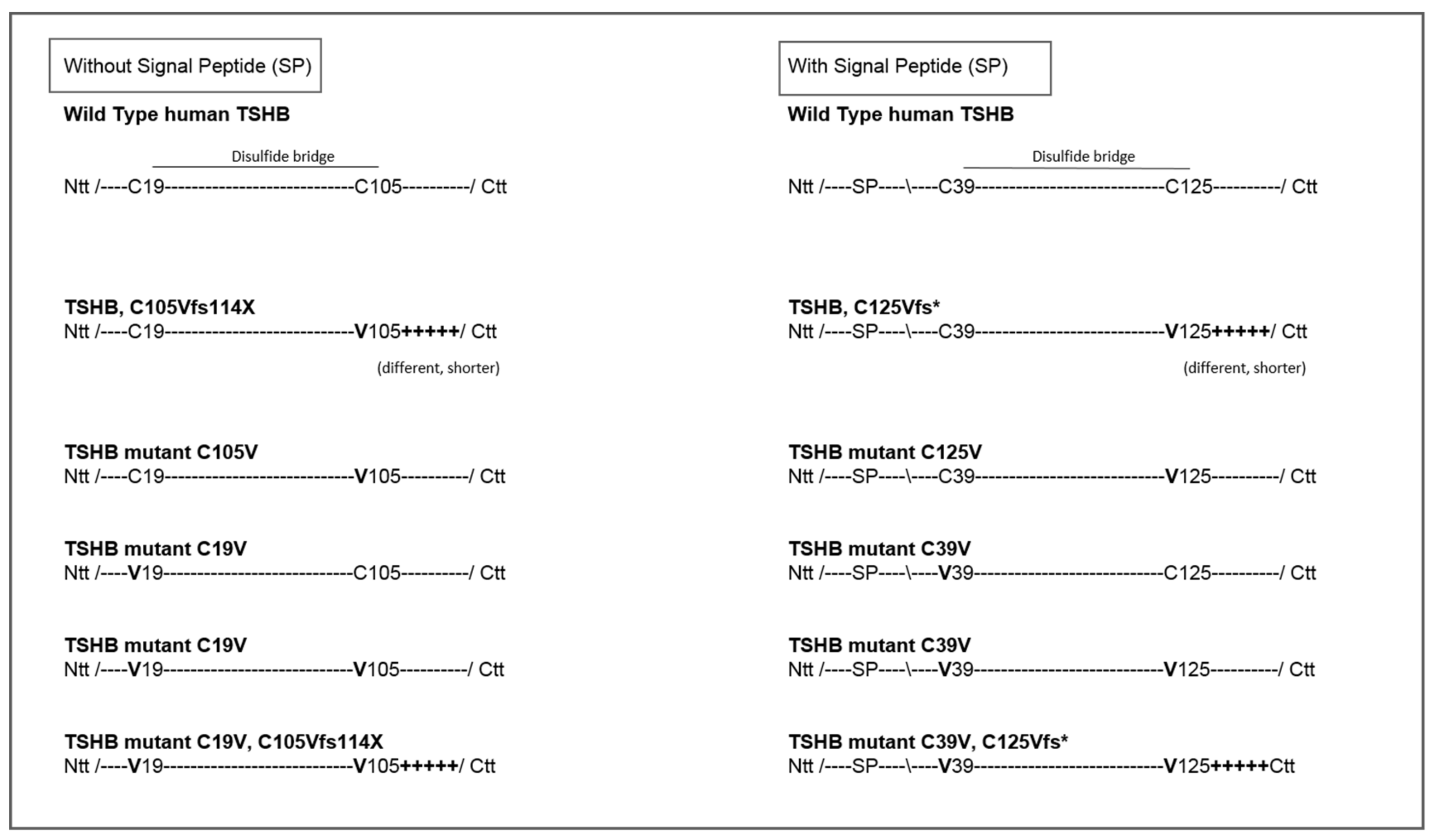
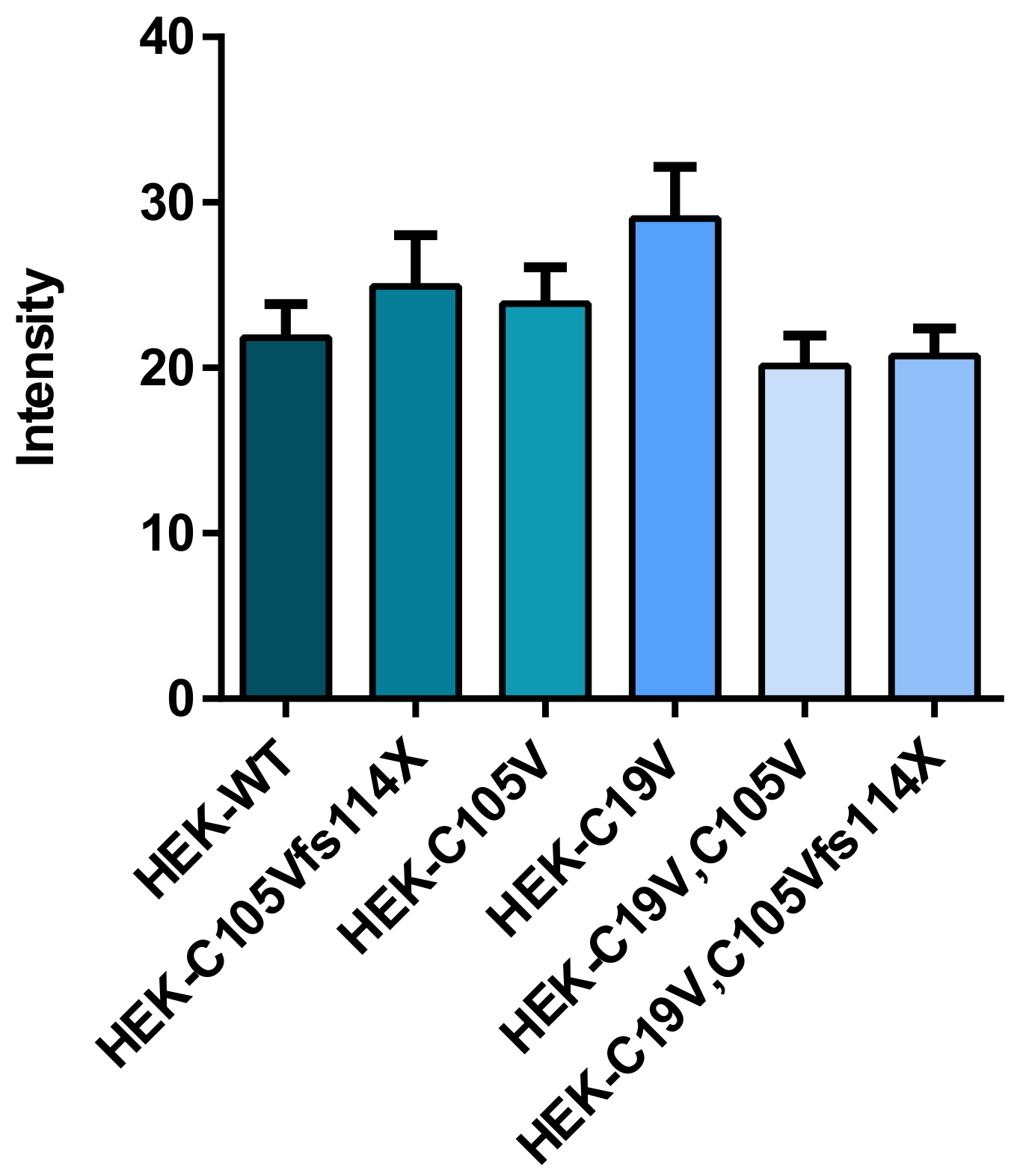
2.1. Quantification of TSH-WT and Mutants
2.2. Functional Characterization
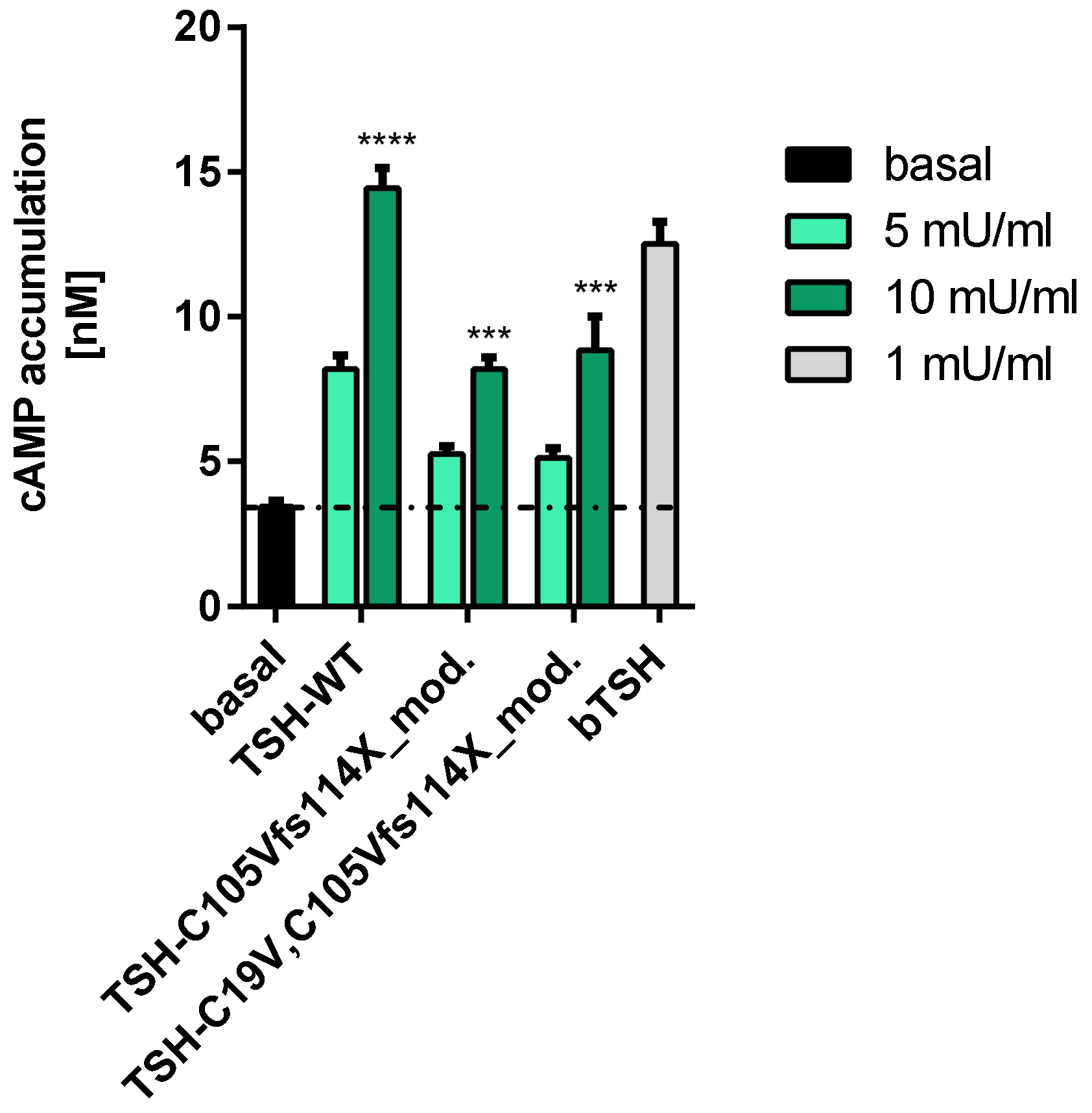
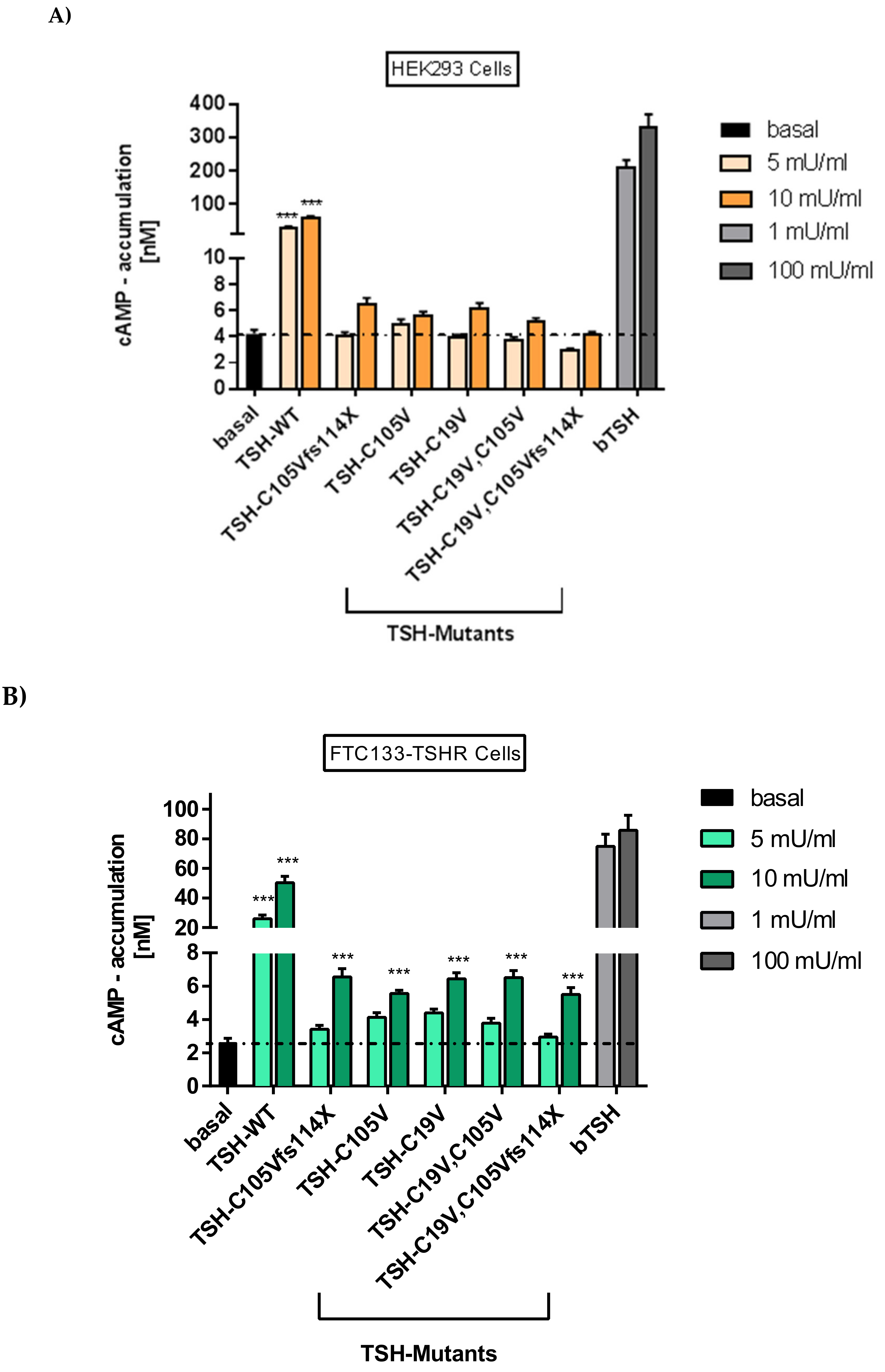
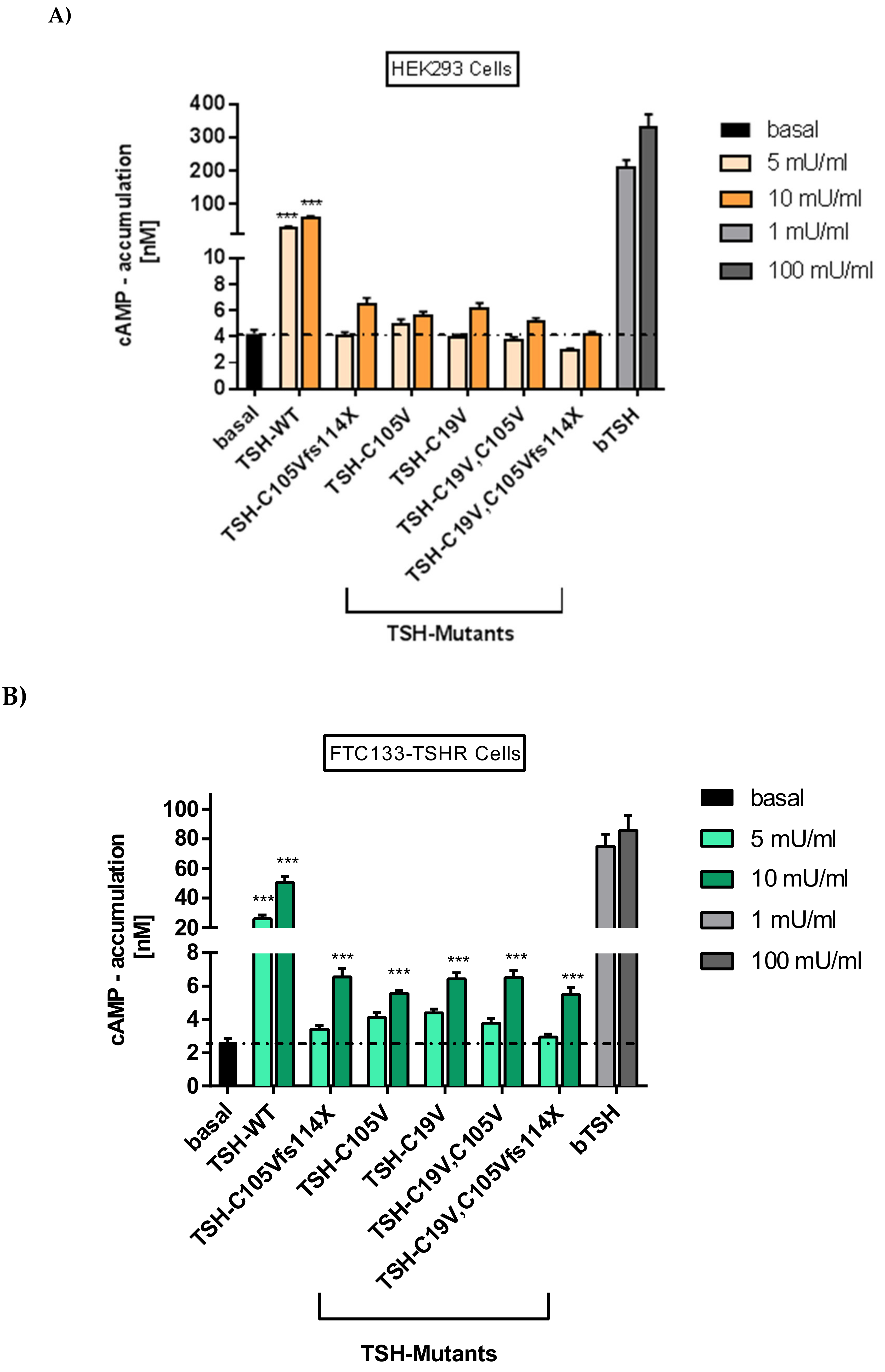
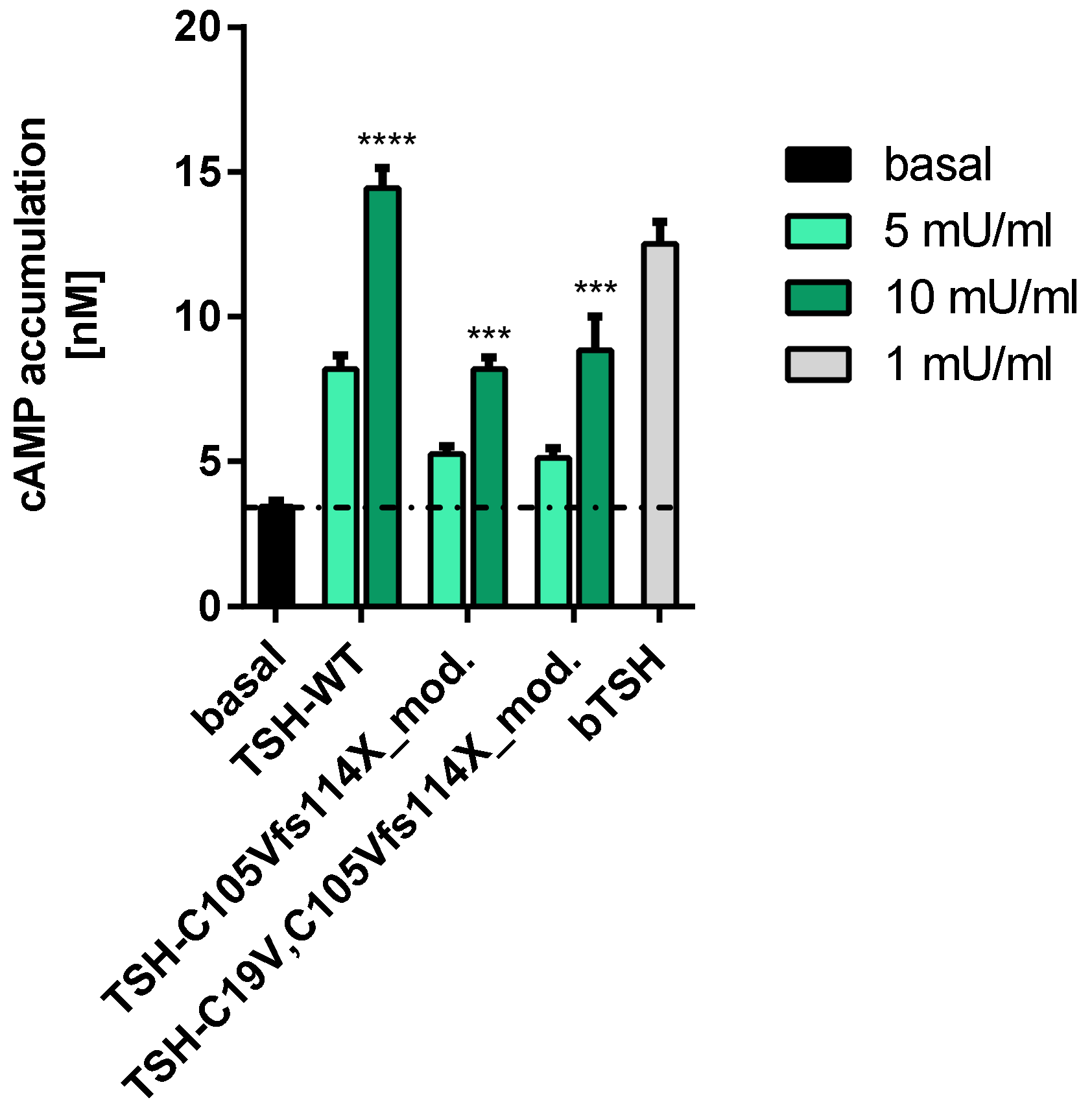
2.2.1. Gs Mediated Signaling in HEK293 Cells
2.2.2. Gs Signaling Pathway in FTC133-Cells
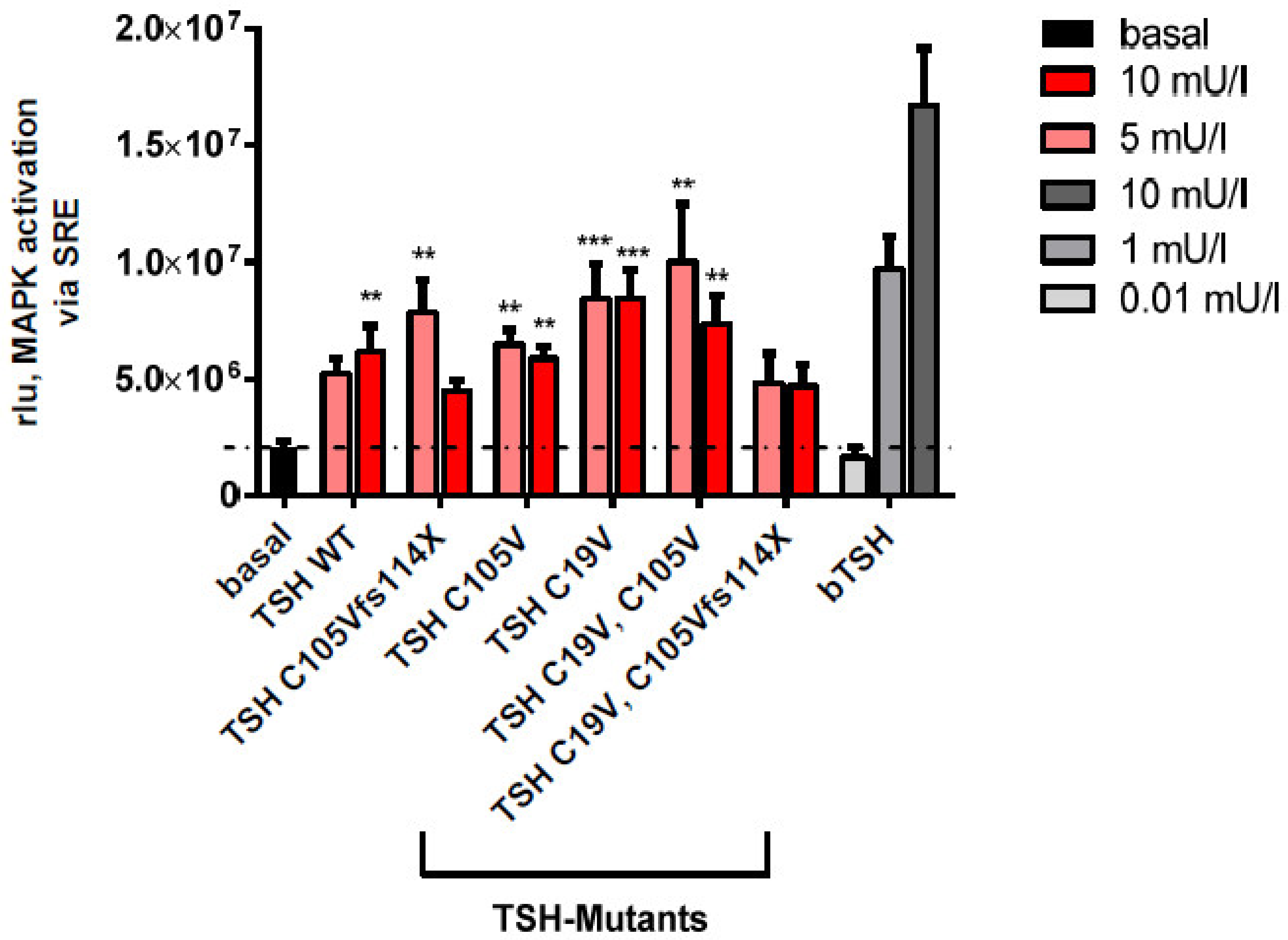
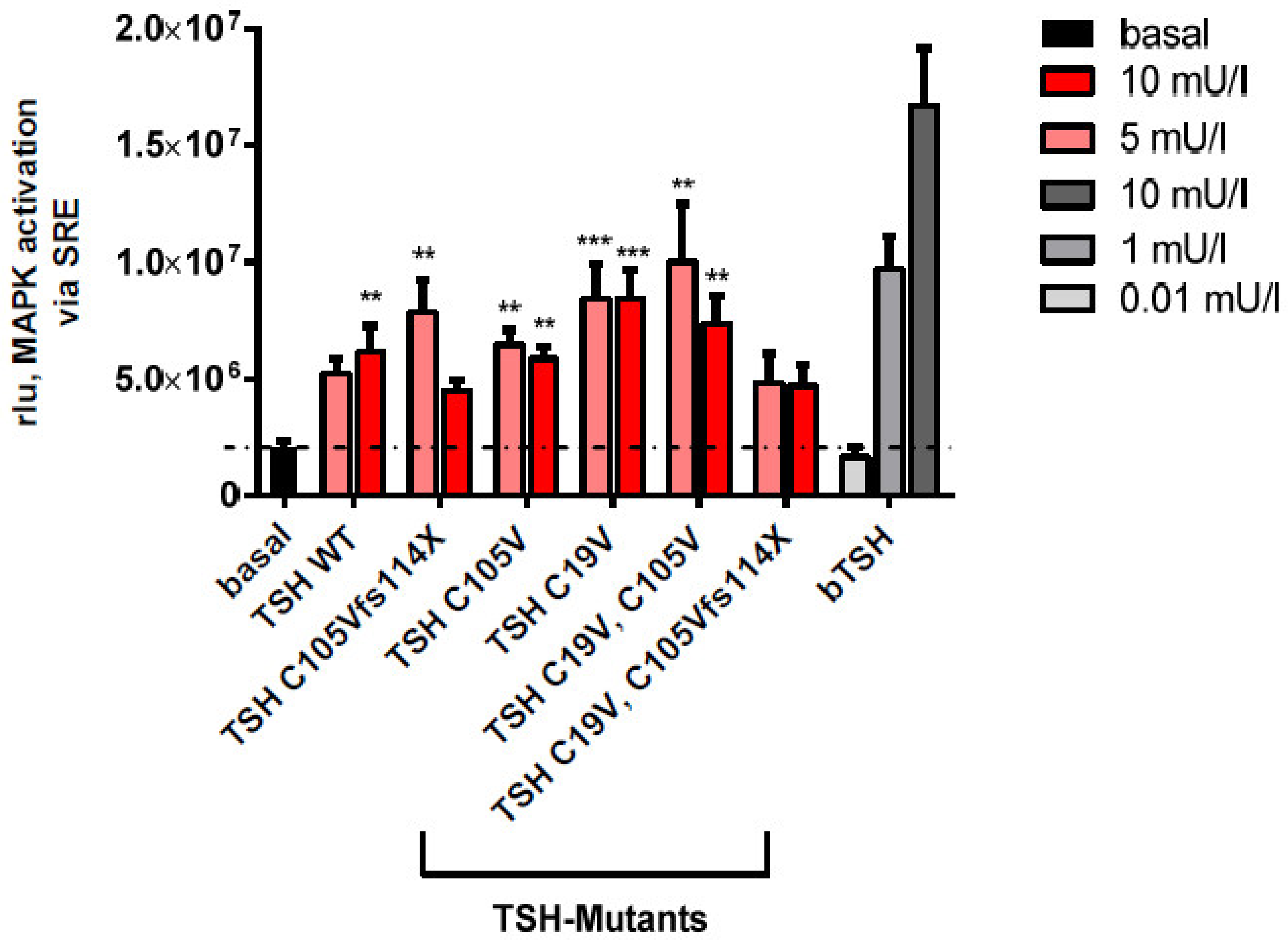
2.2.3. MAPK Signaling Pathway in HEK293 Cells
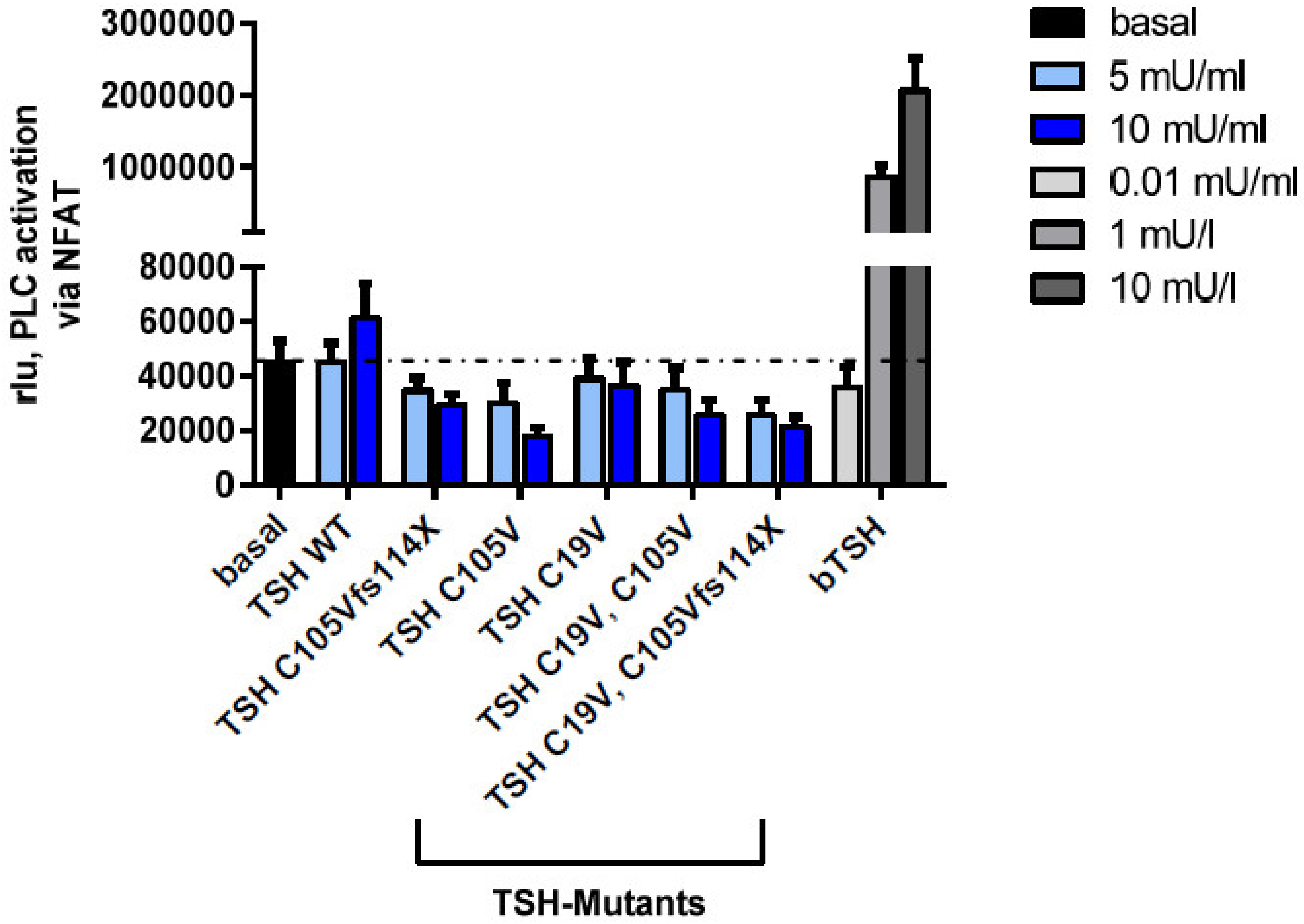
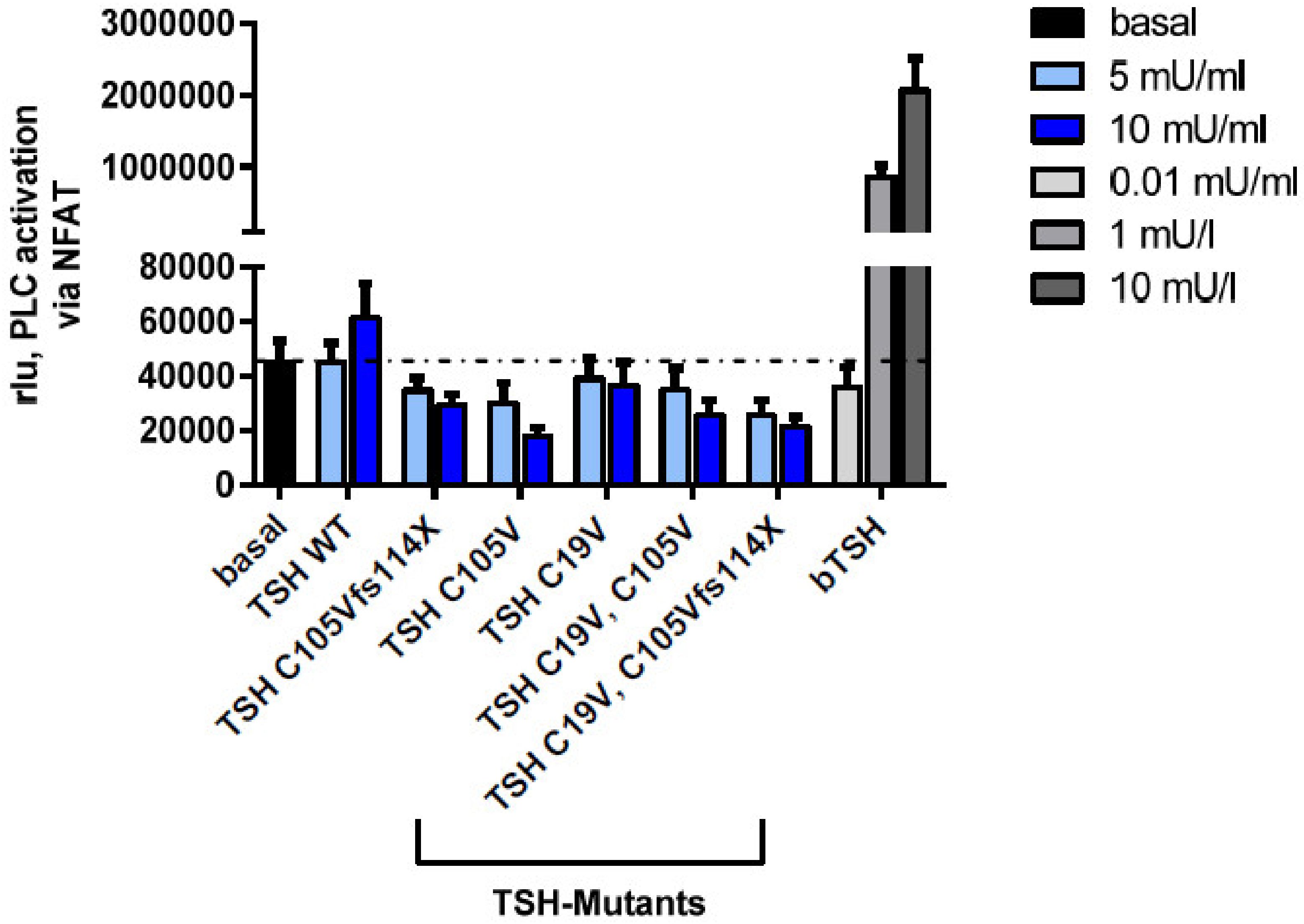
2.2.4. Gq Mediated Signaling Pathway in HEK293 Cells
3. Discussion
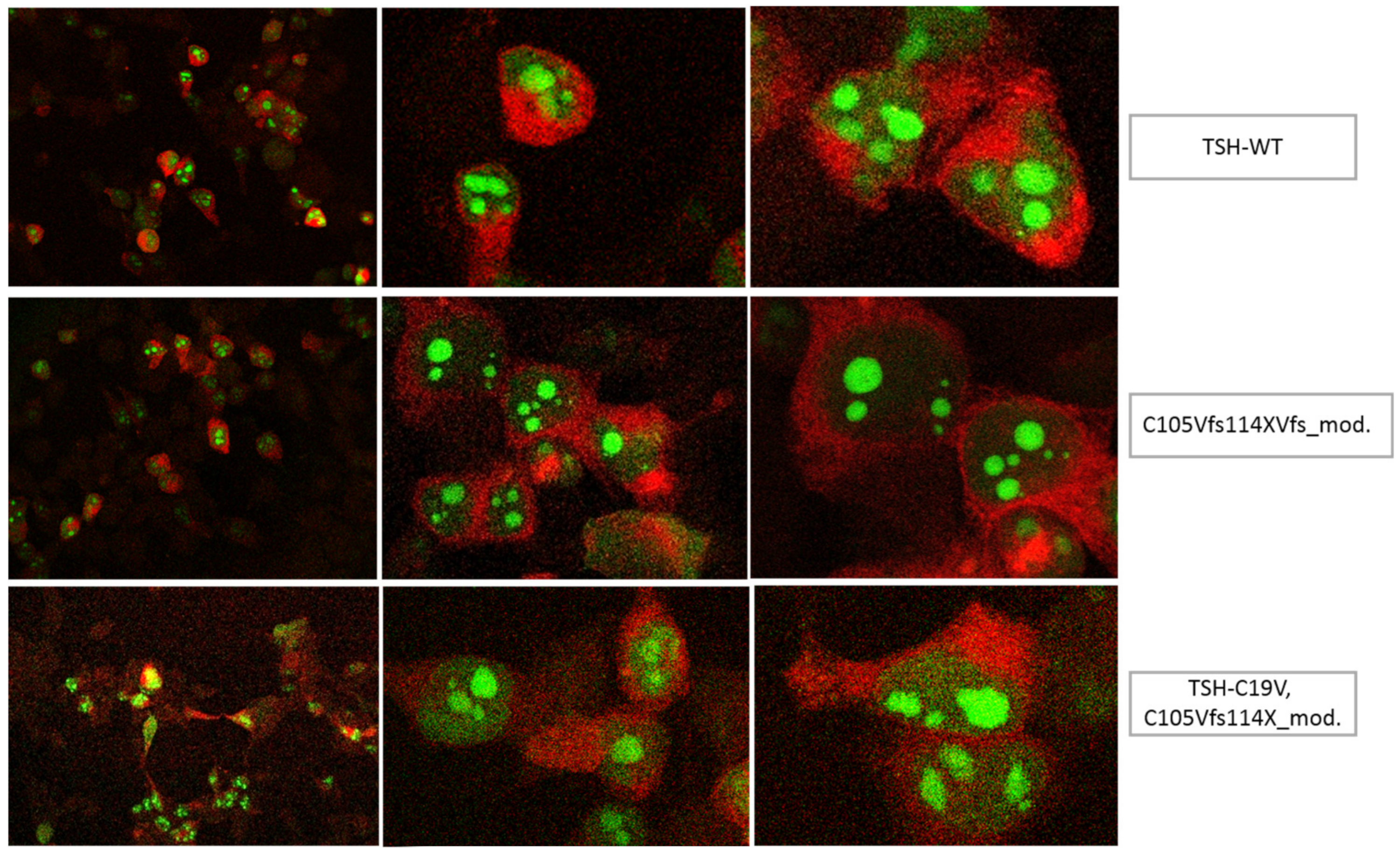
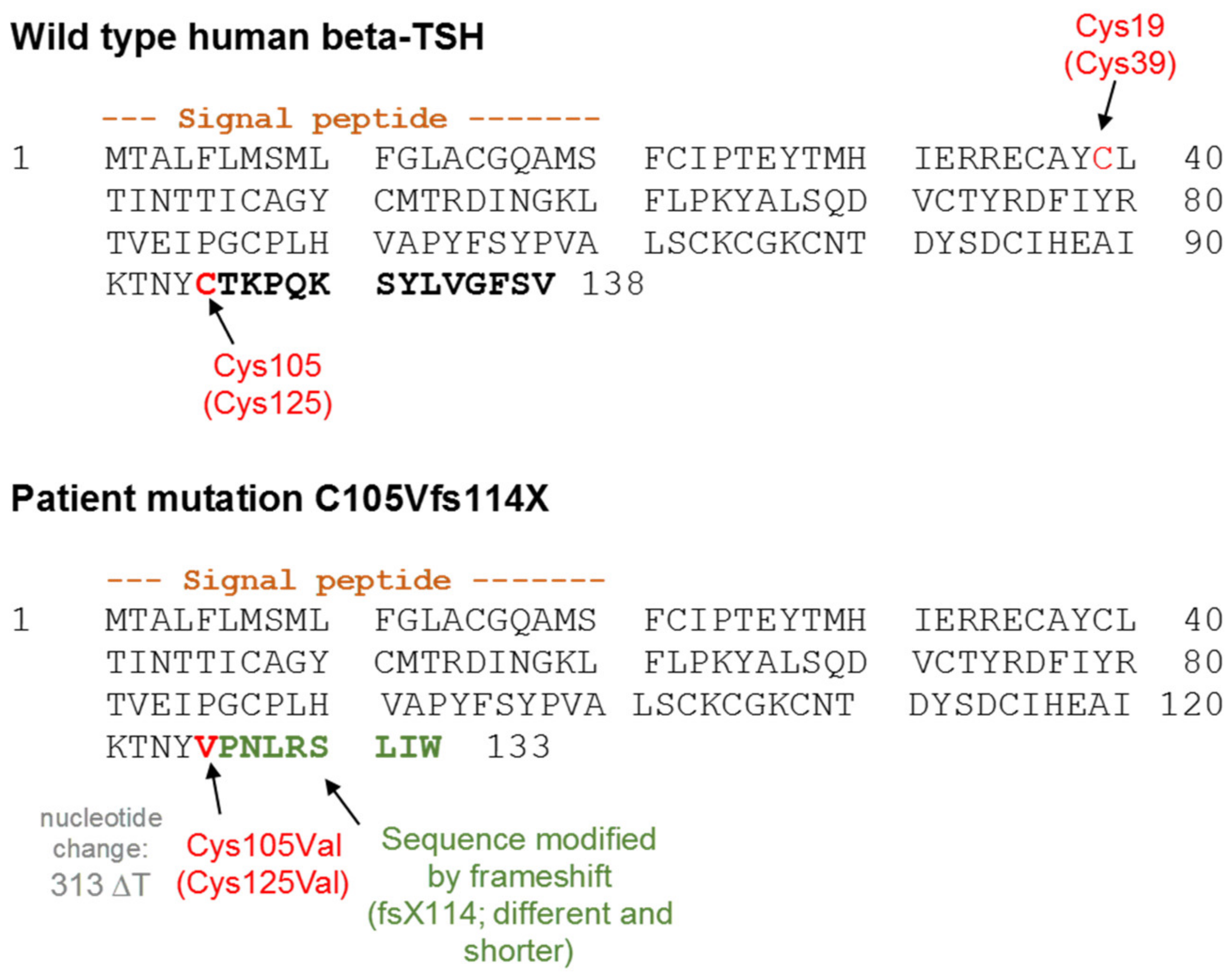
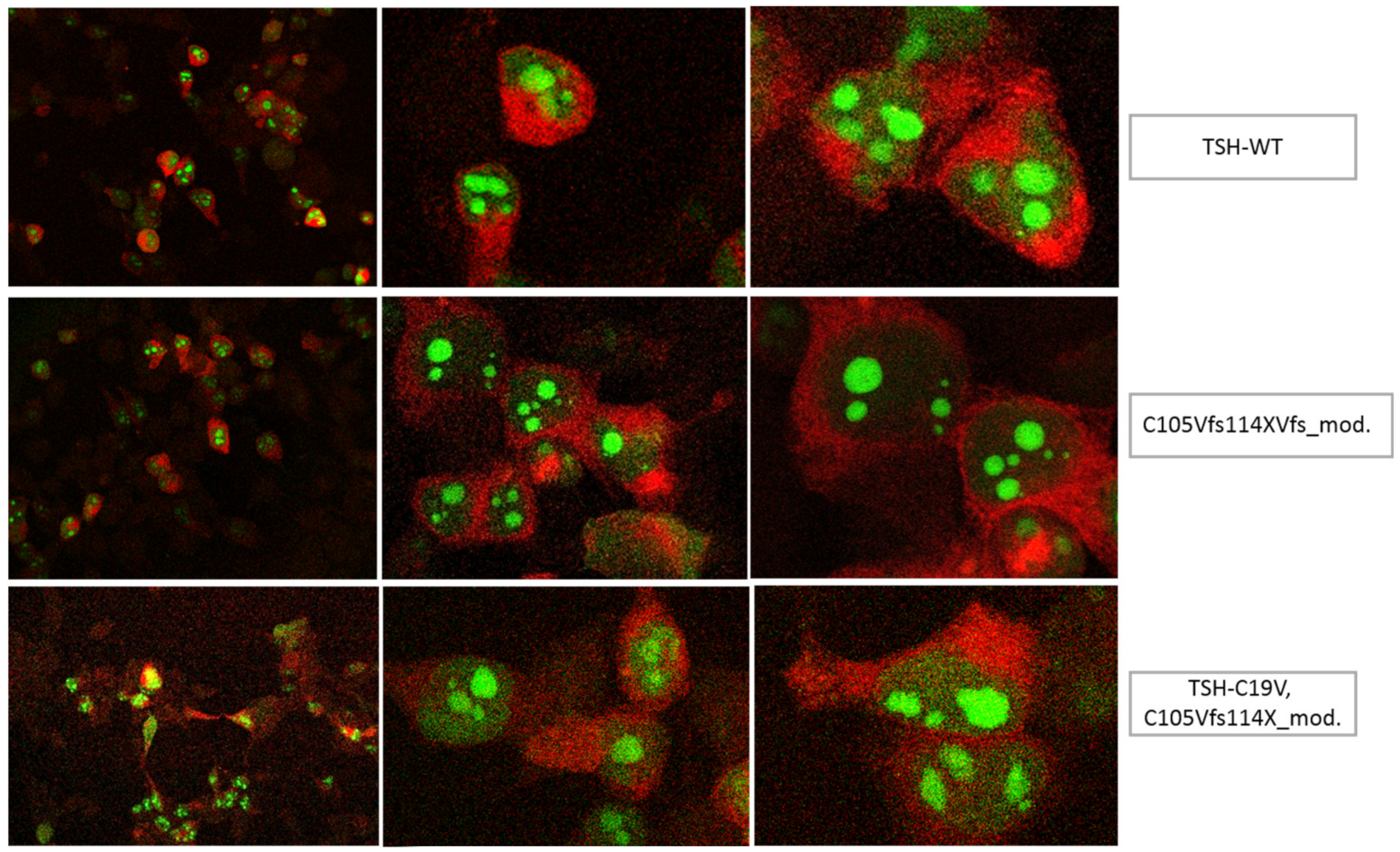
3.1. Altered AmCyan-P2A-mCherry Expression Pattern of the Frameshift Mutations
3.2. Signaling Capacities of the Mutation C105Vfs114X
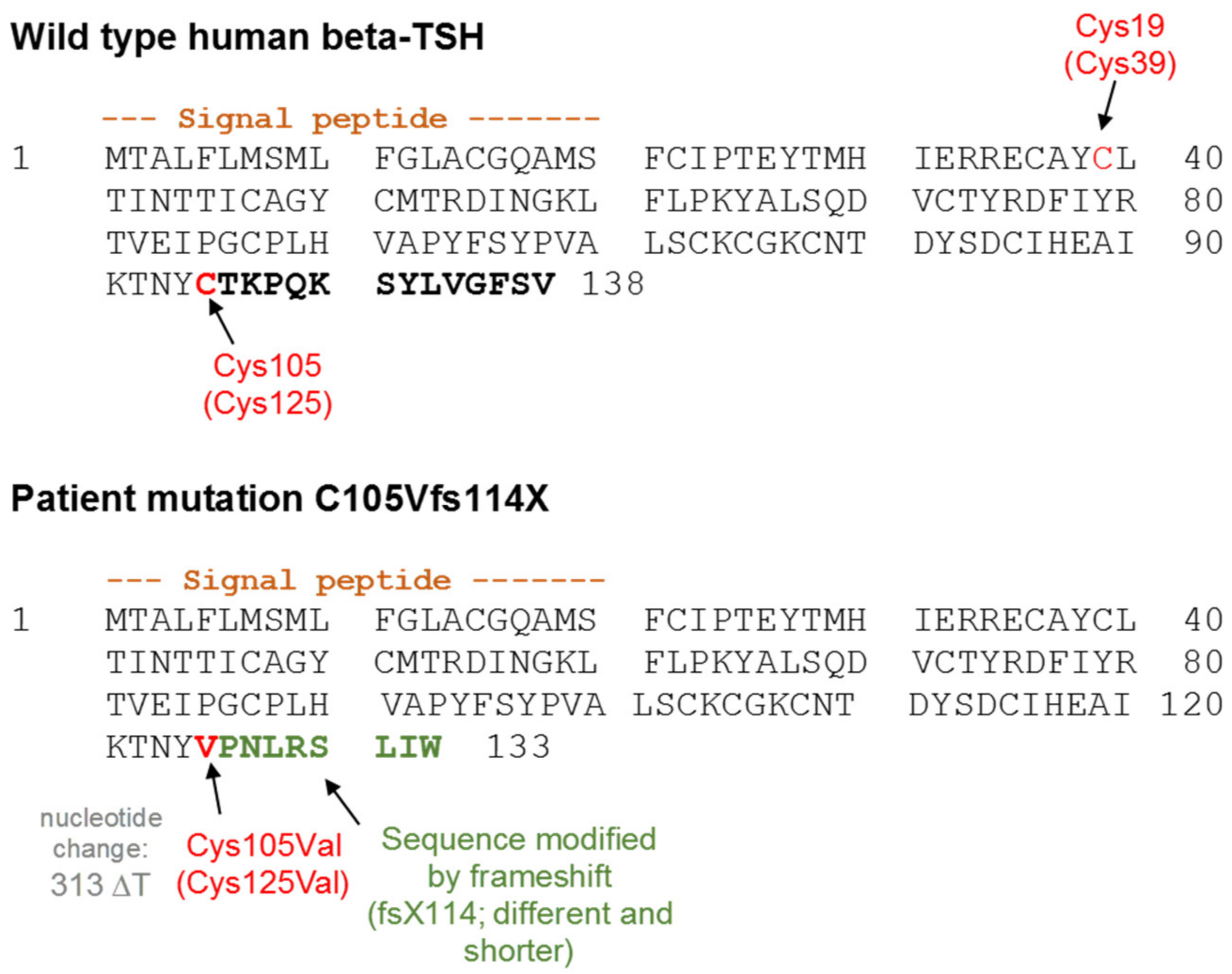
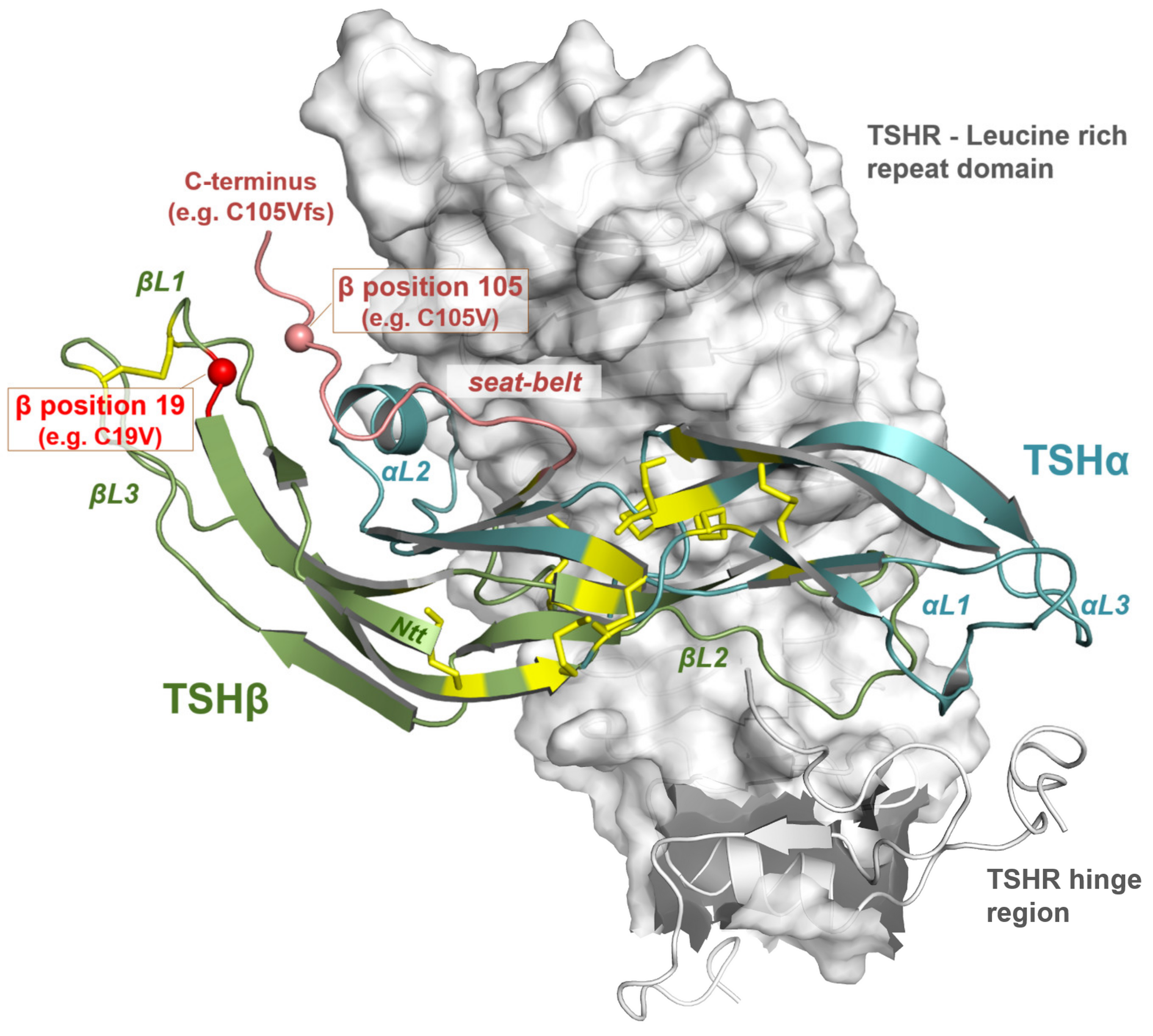
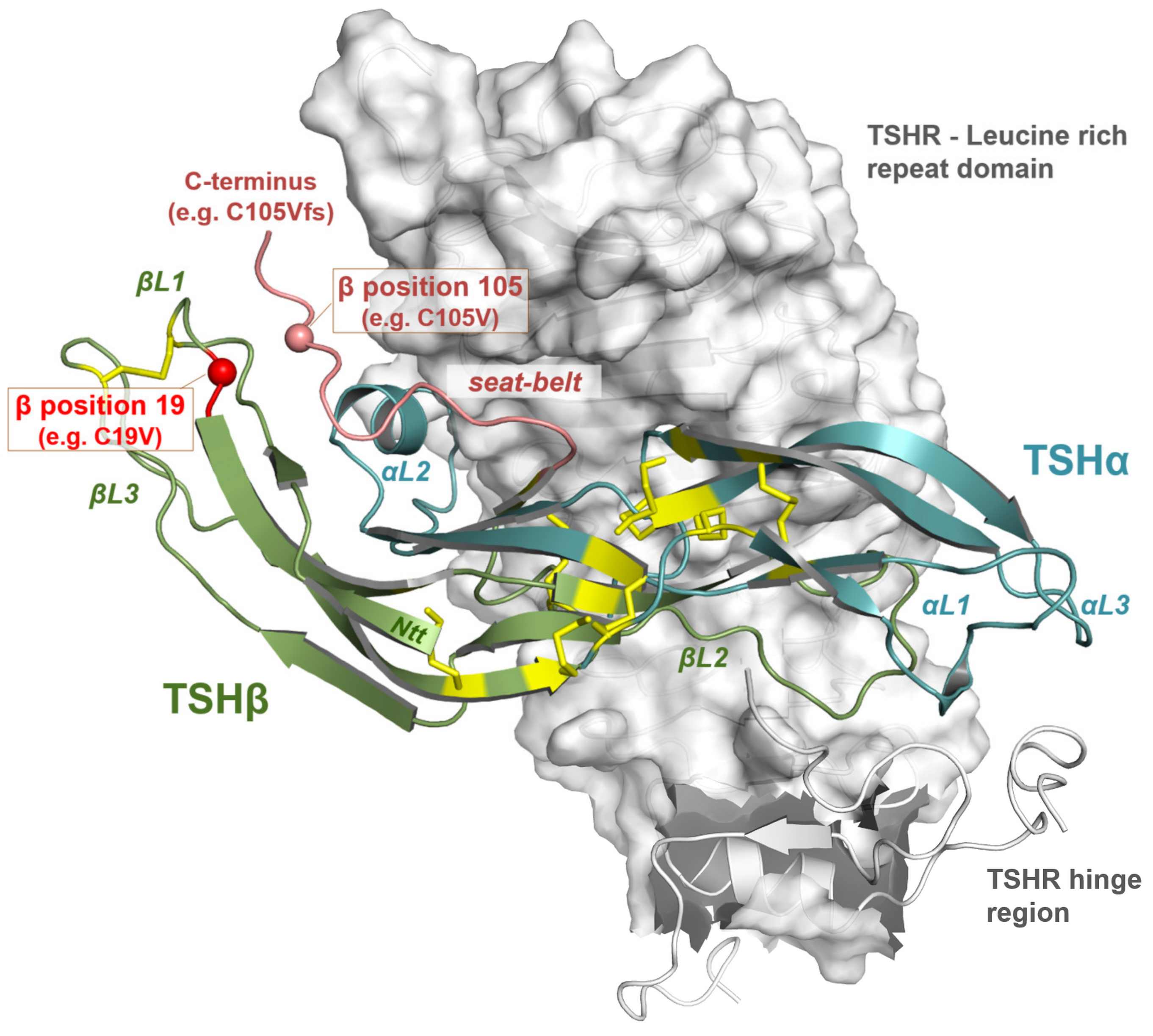
3.3. Impact of Potential TSH Structure Modification by Amino Acid Variations
3.4. Comparison with Other Pathogenic Pituitary Hormone Mutations
3.5. Clinical Features of the Mutation C105Vfs114X in Comparison with Other TSHB Mutations
4. Materials and Methods
4.1. Design of TSH-Mutants and Cloning Strategy
4.2. Cell Culture and Transfection
4.3. Protein Expression and Quantification
4.4. Functional Characterization
4.5. Software
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of Variance |
| bTSH | bovine Thyroid Stimulating Hormone |
| cAMP | cyclic Adenosine Monophosphate |
| CCH | Central Congenital Hypothyroidism |
| CGA | α-Subunit of Glycoproteinhormones (including TSH) |
| CGB | β-subunit of the Chorionic Gonadotropine |
| Ctt | C-Terminus/C-terminal |
| ELISA | Enzyme-linked Immunoabsorbent Assay |
| FSH | Follicle-Stimulating Hormone |
| FSHB | β-subunit of Follicle-Stimulating-Hormone |
| FTC cells | Follicular Thyroid Carcinoma Cells |
| GPCR | G-Protein Coupled Receptor |
| HEK cells | Human Embryonic Kidney Cells |
| hCG | human Chorionic Gonadotropine |
| hTSH | human Thyroid Stimulating Hormone |
| LH | Luteinizing Hormone |
| LHB | β-subunit of Luteinizing Hormone |
| MAPK | Mitogen-Activated Protein Kinases |
| NFAT | Nuclear Factor of Activated T-Cells |
| Ntt | T-Terminus/N-terminal |
| SRE | Serum Response Element |
| TSH | Thyroid Stimulating Hormone |
| TSHB | β-subunit of TSH |
| TSHR | Thyroid Stimulating Hormone Receptor |
