Mouse Models of Human Claudin-Associated Disorders: Benefits and Limitations


Abstract
1. Introduction
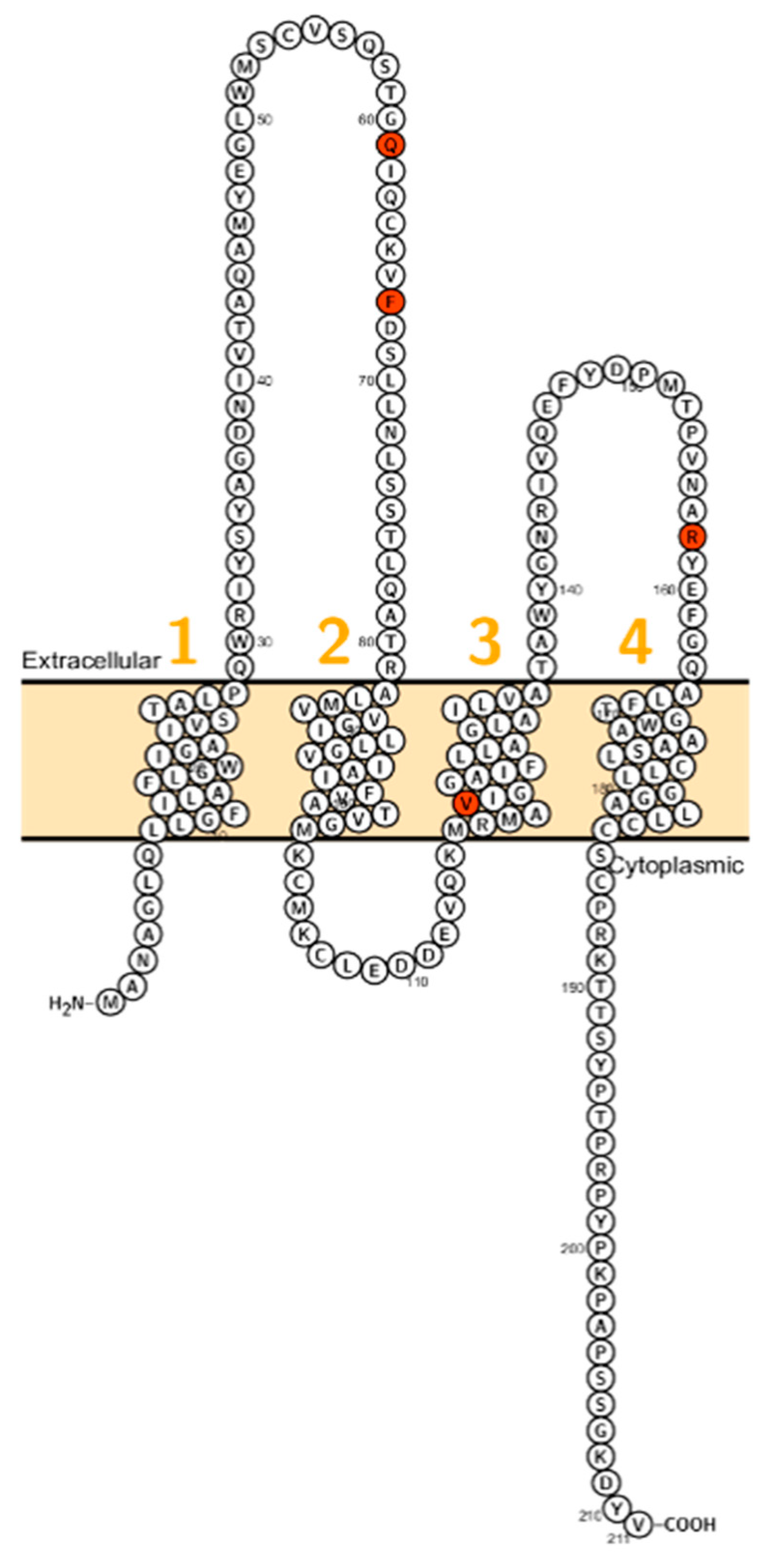
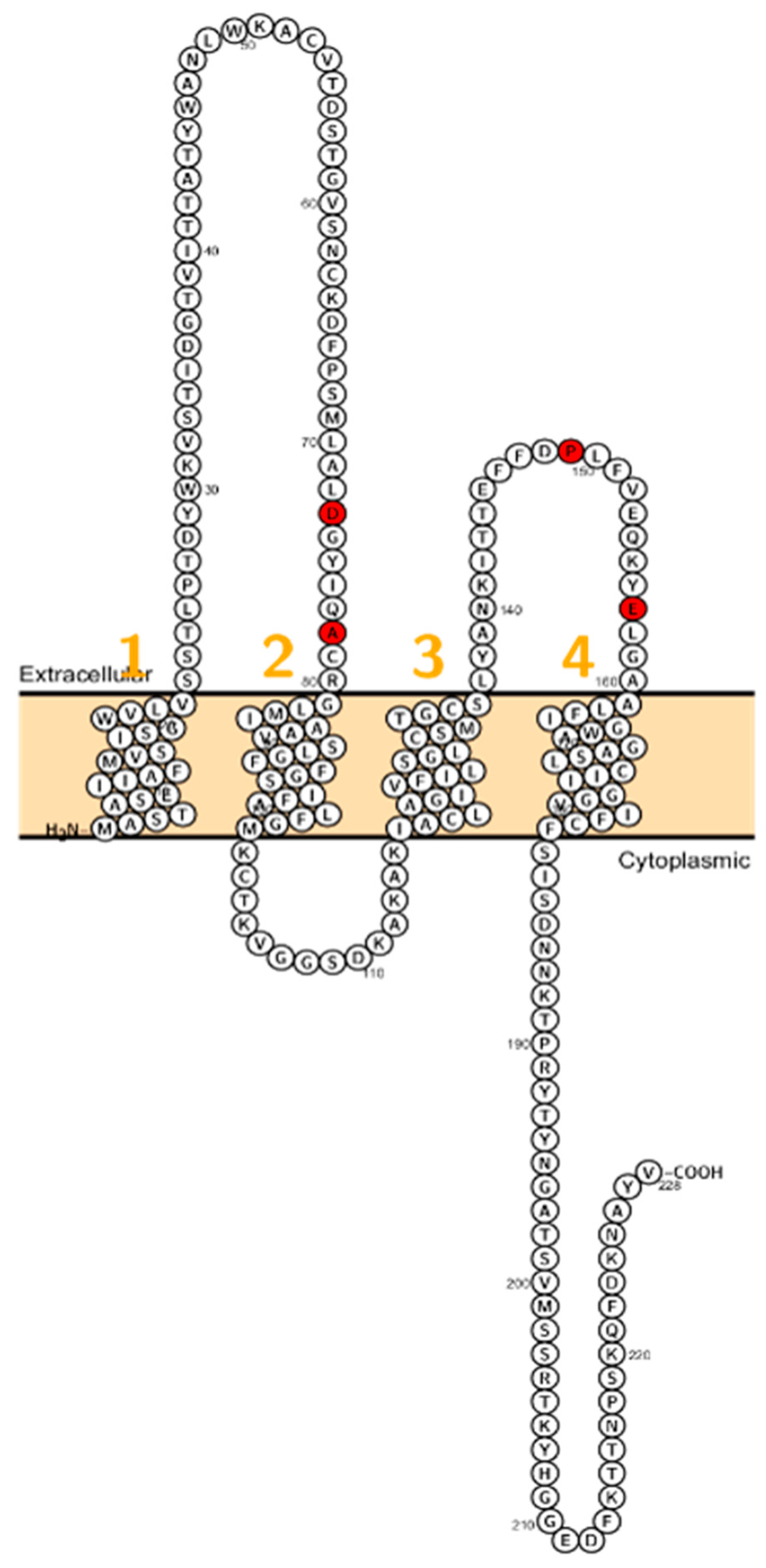
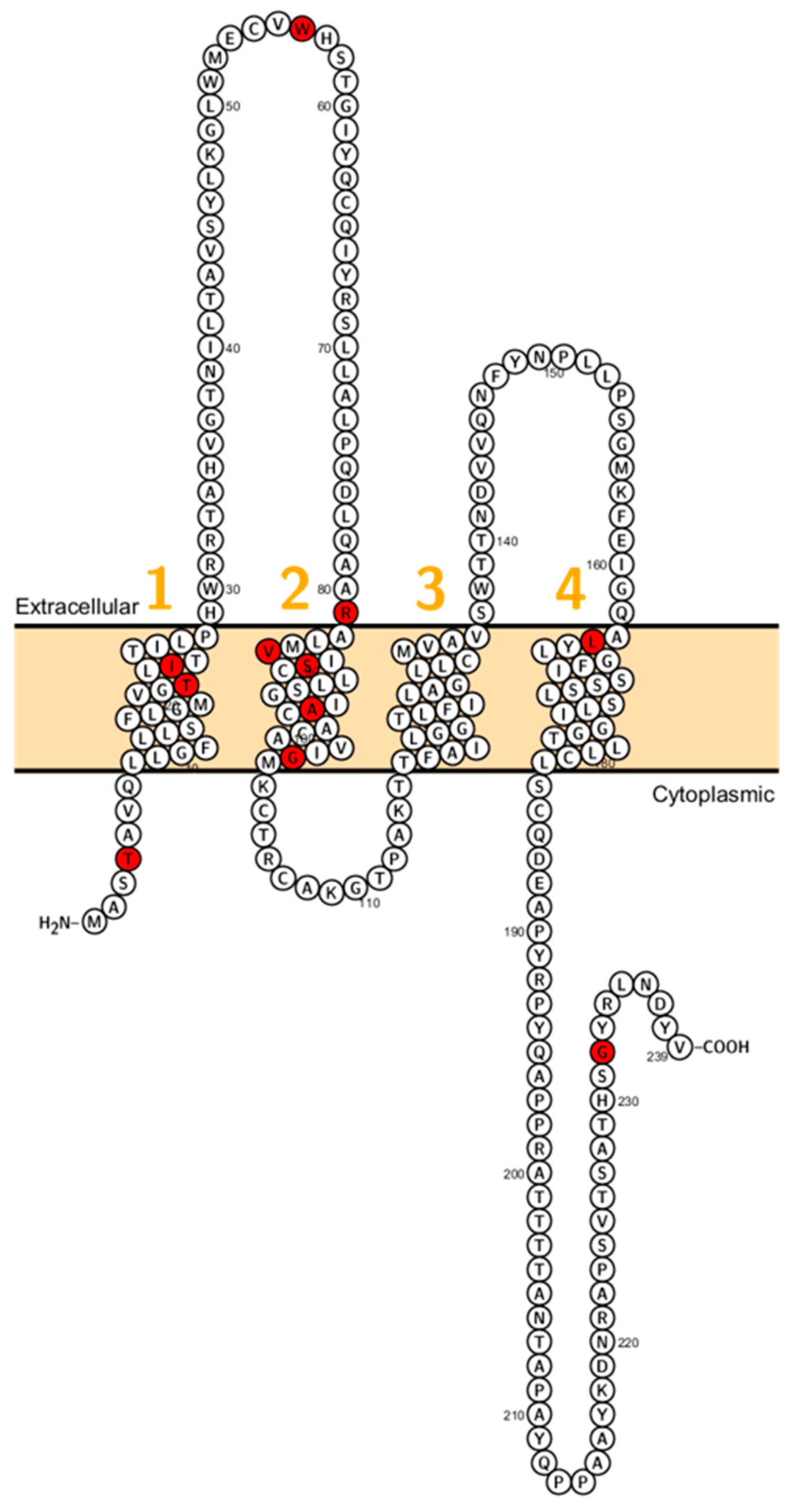
2. Claudin Mutations Causing Human Disorders
2.1. Claudin 1
2.2. Claudin 2
2.3. Claudin 9
2.4. Claudin 10
2.5. Claudin 14
2.6. Claudin 16
2.7. Claudin 19
3. Discussion
Funding
Conflicts of Interest
References
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Baud, L. Renal Epithelial Cells: Differentiation and Plasticity. J. Am. Soc. Nephrol. 2003, 14, S1–S2. [Google Scholar] [CrossRef]
- Macara, I.G.; Guyer, R.; Richardson, G.; Huo, Y.; Ahmed, S.M. Epithelial homeostasis. Curr. Biol. 2014, 24, R815–R825. [Google Scholar] [CrossRef]
- Roignot, J.; Peng, X.; Mostov, K. Polarity in Mammalian Epithelial Morphogenesis. Cold Spring Harb. Perspect. Biol. 2013, 5, a013789. [Google Scholar] [CrossRef] [PubMed]
- Karasov, W.H. Integrative physiology of transcellular and paracellular intestinal absorption. J. Exp. Biol. 2017, 220, 2495–2501. [Google Scholar] [CrossRef]
- Sterling, T.M.; Nemere, I. Calcium uptake and membrane trafficking in response to PTH or 25(OH)D3 in polarized intestinal epithelial cells. Steroids 2007, 72, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Cunha, T.D.S.; Heilberg, I.P. Bartter syndrome: Causes, diagnosis, and treatment. Int. J. Nephrol. Renov. Dis. 2018, 11, 291–301. [Google Scholar] [CrossRef]
- Knoers, N.V.; Levtchenko, E.N. Gitelman syndrome. Orphanet J. Rare Dis. 2008, 3, 22. [Google Scholar] [CrossRef]
- Furuse, M.; Hata, M.; Furuse, K.; Yoshida, Y.; Haratake, A.; Sugitani, Y.; Noda, T.; Kubo, A.; Tsukita, S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: A lesson from claudin-1-deficient mice. J. Cell Biol. 2002, 156, 1099–1111. [Google Scholar] [CrossRef]
- Nakano, Y.; Kim, S.H.; Kim, H.M.; Sanneman, J.D.; Zhang, Y.; Smith, R.J.; Marcus, D.C.; Wangemann, P.; Nessler, R.A.; Banfi, B. A claudin-9-based ion permeability barrier is essential for hearing. PLoS Genet. 2009, 5, e1000610. [Google Scholar] [CrossRef]
- Breiderhoff, T.; Himmerkus, N.; Stuiver, M.; Mutig, K.; Will, C.; Meij, I.C.; Bachmann, S.; Bleich, M.; Willnow, T.E.; Müller, D. Deletion of claudin-10 (Cldn10) in the thick ascending limb impairs paracellular sodium permeability and leads to hypermagnesemia and nephrocalcinosis. Proc. Natl. Acad. Sci. USA 2012, 109, 14241–14246. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yosef, T.; Belyantseva, I.A.; Saunders, T.L.; Hughes, E.D.; Kawamoto, K.; Van Itallie, C.M.; Beyer, L.A.; Halsey, K.; Gardner, D.J.; Wilcox, E.R.; et al. Claudin 14 knockout mice, a model for autosomal recessive deafness DFNB29, are deaf due to cochlear hair cell degeneration. Hum. Mol. Genet. 2003, 12, 2049–2061. [Google Scholar] [CrossRef]
- Will, C.; Breiderhoff, T.; Thumfart, J.; Stuiver, M.; Kopplin, K.; Sommer, K.; Gunzel, D.; Querfeld, U.; Meij, I.C.; Shan, Q.; et al. Targeted deletion of murine Cldn16 identifies extra- and intrarenal compensatory mechanisms of Ca2+ and Mg2+ wasting. Am. J. Physiol. Ren. Physiol. 2010, 298, F1152–F1161. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Furuse, M. The structure and function of claudins, cell adhesion molecules at tight junctions. Ann. N. Y. Acad. Sci. 2000, 915, 129–135. [Google Scholar] [CrossRef]
- Günzel, D.; Yu, A.S.L. Claudins and the Modulation of Tight Junction Permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [PubMed]
- Krause, G.; Winkler, L.; Mueller, S.L.; Haseloff, R.F.; Piontek, J.; Blasig, I.E. Structure and function of claudins. Biochim. Biophys. Acta 2008, 1778, 631–645. [Google Scholar] [CrossRef]
- Sanjad, S.A.; Hariri, A.; Habbal, Z.M.; Lifton, R.P. A novel PCLN-1 gene mutation in familial hypomagnesemia with hypercalciuria and atypical phenotype. Pediatr. Nephrol. 2007, 22, 503–508. [Google Scholar] [CrossRef]
- Hadj-Rabia, S.; Baala, L.; Vabres, P.; Hamel-Teillac, D.; Jacquemin, E.; Fabre, M.; Lyonnet, S.; De Prost, Y.; Munnich, A.; Hadchouel, M.; et al. Claudin-1 gene mutations in neonatal sclerosing cholangitis associated with ichthyosis: A tight junction disease. Gastroenterology 2004, 127, 1386–1390. [Google Scholar] [CrossRef]
- Sineni, C.J.; Yildirim-Baylan, M.; Guo, S.; Camarena, V.; Wang, G.; Tokgoz-Yilmaz, S.; Duman, D.; Bademci, G.; Tekin, M. A truncating CLDN9 variant is associated with autosomal recessive nonsyndromic hearing loss. Hum. Genet. 2019, 138, 1071–1075. [Google Scholar] [CrossRef]
- Hadj-Rabia, S.; Brideau, G.; Al-Sarraj, Y.; Maroun, R.C.; Figueres, M.-L.; Leclerc-Mercier, S.; Olinger, E.; Baron, S.; Chaussain, C.; Nochy, D.; et al. Multiplex epithelium dysfunction due to CLDN10 mutation: The HELIX syndrome. Genet. Med. 2017, 20, 190–201. [Google Scholar] [CrossRef]
- Wilcox, E.R.; Burton, Q.L.; Naz, S.; Riazuddin, S.; Smith, T.N.; Ploplis, B.; Belyantseva, I.; Ben-Yosef, T.; Liburd, N.A.; Morell, R.J.; et al. Mutations in the Gene Encoding Tight Junction Claudin-14 Cause Autosomal Recessive Deafness DFNB29. Cell 2001, 104, 165–172. [Google Scholar] [CrossRef]
- Hampson, G.; A Konrad, M.; Scoble, J. Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHNC): Compound heterozygous mutation in the claudin 16 (CLDN16) gene. BMC Nephrol. 2008, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Konrad, M.; Schaller, A.; Seelow, D.; Pandey, A.V.; Waldegger, S.; Lesslauer, A.; Vitzthum, H.; Suzuki, Y.; Luk, J.M.; Becker, C.; et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am. J. Hum. Genet. 2006, 79, 949–957. [Google Scholar] [CrossRef] [PubMed]
- De Santis, S.; Cavalcanti, E.; Mastronardi, M.; Jirillo, E.; Chieppa, M. Nutritional Keys for Intestinal Barrier Modulation. Front. Immunol. 2015, 6, 612. [Google Scholar] [CrossRef] [PubMed]
- Merikallio, H.; Kaarteenaho, R.; Pääkkö, P.; Lehtonen, S.; Hirvikoski, P.; Mäkitaro, R.; Harju, T.; Soini, Y. Impact of smoking on the expression of claudins in lung carcinoma. Eur. J. Cancer 2011, 47, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, A.; Wang, X.; Gabello, M.; Valenzano, M.C.; Soler, A.P.; Ko, A.; Morin, P.J.; Mullin, J.M. Dietary methionine restriction improves colon tight junction barrier function and alters claudin expression pattern. Am. J. Physiol. Physiol. 2010, 299, C1028–C1035. [Google Scholar] [CrossRef]
- Soini, Y. Claudins in lung diseases. Respir. Res. 2011, 12, 70. [Google Scholar] [CrossRef]
- Barmeyer, C.; Schulzke, J.D.; Fromm, M. Claudin-related intestinal diseases. Semin. Cell Dev. Biol. 2015, 42, 30–38. [Google Scholar] [CrossRef]
- Limaye, A.; Hall, B.; Kulkarni, A.B. Manipulation of mouse embryonic stem cells for knockout mouse production. Curr. Protoc. Cell Biol. 2009, 44, 19.13.1–19.13.24. [Google Scholar]
- Kooij, G.; Kopplin, K.; Blasig, R.; Stuiver, M.; Koning, N.; Goverse, G.; van der Pol, S.M.; van Het Hof, B.; Gollasch, M.; Drexhage, J.A.; et al. Disturbed function of the blood-cerebrospinal fluid barrier aggravates neuro-inflammation. Acta Neuropathol. 2014, 128, 267–277. [Google Scholar] [CrossRef]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, B.L.; Ward, C.; Smith, T.; Ritzenthaler, J.D.; Koval, M. Regulation of Heterotypic Claudin Compatibility. J. Biol. Chem. 2007, 282, 30005–30013. [Google Scholar] [CrossRef] [PubMed]
- Piontek, J.; Fritzsche, S.; Cording, J.; Richter, S.; Hartwig, J.; Walter, M.; Yu, D.; Turner, J.R.; Gehring, C.; Rahn, H.-P.; et al. Elucidating the principles of the molecular organization of heteropolymeric tight junction strands. Cell. Mol. Life Sci. 2011, 68, 3903–3918. [Google Scholar] [CrossRef] [PubMed]
- Zorko, M.S.; Veraniä, P.; Leskovec, N.K.; Pavloviä, M.D.; Pavlović, M.D.; Veranič, P. Expression of tight-junction proteins in the inflamed and clinically uninvolved skin in patients with venous leg ulcers. Clin. Exp. Dermatol. 2009, 34, e949–e952. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.L.; Lamas, M.; Martin, D.; Namorado, M.D.C.; Islas, S.; Luna, J.; Tauc, M.; Gonzalez-Mariscal, L.; Gonz, L. The renal segmental distribution of claudins changes with development. Kidney Int. 2002, 62, 476–487. [Google Scholar] [CrossRef]