| WT | Wildtype |
Appendix A

Appendix B
Appendix C
References
- Hanna, C.E.; Krainz, P.L.; Skeels, M.R.; Miyahira, R.S.; Sesser, D.E.; LaFranchi, S.H. Detection of congenital hypopituitary hypothyroidism: Ten-year experience in the Northwest Regional Screening Program. J. Pediatrics 1986, 109, 959–964. [Google Scholar] [CrossRef]
- Gruters, A.; Biebermann, H.; Krude, H. Neonatal thyroid disorders. Horm. Res. 2003, 59, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Gruters, A. Congenital hypothyroidism. Pediatr. Ann. 1992, 21, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Pfaffle, R.W.; Blankenstein, O.; Wuller, S.; Kentrup, H. Combined pituitary hormone deficiency: Role of Pit-1 and Prop-1. Acta Paediatr. Suppl. 1999, 88, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, R.; Chadli-Chaieb, M.; Vallette-Kasic, S.; Barlier, A.; Sarles, J.; Pellegrini-Bouiller, I.; Enjalbert, A.; Chaieb, L.; Brue, T. A familial form of congenital hypopituitarism due to a PROP1 mutation in a large kindred: Phenotypic and in vitro functional studies. J. Clin. Endocrinol. Metab. 2004, 89, 5779–5786. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, A.L.; Almonte, A.S.; Brown, M.R.; Fisher, D.A.; Baumbach, L.; Parks, J.S. Clinical and biochemical phenotype of familial anterior hypopituitarism from mutation of the PROP1 gene. J. Clin. Endocrinol. Metab. 1999, 84, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Wassner, A.J.; Cohen, L.E.; Hechter, E.; Dauber, A. Isolated central hypothyroidism in young siblings as a manifestation of PROP1 deficiency: Clinical impact of whole exome sequencing. Horm. Res. Paediatr. 2013, 79, 379–386. [Google Scholar] [CrossRef]
- Neumann, S.; Raaka, B.M.; Gershengorn, M.C. Constitutively active thyrotropin and thyrotropin-releasing hormone receptors and their inverse agonists. Methods Enzymol. 2010, 485, 147–160. [Google Scholar]
- Barbesino, G.; Sluss, P.M.; Caturegli, P. Central hypothyroidism in a patient with pituitary autoimmunity: Evidence for TSH-independent thyroid hormone synthesis. J. Clin. Endocrinol. Metab. 2012, 97, 345–350. [Google Scholar] [CrossRef]
- Persani, L. Clinical review: Central hypothyroidism: Pathogenic, diagnostic, and therapeutic challenges. J. Clin. Endocrinol. Metab. 2012, 97, 3068–3078. [Google Scholar] [CrossRef]