- Laurila, J.J.; Karttunen, T.; Koivukangas, V.; Laurila, P.A.; Syrjala, H.; Saarnio, J.; Soini, Y.; Ala-Kokko, T.I. Tight junction proteins in gallbladder epithelium: Different expression in acute acalculous and calculous cholecystitis. J. Histochem. Cytochem. 2007, 55, 567–573. [Google Scholar] [CrossRef]
- Zhu, Y.; Brannstrom, M.; Janson, P.O.; Sundfeldt, K. Differences in expression patterns of the tight junction proteins, claudin 1, 3, 4 and 5, in human ovarian surface epithelium as compared to epithelia in inclusion cysts and epithelial ovarian tumours. Int. J. Cancer 2006, 118, 1884–1891. [Google Scholar] [CrossRef]
- Kitajiri, S.-I.; Furuse, M.; Morita, K.; Saishin-Kiuchi, Y.; Kido, H.; Ito, J.; Tsukita, S. Expression patterns of claudins, tight junction adhesion molecules, in the inner ear. Hear. Res. 2004, 187, 25–34. [Google Scholar] [CrossRef]
- Mori, M.; Nakagawa, S.; Miyoshi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Sekimoto, M.; Doki, Y. Expression of CLDN1 in colorectal cancer: A novel marker for prognosis. Int. J. Oncol. 2011, 39, 791–796. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, A.; Sun, B.; Zhan, Z.; Chen, K.; Wang, C. Expression of CLDN1 and CLDN10 in lung adenocarcinoma in situ and invasive lepidic predominant adenocarcinoma. J. Cardiothorac. Surg. 2013, 8, 95. [Google Scholar] [CrossRef]
- Zhang, W.-N.; Li, W.; Wang, X.-L.; Hu, Z.; Zhu, D.; Ding, W.-C.; Liu, D.; Li, K.-Z.; Ma, D.; Wang, H. CLDN1 expression in cervical cancer cells is related to tumor invasion and metastasis. Oncotarget 2016, 7, 87449–87461. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Piao, X.; Wang, C.; Wang, R.; Song, Z. Identification of claudin1, 3, 7 and 8 as prognostic markers in human laryngeal carcinoma. Mol. Med. Rep. 2019, 20, 393–400. [Google Scholar] [PubMed]
- Evans, M.J.; Von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wölk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef]
- Baala, L.; Hadj-Rabia, S.; Hamel-Teillac, D.; Hadchouel, M.; Prost, C.; Leal, S.M.; Jacquemin, E.; Sefiani, A.; De Prost, Y.; Courtois, G.; et al. Homozygosity mapping of a locus for a novel syndromic ichthyosis to chromosome 3q27-q28. J. Investig. Dermatol. 2002, 119, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Kirchmeier, P.; Sayar, E.; Hotz, A.; Hausser, I.; Islek, A.; Yilmaz, A.; Artan, R.; Fischer, J. Novel mutation in the CLDN1 gene in a Turkish family with neonatal ichthyosis sclerosing cholangitis (NISCH) syndrome. Br. J. Dermatol. 2014, 170, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Vreeburg, M.; Wagner, A.; Van Geel, M.; Nagtzaam, I.F.; Peeters, V.P.M.; Steijlen, P.M.; Van Steensel, M.A.M. Novel CLDN1 mutation in ichthyosis-hypotrichosis-sclerosing cholangitis syndrome without signs of liver disease. Br. J. Dermatol. 2018, 178, e202–e203. [Google Scholar]
- Tokumasu, R.; Yamaga, K.; Yamazaki, Y.; Murota, H.; Suzuki, K.; Tamura, A.; Bando, K.; Furuta, Y.; Katayama, I.; Tsukita, S. Dose-dependent role of claudin-1 in vivo in orchestrating features of atopic dermatitis. Proc. Natl. Acad. Sci. USA 2016, 113, E4061–E4068. [Google Scholar] [CrossRef]
- Hirano, T.; Yokouchi, M.; Atsugi, T.; Amagai, M.; Kubo, A. Epidermis-specific ablation of claudin-1 in adult mice demonstrates the essential role of a tight junction barrier in skin homeostasis. J. Dermatol. Sci. 2016, 84, e37. [Google Scholar] [CrossRef]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef]
- Amasheh, S.; Meiri, N.; Gitter, A.H.; Schöneberg, T.; Mankertz, J.; Schulzke, J.D.; Fromm, M. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J. Cell Sci. 2002, 115, 4969–4976. [Google Scholar] [CrossRef]