- Medeiros-Neto, G.; Herodotou, D.T.; Rajan, S.; Kommareddi, S.; de Lacerda, L.; Sandrini, R.; Boguszewski, M.C.; Hollenberg, A.N.; Radovick, S.; Wondisford, F.E. A circulating, biologically inactive thyrotropin caused by a mutation in the beta subunit gene. J. Clin. Investig. 1996, 97, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Morales, A.E.; Shi, J.D.; Wang, C.Y.; She, J.X.; Muir, A. Novel TSHbeta subunit gene mutation causing congenital central hypothyroidism in a newborn male. J. Pediatric Endocrinol. Metab. 2004, 17, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Baquedano, M.S.; Ciaccio, M.; Dujovne, N.; Herzovich, V.; Longueira, Y.; Warman, D.M.; Rivarola, M.A.; Belgorosky, A. Two novel mutations of the TSH-beta subunit gene underlying congenital central hypothyroidism undetectable in neonatal TSH screening. J. Clin. Endocrinol. Metab. 2010, 95, E98–E103. [Google Scholar] [CrossRef] [PubMed]
- Doeker, B.M.; Pfaffle, R.W.; Pohlenz, J.; Andler, W. Congenital central hypothyroidism due to a homozygous mutation in the thyrotropin beta-subunit gene follows an autosomal recessive inheritance. J. Clin. Endocrinol. Metab. 1998, 83, 1762–1765. [Google Scholar]
- Heinrichs, C.; Parma, J.; Scherberg, N.H.; Delange, F.; Van Vliet, G.; Duprez, L.; Bourdoux, P.; Bergmann, P.; Vassart, G.; Refetoff, S. Congenital central isolated hypothyroidism caused by a homozygous mutation in the TSH-beta subunit gene. Thyroid Off. J. Am. Thyroid Assoc. 2000, 10, 387–391. [Google Scholar] [CrossRef]
- Brumm, H.; Pfeufer, A.; Biebermann, H.; Schnabel, D.; Deiss, D.; Gruters, A. Congenital central hypothyroidism due to homozygous thyrotropin beta 313 Delta T mutation is caused by a Founder effect. J. Clin. Endocrinol. Metab. 2002, 87, 4811–4816. [Google Scholar] [CrossRef]
- Felner, E.I.; Dickson, B.A.; White, P.C. Hypothyroidism in siblings due to a homozygous mutation of the TSH-beta subunit gene. J. Pediatric Endocrinol. Metab. 2004, 17, 669–672. [Google Scholar] [CrossRef]
- Domene, H.M.; Gruneiro-Papendieck, L.; Chiesa, A.; Iorcansky, S.; Herzovich, V.C.; Papazian, R.; Forclaz, V.; Prieto, L.; Sanso, G.; Scaglia, P.; et al. The C105fs114X is the prevalent thyrotropin beta-subunit gene mutation in Argentinean patients with congenital central hypothyroidism. Horm. Res. 2004, 61, 41–46. [Google Scholar] [CrossRef]
- Karges, B.; LeHeup, B.; Schoenle, E.; Castro-Correia, C.; Fontoura, M.; Pfaffle, R.; Andler, W.; Debatin, K.M.; Karges, W. Compound heterozygous and homozygous mutations of the TSHbeta gene as a cause of congenital central hypothyroidism in Europe. Horm. Res. 2004, 62, 149–155. [Google Scholar]