- Lamas, M.; González-Mariscal, L.; Gutiérrez, R. Presence of claudins mRNA in the brain. Selective modulation of expression by kindling epilepsy. Mol. Brain Res. 2002, 104, 250–254. [Google Scholar] [CrossRef]
- Enck, A.H.; Berger, U.V.; Yu, A.S.L. Claudin-2 is selectively expressed in proximal nephron in mouse kidney. Am. J. Physiol. Physiol. 2001, 281, F966–F974. [Google Scholar] [CrossRef]
- Guan, X.; Inai, T.; Shibata, Y. Segment-specific expression of tight junction proteins, claudin-2 and -10, in the rat epididymal epithelium. Arch. Histol. Cytol. 2005, 68, 213–225. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kinugasa, T.; Huo, Q.; Higashi, D.; Shibaguchi, H.; Kuroki, M.; Tanaka, T.; Futami, K.; Yamashita, Y.; Hachimine, K.; Maekawa, S.; et al. Selective up-regulation of claudin-1 and claudin-2 in colorectal cancer. Anticancer Res. 2007, 27, 3729–3734. [Google Scholar] [CrossRef]
- Oshima, T.; Miwa, H.; Joh, T. Changes in the expression of claudins in active ulcerative colitis. J. Gastroenterol. Hepatol. 2008, 23, S146–S150. [Google Scholar] [CrossRef]
- Szakál, D.N.; Győrffy, H.; Arató, A.; Cseh, Á.; Molnár, K.; Papp, M.; Dezsofi, A.; Veres, G. Mucosal expression of claudins 2, 3 and 4 in proximal and distal part of duodenum in children with coeliac disease. Virchows Archiv. 2010, 456, 245–250. [Google Scholar] [CrossRef]
- Weber, C.R.; Nalle, S.C.; Tretiakova, M.; Rubin, D.T.; Turner, J.R. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab. Investig. 2008, 88, 1110–1120. [Google Scholar] [CrossRef]
- Kim, T.H.; Huh, J.H.; Lee, S.; Kang, H.; I Kim, G.; An, H.J. Down-regulation of claudin-2 in breast carcinomas is associated with advanced disease. Histopathology 2008, 53, 48–55. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, H.; Li, Q.; Li, T. CLDN2 inhibits the metastasis of osteosarcoma cells via down-regulating the afadin/ERK signaling pathway. Cancer Cell Int. 2018, 18, 160. [Google Scholar] [CrossRef]
- Martínez, C.; Rodiño-Janeiro, B.K.; Lobo, B.; Stanifer, M.L.; Klaus, B.; Granzow, M.; González-Castro, A.M.; Salvo-Romero, E.; Alonso-Cotoner, C.; Pigrau, M.; et al. miR-16 and miR-125b are involved in barrier function dysregulation through the modulation of claudin-2 and cingulin expression in the jejunum in IBS with diarrhoea. Gut 2017, 66, 1537–1538. [Google Scholar] [CrossRef]
- Muto, S.; Hata, M.; Taniguchi, J.; Tsuruoka, S.; Moriwaki, K.; Saitou, M.; Furuse, K.; Sasaki, H.; Fujimura, A.; Imai, M.; et al. Claudin-2–deficient mice are defective in the leaky and cation-selective paracellular permeability properties of renal proximal tubules. Proc. Natl. Acad. Sci. USA 2010, 107, 8011–8016. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Imasato, M.; Yamazaki, Y.; Tanaka, H.; Watanabe, M.; Eguchi, H.; Nagano, H.; Hikita, H.; Tatsumi, T.; Takehara, T.; et al. Claudin 2 Deficiency Reduces Bile Flow and Increases Susceptibility to Cholesterol Gallstone Disease in Mice. Gastroenterology 2014, 147, 1134–1145.e10. [Google Scholar] [CrossRef] [PubMed]
- Wada, M.; Tamura, A.; Takahashi, N.; Tsukita, S. Loss of claudins 2 and 15 from mice causes defects in paracellular Na+ flow and nutrient transport in gut and leads to death from malnutrition. Gastroenterology 2013, 144, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Chaturvedi, R.; Olivares-Villagómez, D.; Habib, T.; Asim, M.; Shivesh, P.; Polk, D.B.; Wilson, K.T.; Washington, M.K.; Van Kaer, L.; et al. Targeted colonic claudin-2 expression renders resistance to epithelial injury, induces immune suppression, and protects from colitis. Mucosal Immunol. 2014, 7, 1340–1353. [Google Scholar] [CrossRef] [PubMed]
- Dubé, É.; Dufresne, J.; Chan, P.T.; Hermo, L.; Cyr, D.G. Assessing the Role of Claudins in Maintaining the Integrity of Epididymal Tight Junctions Using Novel Human Epididymal Cell Lines1. Biol. Reprod. 2010, 82, 1119–1128. [Google Scholar] [CrossRef]
- Zheng, A.; Yuan, F.; Li, Y.; Zhu, F.; Hou, P.; Li, J.; Song, X.; Ding, M.; Deng, H. Claudin-6 and Claudin-9 Function as Additional Coreceptors for Hepatitis C Virus. J. Virol. 2007, 81, 12465–12471. [Google Scholar] [CrossRef]
- Abuazza, G.; Becker, A.; Williams, S.S.; Chakravarty, S.; Truong, H.-T.; Lin, F.; Baum, M. Claudins 6, 9, and 13 are developmentally expressed renal tight junction proteins. Am. J. Physiol. Physiol. 2006, 291, F1132–F1141. [Google Scholar] [CrossRef]
- Fofana, I.; Zona, L.; Thumann, C.; Heydmann, L.; Durand, S.C.; Lupberger, J.; Blum, H.E.; Pessaux, P.; Gondeau, C.; Reynolds, G.M.; et al. Functional Analysis of Claudin-6 and Claudin-9 as Entry Factors for Hepatitis C Virus Infection of Human Hepatocytes by Using Monoclonal Antibodies. J. Virol. 2013, 87, 10405–10410. [Google Scholar] [CrossRef]
- Zavala-Zendejas, V.E.; Torres-Martinez, A.C.; Salas-Morales, B.; Fortoul, T.I.; Montano, L.F.; Rendon-Huerta, E.P. Claudin-6, 7, or 9 overexpression in the human gastric adenocarcinoma cell line AGS increases its invasiveness, migration, and proliferation rate. Cancer Investig. 2011, 29, 1–11. [Google Scholar] [CrossRef]
- Sharma, R.K.; Chheda, Z.S.; Das Purkayastha, B.P.; Gomez-Gutierrez, J.G.; Jala, V.R.; Haribabu, B. A spontaneous metastasis model reveals the significance of claudin-9 overexpression in lung cancer metastasis. Clin. Exp. Metastasis 2016, 33, 263–275. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, R.; Cao, H.; Zhang, H.; Xu, S.; Wang, A.; Liu, B.; Wang, Y.; Wang, R. Expression of claudin-5, -7, -8 and -9 in cervical carcinoma tissues and adjacent non-neoplastic tissues. Int. J. Clin. Exp. Pathol. 2015, 8, 9479–9486. [Google Scholar] [PubMed]
- Birkenhäger, R.; Otto, E.; Schürmann, M.J.; Vollmer, M.; Ruf, E.-M.; Maier-Lutz, I.; Beekmann, F.; Fekete, A.; Omran, H.; Feldmann, D.; et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat. Genet. 2001, 29, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Rogan, S.; Yu, A.; Vidal, L.S.; Holmes, J.; Anderson, J.M. Two splice variants of claudin-10 in the kidney create paracellular pores with different ion selectivities. Am. J. Physiol. Physiol. 2006, 291, F1288–F1299. [Google Scholar] [CrossRef] [PubMed]
- Günzel, D.; Stuiver, M.; Kausalya, P.J.; Haisch, L.; Krug, S.; Rosenthal, R.; Meij, I.C.; Hunziker, W.; Fromm, M.; Muller, D. Claudin-10 exists in six alternatively spliced isoforms that exhibit distinct localization and function. J. Cell Sci. 2009, 122, 1507–1517. [Google Scholar] [CrossRef]
- Zemke, A.C.; Snyder, J.C.; Brockway, B.L.; Drake, J.A.; Reynolds, S.D.; Kaminski, N.; Stripp, B.R. Molecular staging of epithelial maturation using secretory cell-specific genes as markers. Am. J. Respir. Cell Mol. Biol. 2009, 40, 340–348. [Google Scholar] [CrossRef]
- Hashizume, A.; Ueno, T.; Furuse, M.; Tsukita, S.; Nakanishi, Y.; Hieda, Y. Expression patterns of claudin family of tight junction membrane proteins in developing mouse submandibular gland. Dev. Dyn. 2004, 231, 425–431. [Google Scholar] [CrossRef]
- Jakab, C.; Halász, J.; Szasz, A.M.; Batmunkh, E.; Kiss, A.; Schaff, Z.; Rusvai, M.; Galfi, P.; Kulka, J. Expression and localisation of claudin-1,-2,-3,-4,-5,-7 and-10 proteins in the normal canine mammary gland. Acta Vet. Hung. 2008, 56, 341–352. [Google Scholar] [CrossRef]
- Hou, J.; Renigunta, A.; Gomes, A.S.; Hou, M.; Paul, D.L.; Waldegger, S.; Goodenough, D.A. Claudin-16 and claudin-19 interaction is required for their assembly into tight junctions and for renal reabsorption of magnesium. Proc. Natl. Acad. Sci. USA 2009, 106, 15350–15355. [Google Scholar] [CrossRef]
- Milatz, S.; Himmerkus, N.; Wulfmeyer, V.C.; Drewell, H.; Mutig, K.; Hou, J.; Breiderhoff, T.; Muller, D.; Fromm, M.; Bleich, M.; et al. Mosaic expression of claudins in thick ascending limbs of Henle results in spatial separation of paracellular Na+ and Mg2+ transport. Proc. Natl. Acad. Sci. USA 2017, 114, e219–e227. [Google Scholar] [CrossRef]