- Partsch, C.J.; Riepe, F.G.; Krone, N.; Sippell, W.G.; Pohlenz, J. Initially elevated TSH and congenital central hypothyroidism due to a homozygous mutation of the TSH beta subunit gene: Case report and review of the literature. Exp. Clin. Endocrinol. Diabetes 2006, 114, 227–234. [Google Scholar] [CrossRef]
- Biebermann, H.; Liesenkotter, K.P.; Emeis, M.; Oblanden, M.; Gruters, A. Severe congenital hypothyroidism due to a homozygous mutation of the betaTSH gene. Pediatric Res. 1999, 46, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Ramos, H.E.; Labedan, I.; Carre, A.; Castanet, M.; Guemas, I.; Tron, E.; Madhi, F.; Delacourt, C.; Maciel, R.M.; Polak, M. New cases of isolated congenital central hypothyroidism due to homozygous thyrotropin beta gene mutations: A pitfall to neonatal screening. Thyroid Off. J. Am. Thyroid Assoc. 2010, 20, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, A.K.; Jaleel, S.; Lyons, G.; Schoenmakers, E.; Dattani, M.T.; Crowne, E.; Bernhard, B.; Kirk, J.; Roche, E.F.; Chatterjee, V.K.; et al. Molecular spectrum of TSHbeta subunit gene defects in central hypothyroidism in the UK and Ireland. Clin. Endocrinol. 2017, 86, 410–418. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.T.; Haugen, B.R.; Black, J.N.; Wood, W.M.; Gordon, D.F.; Ridgway, E.C. Congenital isolated central hypothyroidism caused by a “hot spot” mutation in the thyrotropin-beta gene. Thyroid Off. J. Am. Thyroid Assoc. 2002, 12, 1141–1146. [Google Scholar] [CrossRef]
- Deladoey, J.; Vuissoz, J.M.; Domene, H.M.; Malik, N.; Gruneiro-Papendieck, L.; Chiesa, A.; Heinrich, J.J.; Mullis, P.E. Congenital secondary hypothyroidism due to a mutation C105Vfs114X thyrotropin-beta mutation: Genetic study of five unrelated families from Switzerland and Argentina. Thyroid Off. J. Am. Thyroid Assoc. 2003, 13, 553–559. [Google Scholar] [CrossRef]
- Kleinau, G.; Kalveram, L.; Kohrle, J.; Szkudlinski, M.; Schomburg, L.; Biebermann, H.; Gruters-Kieslich, A. Minireview: Insights Into the Structural and Molecular Consequences of the TSH-beta Mutation C105Vfs114X. Mol. Endocrinol. 2016, 30, 954–964. [Google Scholar] [CrossRef]
- Grant, D.B.; Smith, I.; Fuggle, P.W.; Tokar, S.; Chapple, J. Congenital hypothyroidism detected by neonatal screening: Relationship between biochemical severity and early clinical features. Arch Dis. Child 1992, 67, 87–90. [Google Scholar] [CrossRef]
- Szkudlinski, M.W.; Fremont, V.; Ronin, C.; Weintraub, B.D. Thyroid-stimulating hormone and thyroid-stimulating hormone receptor structure-function relationships. Physiol. Rev. 2002, 82, 473–502. [Google Scholar] [CrossRef]