- Schumann, S.; Buck, V.U.; Classen-Linke, I.; Wennemuth, G.; Grümmer, R. Claudin-3, claudin-7, and claudin-10 show different distribution patterns during decidualization and trophoblast invasion in mouse and human. Histochem. Cell Biol. 2015, 144, 571–585. [Google Scholar] [CrossRef]
- Klar, J.; Piontek, J.; Milatz, S.; Tariq, M.; Jameel, M.; Breiderhoff, T.; Schuster, J.; Fatima, A.; Asif, M.; Sher, M.; et al. Altered paracellular cation permeability due to a rare CLDN10B variant causes anhidrosis and kidney damage. PLoS Genet. 2017, 13, e1006897. [Google Scholar] [CrossRef] [PubMed]
- Meyers, N.; Nelson-Williams, C.; Malaga-Dieguez, L.; Kaufmann, H.; Loring, E.; Knight, J.; Lifton, R.P.; Trachtman, H. Hypokalemia Associated with a Claudin 10 Mutation: A Case Report. Am. J. Kidney Dis. 2019, 73, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Breiderhoff, T.; Himmerkus, N.; Drewell, H.; Plain, A.; Gunzel, D.; Mutig, K.; Willnow, T.E.; Muller, D.; Bleich, M. Deletion of claudin-10 rescues claudin-16-deficient mice from hypomagnesemia and hypercalciuria. Kidney Int. 2018, 93, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Li, W.; Wang, H.; Wang, G. The distinct expression patterns of claudin-10, -14, -17 and E-cadherin between adjacent non-neoplastic tissues and gastric cancer tissues. Diagn. Pathol. 2013, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Renigunta, V.; Himmerkus, N.; Zhang, J.; Renigunta, A.; Bleich, M.; Hou, J. Claudin-14 regulates renal Ca++ transport in response to CaSR signalling via a novel microRNA pathway. EMBO J. 2012, 31, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Ansar, M.; Andrade, P.B.; Khan, B.; Santos-Cortez, R.L.P.; Ahmad, W.; Leal, S.M. Novel CLDN14 mutations in Pakistani families with autosomal recessive non-syndromic hearing loss. Am. J. Med Genet. Part A 2012, 158, 315–321. [Google Scholar] [CrossRef]
- Wattenhofer, M.; Reymond, A.; Falciola, V.; Charollais, A.; Caille, D.; Borel, C.; Lyle, R.; Estivill, X.; Petersen, M.B.; Meda, P.; et al. Different mechanisms preclude mutant CLDN14 proteins from forming tight junctions in vitro. Hum. Mutat. 2005, 25, 543–549. [Google Scholar] [CrossRef]
- Gong, Y.; Hou, J. Claudin-14 underlies Ca(+)(+)-sensing receptor-mediated Ca(+)(+) metabolism via NFAT-microRNA-based mechanisms. J. Am. Soc. Nephrol. 2014, 25, 745–760. [Google Scholar] [CrossRef]
- Pater, J.A.; Benteau, T.; Griffin, A.; Penney, C.; Stanton, S.G.; Predham, S.; Kielley, B.; Squires, J.; Zhou, J.; Li, Q.; et al. A common variant in CLDN14 causes precipitous, prelingual sensorineural hearing loss in multiple families due to founder effect. Hum. Genet. 2017, 136, 107–118. [Google Scholar] [CrossRef]
- Hou, J. Claudins and mineral metabolism. Curr. Opin. Nephrol. Hypertens. 2016, 25, 308–313. [Google Scholar] [CrossRef]
- Bardet, C.; Courson, F.; Wu, Y.; Khaddam, M.; Salmon, B.; Ribes, S.; Thumfart, J.; Yamaguti, P.M.; Rochefort, G.Y.; Figueres, M.L.; et al. Claudin-16 Deficiency Impairs Tight Junction Function in Ameloblasts, Leading to Abnormal Enamel Formation. J. Bone Miner. Res. 2016, 31, 498–513. [Google Scholar] [CrossRef] [PubMed]
- Kriegs, J.O.; Homann, V.; Kinne-Saffran, E.; Kinne, R.K.H. Identification and subcellular localization of paracellin-1 (claudin-16) in human salivary glands. Histochem. Cell Biol. 2007, 128, 45–53. [Google Scholar] [CrossRef]
- Runggaldier, D.; Pradas, L.G.; Neckel, P.H.; Mack, A.F.; Hirt, B.; Gleiser, C. Claudin expression in the rat endolymphatic duct and sac—First insights into regulation of the paracellular barrier by vasopressin. Sci. Rep. 2017, 7, 45482. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Schlingmann, K.P.; Peters, M.; Nejsum, L.N.; Nielsen, S.; Engel, H.; Grzeschik, K.H.; Seyberth, H.W.; Gröne, H.J.; Nüsing, R.; et al. Primary gene structure and expression studies of rodent paracellin-1. J. Am. Soc. Nephrol. 2001, 12, 2664–2672. [Google Scholar] [PubMed]
- Rangel, L.B.; Sherman-Baust, C.A.; Wernyj, R.P.; Schwartz, D.R.; Cho, K.R.; Morin, P.J. Characterization of novel human ovarian cancer-specific transcripts (HOSTs) identified by serial analysis of gene expression. Oncogene 2003, 22, 7225–7232. [Google Scholar] [CrossRef] [PubMed]
- Fluge, Ø.; Bruland, O.; Akslen, L.A.; Lillehaug, J.R.; Varhaug, J.E. Gene Expression in Poorly Differentiated Papillary Thyroid Carcinomas. Thyroid 2006, 16, 161–175. [Google Scholar]
- Lee, N.P.; Tong, M.K.; Leung, P.P.; Chan, V.W.; Leung, S.; Tam, P.-C.; Chan, K.-W.; Lee, K.-F.; Yeung, W.S.; Luk, J.M. Kidney claudin-19: Localization in distal tubules and collecting ducts and dysregulation in polycystic renal disease. FEBS Lett. 2006, 580, 923–931. [Google Scholar] [CrossRef]
- Martin, T.A.; Harrison, G.M.; Watkins, G.; Jiang, W.G. Claudin-16 reduces the aggressive behavior of human breast cancer cells. J. Cell. Biochem. 2008, 105, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.B.; Lu, Y.; Choate, K.A.; Velazquez, H.; Al-Sabban, E.; Praga, M.; Casari, G.; Bettinelli, A.; Colussi, G.; Rodriguez-Soriano, J.; et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 1999, 285, 103–106. [Google Scholar] [CrossRef]
- Hou, J.; Shan, Q.; Wang, T.; Gomes, A.S.; Yan, Q.; Paul, D.L.; Bleich, M.; Goodenough, D.A. Transgenic RNAi Depletion of Claudin-16 and the Renal Handling of Magnesium. J. Biol. Chem. 2007, 282, 17114–17122. [Google Scholar] [CrossRef]
- Kausalya, P.J.; Amasheh, S.; Gunzel, D.; Wurps, H.; Muller, D.; Fromm, M.; Hunziker, W. Disease-associated mutations affect intracellular traffic and paracellular Mg2+ transport function of Claudin-16. J. Clin. Investig. 2006, 116, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Hirai, N.; Shiroma, M.; Harada, H.; Sakai, H.; Hayashi, H.; Suzuki, Y.; Degawa, M.; Takagi, K. Association of Paracellin-1 with ZO-1 Augments the Reabsorption of Divalent Cations in Renal Epithelial Cells. J. Biol. Chem. 2004, 279, 54826–54832. [Google Scholar] [CrossRef] [PubMed]
- Marunaka, K.; Furukawa, C.; Fujii, N.; Kimura, T.; Furuta, T.; Matsunaga, T.; Endo, S.; Hasegawa, H.; Anzai, N.; Yamazaki, Y.; et al. The RING finger- and PDZ domain-containing protein PDZRN3 controls localization of the Mg(2+) regulator claudin-16 in renal tube epithelial cells. J. Biol. Chem. 2017, 292, 13034–13044. [Google Scholar] [CrossRef]
- Hou, J.; Renigunta, V.; Nie, M.; Sunq, A.; Himmerkus, N.; Quintanova, C.; Bleich, M.; Renigunta, A.; Wolf, M.T.F. Phosphorylated claudin-16 interacts with Trpv5 and regulates transcellular calcium transport in the kidney. Proc. Natl. Acad. Sci. USA 2019, 116, 19176–19186. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Mascarell, P.; Schirrmacher, C.E.; Martínez-Cruz, L.A.; Muller, D. Novel Aspects of Renal Magnesium Homeostasis. Front. Pediatr. 2018, 6, 77. [Google Scholar] [CrossRef]
- Yamaguti, P.M.; Dos Santos, P.A.C.; Leal, B.S.; Santana, V.B.B.D.M.; Mazzeu, J.F.; Acevedo, A.C.; Neves, F.D.A.R. Identification of the first large deletion in the CLDN16 gene in a patient with FHHNC and late-onset of chronic kidney disease: Case report. BMC Nephrol. 2015, 16, 92. [Google Scholar] [CrossRef]
- Konrad, M.; Hou, J.; Weber, S.; Dötsch, J.; Kari, J.A.; Seeman, T.; Kuwertz-Bröking, E.; Peco-Antic, A.; Tasic, V.; Dittrich, K.; et al. CLDN16 genotype predicts renal decline in familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J. Am. Soc. Nephrol. 2008, 19, 171–181. [Google Scholar] [CrossRef]
- Niimi, T.; Nagashima, K.; Ward, J.M.; Minoo, P.; Zimonjic, D.B.; Popescu, N.C.; Kimura, S. claudin-18, a Novel Downstream Target Gene for the T/EBP/NKX2.1 Homeodomain Transcription Factor, Encodes Lung- and Stomach-Specific Isoforms through Alternative Splicing. Mol. Cell. Biol. 2001, 21, 7380–7390. [Google Scholar] [CrossRef]
- Wolburg, H.; Wolburg-Buchholz, K.; Liebner, S.; Engelhardt, B. Claudin-1, claudin-2 and claudin-11 are present in tight junctions of choroid plexus epithelium of the mouse. Neurosci. Lett. 2001, 307, 77–80. [Google Scholar] [CrossRef]