- Biebermann, H.; Winkler, F.; Handke, D.; Gruters, A.; Krude, H.; Kleinau, G. Molecular description of non-autoimmune hyperthyroidism at a neonate caused by a new thyrotropin receptor germline mutation. Thyroid Res. 2011, 4, S8. [Google Scholar] [CrossRef]
- Kero, J.; Ahmed, K.; Wettschureck, N.; Tunaru, S.; Wintermantel, T.; Greiner, E.; Schutz, G.; Offermanns, S. Thyrocyte-specific Gq/G11 deficiency impairs thyroid function and prevents goiter development. J. Clin. Investig. 2007, 117, 2399–2407. [Google Scholar] [CrossRef] [Green Version]
- Allgeier, A.; Offermanns, S.; Van Sande, J.; Spicher, K.; Schultz, G.; Dumont, J.E. The human thyrotropin receptor activates G-proteins Gs and Gq/11. J. Biol. Chem. 1994, 269, 13733–13735. [Google Scholar] [PubMed]
- Laugwitz, K.L.; Allgeier, A.; Offermanns, S.; Spicher, K.; Van Sande, J.; Dumont, J.E.; Schultz, G. The human thyrotropin receptor: A heptahelical receptor capable of stimulating members of all four G protein families. Proc. Natl. Acad. Sci. USA 1996, 93, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Van Sande, J.; Raspe, E.; Perret, J.; Lejeune, C.; Maenhaut, C.; Vassart, G.; Dumont, J.E. Thyrotropin activates both the cyclic AMP and the PIP2 cascades in CHO cells expressing the human cDNA of TSH receptor. Mol. Cell. Endocrinol. 1990, 74, R1–R6. [Google Scholar] [CrossRef]
- Potorac, I.; Rivero-Muller, A.; Trehan, A.; Kielbus, M.; Jozwiak, K.; Pralong, F.; Hafidi, A.; Thiry, A.; Menage, J.J.; Huhtaniemi, I.; et al. A vital region for human glycoprotein hormone trafficking revealed by an LHB mutation. J. Endocrinol. 2016, 231, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Kleinau, G.; Neumann, S.; Gruters, A.; Krude, H.; Biebermann, H. Novel Insights on Thyroid Stimulating Hormone Receptor Signal Transduction. Endocr. Rev. 2013, 34, 691–724. [Google Scholar] [CrossRef]
- Krause, G.; Kreuchwig, A.; Kleinau, G. Extended and structurally supported insights into extracellular hormone binding, signal transduction and organization of the thyrotropin receptor. PLoS ONE 2012, 7, e52920. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, H.; Chen, X.; Chen, P.H.; Fischer, D.; Sriraman, V.; Yu, H.N.; Arkinstall, S.; He, X. Structure of follicle-stimulating hormone in complex with the entire ectodomain of its receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 12491–12496. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, K.; Sato, K.; Shizume, K.; Kanaji, Y.; Ito, Y.; Obara, T.; Nakagawa, T.; Koizumi, T.; Nishimura, R. Potent thyrotropic activity of human chorionic gonadotropin variants in terms of 125I incorporation and de novo synthesized thyroid hormone release in human thyroid follicles. J. Clin. Endocrinol. Metab. 1995, 80, 473–479. [Google Scholar]