- Fromm, M.; Piontek, J.; Rosenthal, R.; Gunzel, D.; Krug, S.M. Tight junctions of the proximal tubule and their channel proteins. Pflüg. Arch. 2017, 469, 877–887. [Google Scholar] [CrossRef]
- Gong, Y.; Renigunta, V.; Zhou, Y.; Sunq, A.; Wang, J.; Yang, J.; Renigunta, A.; Baker, L.A.; Hou, J. Biochemical and biophysical analyses of tight junction permeability made of claudin-16 and claudin-19 dimerization. Mol. Biol. Cell 2015, 26, 4333–4346. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Renigunta, A.; Konrad, M.; Gomes, A.S.; Schneeberger, E.E.; Paul, D.L.; Waldegger, S.; Goodenough, D.A. Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J. Clin. Investig. 2008, 118, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Meier, W.; Blumberg, A.; Imahorn, W.; De Luca, F.; Wildberger, H.; Oetliker, O. Idiopathic hypercalciuria with bilateral macular colobomata: A new variant of oculo-renal syndrome. Helvetica Paediatr. Acta 1979, 34, 257–269. [Google Scholar]
- Rodriguez-Soriano, J.; Vallo, A. Pathophysiology of the renal acidification defect present in the syndrome of familial hypomagnesaemia-hypercalciuria. Pediatr. Nephrol. 1994, 8, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Kondoh, M.; Suzuki, H.; Yagi, K. Claudin as a target for drug development. Curr. Med. Chem. 2011, 18, 1861–1865. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A.S.; Thongprayoon, C.; Cheungpasitporn, W.; Sakhuja, A.; Mao, M.A.; Erickson, S.B. Incidence and characteristics of kidney stones in patients with horseshoe kidney: A systematic review and meta-analysis. Urol. Ann. 2018, 10, 87–93. [Google Scholar]
- Alelign, T.; Petros, B. Kidney Stone Disease: An Update on Current Concepts. Adv. Urol. 2018, 2018, 3068365. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human | Mouse | |||||
|---|---|---|---|---|---|---|
| Claudin | Human Disorder | Hallmarks | Knockout | Knockdown | Conditional Knockout | Transgenic Overexpression |
| 1 | NISCH Syndrome | Ichthyosis | Ichthyosis | Ichthyosis | + | + |
| 2 | Obstructive azoospermia | Male infertility | (Cldn2−/−) Larger intestine (Cldn2−/− Cldn15−/−) Hypoglycemia Perinatal lethality | − | − | Larger colon |
| 9 | Nonsyndromic sensorioneural deafness | Deafness | Deafness | − | − | − |
| 10 | HELIX syndrome | Hypokalemia Hypermagnesemia Nephrocalcinosis | Perinatal lethality | − | Hypokalemi Hypermagnesemia Nephrocalcinosis | − |
| 14 | Nonsyndromic sensorioneural deafness | Deafness | Deafness | − | − | Hypercalcuria Hypermagnesemia |
| 16 | FHHNC | Hypercalciuria Renal insufficiency Hypomagnesemia Nephrocalcinosis | Hypercalciuria Hypomagnesemia | Hypercalciuria Hypomagnesemia | − | − |
| 19 | FHHNC + Eye Involvement | Hypercalciuria Renal insufficiency Hypomagnesemia Nephrocalcinosis Eye involvement | Hypercalciuria Hypomagnesemia | Hypercalciuria Hypomagnesemia Eye abnormalities | − | − |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seker, M.; Fernández-Rodríguez, C.; Martínez-Cruz, L.A.; Müller, D. Mouse Models of Human Claudin-Associated Disorders: Benefits and Limitations. Int. J. Mol. Sci. 2019, 20, 5504. https://doi.org/10.3390/ijms20215504
Seker M, Fernández-Rodríguez C, Martínez-Cruz LA, Müller D. Mouse Models of Human Claudin-Associated Disorders: Benefits and Limitations. International Journal of Molecular Sciences. 2019; 20(21):5504. https://doi.org/10.3390/ijms20215504
Chicago/Turabian StyleSeker, Murat, Cármen Fernández-Rodríguez, Luis Alfonso Martínez-Cruz, and Dominik Müller. 2019. "Mouse Models of Human Claudin-Associated Disorders: Benefits and Limitations" International Journal of Molecular Sciences 20, no. 21: 5504. https://doi.org/10.3390/ijms20215504
APA StyleSeker, M., Fernández-Rodríguez, C., Martínez-Cruz, L. A., & Müller, D. (2019). Mouse Models of Human Claudin-Associated Disorders: Benefits and Limitations. International Journal of Molecular Sciences, 20(21), 5504. https://doi.org/10.3390/ijms20215504