- Szkudlinski, M.W.; Teh, N.G.; Grossmann, M.; Tropea, J.E.; Weintraub, B.D. Engineering human glycoprotein hormone superactive analogues. Nat. Biotechnol. 1996, 14, 1257–1263. [Google Scholar] [CrossRef]
- Mueller, S.; Kleinau, G.; Jaeschke, H.; Paschke, R.; Krause, G. Extended hormone binding site of the human thyroid stimulating hormone receptor: Distinctive acidic residues in the hinge region are involved in bovine thyroid stimulating hormone binding and receptor activation. J. Biol. Chem. 2008, 283, 18048–18055. [Google Scholar] [CrossRef]
- Derwahl, M.; Kuemmel, M.; Goretzki, P.; Schatz, H.; Broecker, M. Expression of the human TSH receptor in a human thyroid carcinoma cell line that lacks an endogenous TSH receptor: Growth inhibition by cAMP. Biochem. Biophys. Res. Commun. 1993, 191, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F.; Kleinau, G.; Tarnow, P.; Rediger, A.; Grohmann, L.; Gaetjens, I.; Krause, G.; L’Allemand, D.; Gruters, A.; Krude, H.; et al. A new phenotype of nongoitrous and nonautoimmune hyperthyroidism caused by a heterozygous thyrotropin receptor mutation in transmembrane helix 6. J. Clin. Endocrinol. Metab. 2010, 95, 3605–3610. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Puett, D. Delineation via site-directed mutagenesis of the carboxyl-terminal region of human choriogonadotropin beta required for subunit assembly and biological activity. J. Biol. Chem. 1991, 266, 6904–6908. [Google Scholar] [PubMed]
- Mishra, A.K.; Mahale, S.D.; Iyer, K.S. Disulfide bonds Cys(9)-Cys(57), Cys(34)-Cys(88) and Cys(38)-Cys(90) of the beta-subunit of human chorionic gonadotropin are crucial for heterodimer formation with the alpha-subunit: Experimental evidence for the conclusions from the crystal structure of hCG. Biochim. Biophys. Acta 2003, 1645, 49–55. [Google Scholar] [PubMed]
- Riccetti, L.; Yvinec, R.; Klett, D.; Gallay, N.; Combarnous, Y.; Reiter, E.; Simoni, M.; Casarini, L.; Ayoub, M.A. Human Luteinizing Hormone and Chorionic Gonadotropin Display Biased Agonism at the LH and LH/CG Receptors. Sci. Rep. 2017, 7, 940. [Google Scholar] [CrossRef] [PubMed]
- Okajima, Y.; Nagasaki, H.; Suzuki, C.; Suga, H.; Ozaki, N.; Arima, H.; Hamada, Y.; Civelli, O.; Oiso, Y. Biochemical roles of the oligosaccharide chains in thyrostimulin, a heterodimeric hormone of glycoprotein hormone subunits alpha 2 (GPA2) and beta 5 (GPB5). Regul. Pept. 2008, 148, 62–67. [Google Scholar] [CrossRef]
- Themmen, A.P. An update of the pathophysiology of human gonadotrophin subunit and receptor gene mutations and polymorphisms. Reproduction 2005, 130, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Misgar, R.A.; Wani, A.I.; Bankura, B.; Bashir, M.I.; Roy, A.; Das, M. FSH beta-subunit mutations in two sisters: The first report from the Indian sub-continent and review of previous cases. Gynecol. Endocrinol. 2019, 35, 290–293. [Google Scholar] [CrossRef]
- Nagirnaja, L.; Venclovas, C.; Rull, K.; Jonas, K.C.; Peltoketo, H.; Christiansen, O.B.; Kairys, V.; Kivi, G.; Steffensen, R.; Huhtaniemi, I.T.; et al. Structural and functional analysis of rare missense mutations in human chorionic gonadotrophin beta-subunit. Mol. Hum. Reprod. 2012, 18, 379–390. [Google Scholar] [CrossRef]
- Vuissoz, J.M.; Deladoey, J.; Buyukgebiz, A.; Cemeroglu, P.; Gex, G.; Gallati, S.; Mullis, P.E. New autosomal recessive mutation of the TSH-beta subunit gene causing central isolated hypothyroidism. J. Clin. Endocrinol. Metab. 2001, 86, 4468–4471. [Google Scholar]
- Davies, T.F.; Ando, T.; Lin, R.Y.; Tomer, Y.; Latif, R. Thyrotropin receptor-associated diseases: From adenomata to Graves disease. J. Clin. Investig. 2005, 115, 1972–1983. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, B.; Chazenbalk, G.D.; Jaume, J.C.; McLachlan, S.M. The thyrotropin (TSH) receptor: Interaction with TSH and autoantibodies. Endocr. Rev. 1998, 19, 673–716. [Google Scholar] [PubMed]
- Dacou-Voutetakis, C.; Feltquate, D.M.; Drakopoulou, M.; Kourides, I.A.; Dracopoli, N.C. Familial hypothyroidism caused by a nonsense mutation in the thyroid-stimulating hormone beta-subunit gene. Am. J. Hum. Genet. 1990, 46, 988–993. [Google Scholar] [PubMed]
- Trehan, A.; Kielbus, M.; Czapinski, J.; Stepulak, A.; Huhtaniemi, I.; Rivero-Muller, A. REPLACR-mutagenesis, a one-step method for site-directed mutagenesis by recombineering. Sci. Rep. 2016, 6, 19121. [Google Scholar] [CrossRef] [PubMed]
- Derwahl, M.; Hamacher, C.; Russo, D.; Broecker, M.; Manole, D.; Schatz, H.; Kopp, P.; Filetti, S. Constitutive activation of the Gs alpha protein-adenylate cyclase pathway may not be sufficient to generate toxic thyroid adenomas. J. Clin. Endocrinol. Metab. 1996, 81, 1898–1904. [Google Scholar] [PubMed]
- Fischer, J.; Kleinau, G.; Rutz, C.; Zwanziger, D.; Khajavi, N.; Muller, A.; Rehders, M.; Brix, K.; Worth, C.L.; Fuhrer, D.; et al. Evidence of G-protein-coupled receptor and substrate transporter heteromerization at a single molecule level. Cell Mol Life Sci 2018, 75, 2227–2239. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Forward Primer | Reverse Primer | |
|---|---|---|---|
| 1 | TSHB_C105Vfs114X | TSHB_C105Vfs114X_FP CCATCAAGACAAACTACGTACCAAACCTC | TSHB_C105Vfs114X_C_V_RP GTAGTTTGTCTTGATGGCTTCATGTATGC |
| 2 | TSHB_C105V | TSHB_C105V_FP CCATCAAGACAAACTACGTTACCAAACCTC | TSHB_C105V_C_V_RP GTAGTTTGTCTTGATGGCTTCATGTATGC |
| 3 | TSHB_C19V | TSHB_C19V_FP GGAGAGAGTGTGCTTATGTCCTAACCATC | TSHB_C19V_RP GACATAAGCACACTCTCTCCTTTCGATG (CATCGAAAGGAGAGAGTGTGCTTATGTC) |
| 4 | Lin_TSHB (Linearized vector for Gibson Assembly cloning) | Lin_TSHB_Gibs_FP ACCAAACCTCAGAAGTCTTATCTGG | Lin_TSHB_Gibs_RP ATAAGCACACTCTCTCCTTTCGATG (CATCGAAAGGAGAGAGTGTGCTTAT) |
| 5 | TSHB_C19V_ C105V (Insert) | TSHB_C19V_FP GGAGAGAGTGTGCTTATGTCCTAACCATC | TSHB_C19V_ C105V_RP GACTTCTGAGGTTTGGTAACGTAGTTTGTC (GACAAACTACGTTACCAAACCTCAGAAGTC) |
| 6 | TSHB_ C19V_C105Vfs114X (Insert) | TSHB_C19V_FP GGAGAGAGTGTGCTTATGTCCTAACCATC | TSHB_ C19V_C105Vfs114X_RP GACTTCTGAGGTTTGGTACGTAGTTTGTC (GACAAACTACGTACCAAACCTCAGAAGTC) |
| 7 | TSHB_C105Vfs114X_mod. (Insert) | TSHB_C105Vfs114X_mod.FP CCTCAGAAGTCTTATCTGGTATGGATTTTCTGTCGTGAGCAAG | TSHB_C105Vfs114X_mod.RP CTTGCTCACGACAGAAAATCCATACCAGATAAGACTTCTGAGG |
| 8 | TSHB_C19V_C105Vfs114X_mod(Insert) | TSHB_C19V, C105Vfs114X_mod.FP CCTCAGAAGTCTTATCTGGTATGGATTTTCTGTCGTGAGCAAG | TSHB_C19V, C105Vfs114X_mod.RP CTTGCTCACGACAGAAAATCCATACCAGATAAGACTTCTGAGG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalveram, L.; Kleinau, G.; Szymańska, K.; Scheerer, P.; Rivero-Müller, A.; Grüters-Kieslich, A.; Biebermann, H. The Pathogenic TSH β-Subunit Variant C105Vfs114X Causes a Modified Signaling Profile at TSHR. Int. J. Mol. Sci. 2019, 20, 5564. https://doi.org/10.3390/ijms20225564
Kalveram L, Kleinau G, Szymańska K, Scheerer P, Rivero-Müller A, Grüters-Kieslich A, Biebermann H. The Pathogenic TSH β-Subunit Variant C105Vfs114X Causes a Modified Signaling Profile at TSHR. International Journal of Molecular Sciences. 2019; 20(22):5564. https://doi.org/10.3390/ijms20225564
Chicago/Turabian StyleKalveram, Laura, Gunnar Kleinau, Kamila Szymańska, Patrick Scheerer, Adolfo Rivero-Müller, Annette Grüters-Kieslich, and Heike Biebermann. 2019. "The Pathogenic TSH β-Subunit Variant C105Vfs114X Causes a Modified Signaling Profile at TSHR" International Journal of Molecular Sciences 20, no. 22: 5564. https://doi.org/10.3390/ijms20225564