The Systemic Immune Response to Collagen-Induced Arthritis and the Impact of Bone Injury in Inflammatory Conditions

 ,
,  , , ,
, , , 
Abstract
1. Introduction
2. Results
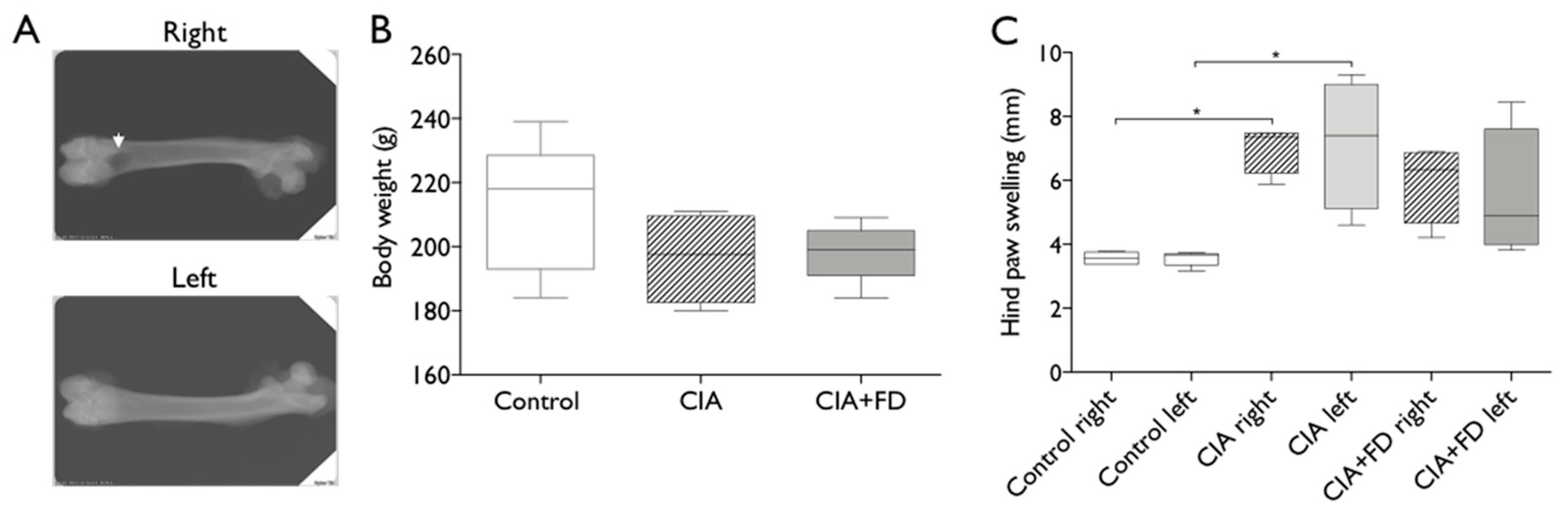
2.1. Collagen-induced arthritis (CIA) as a Model to Study Bone Injury in Inflammatory Conditions
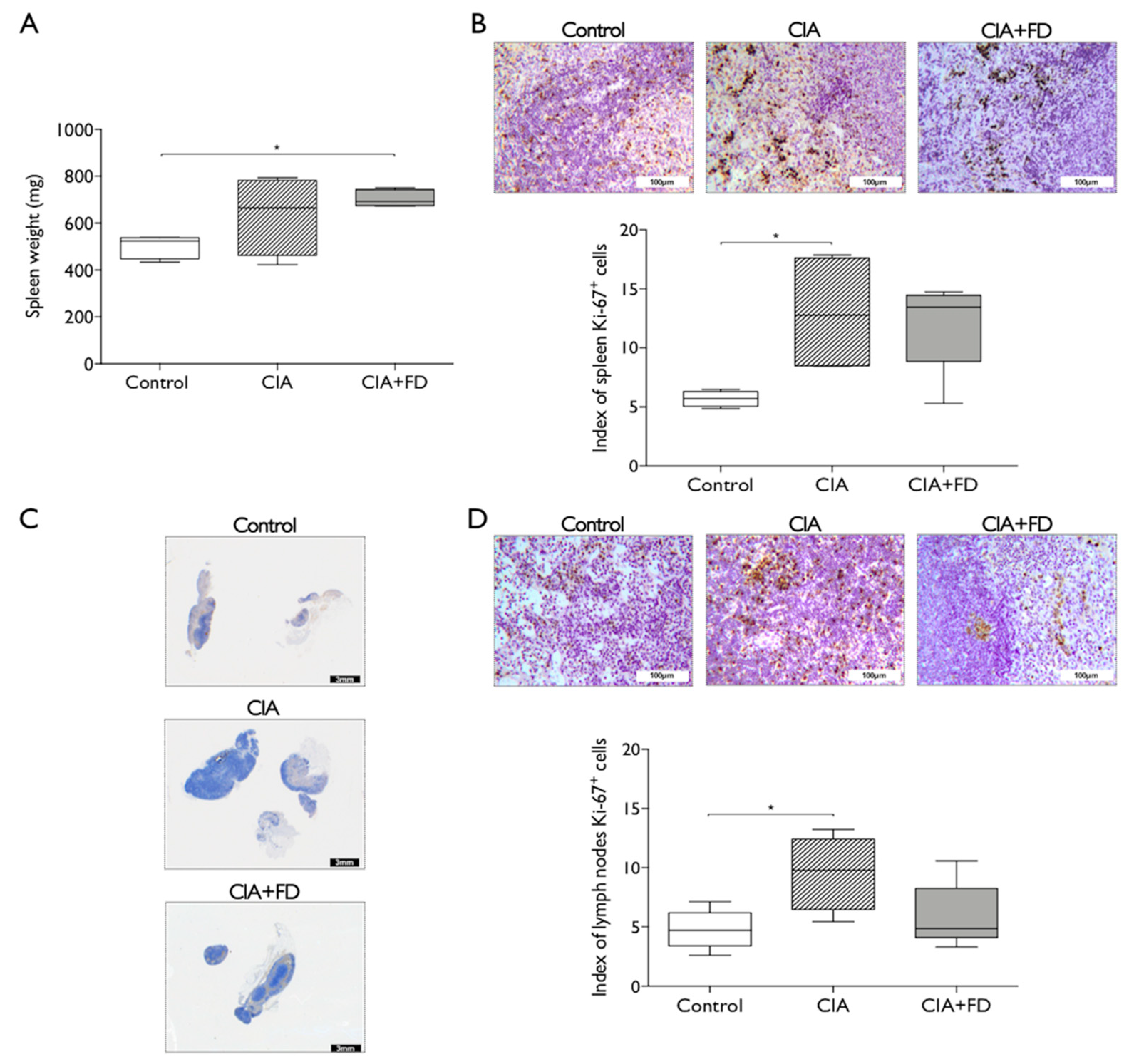
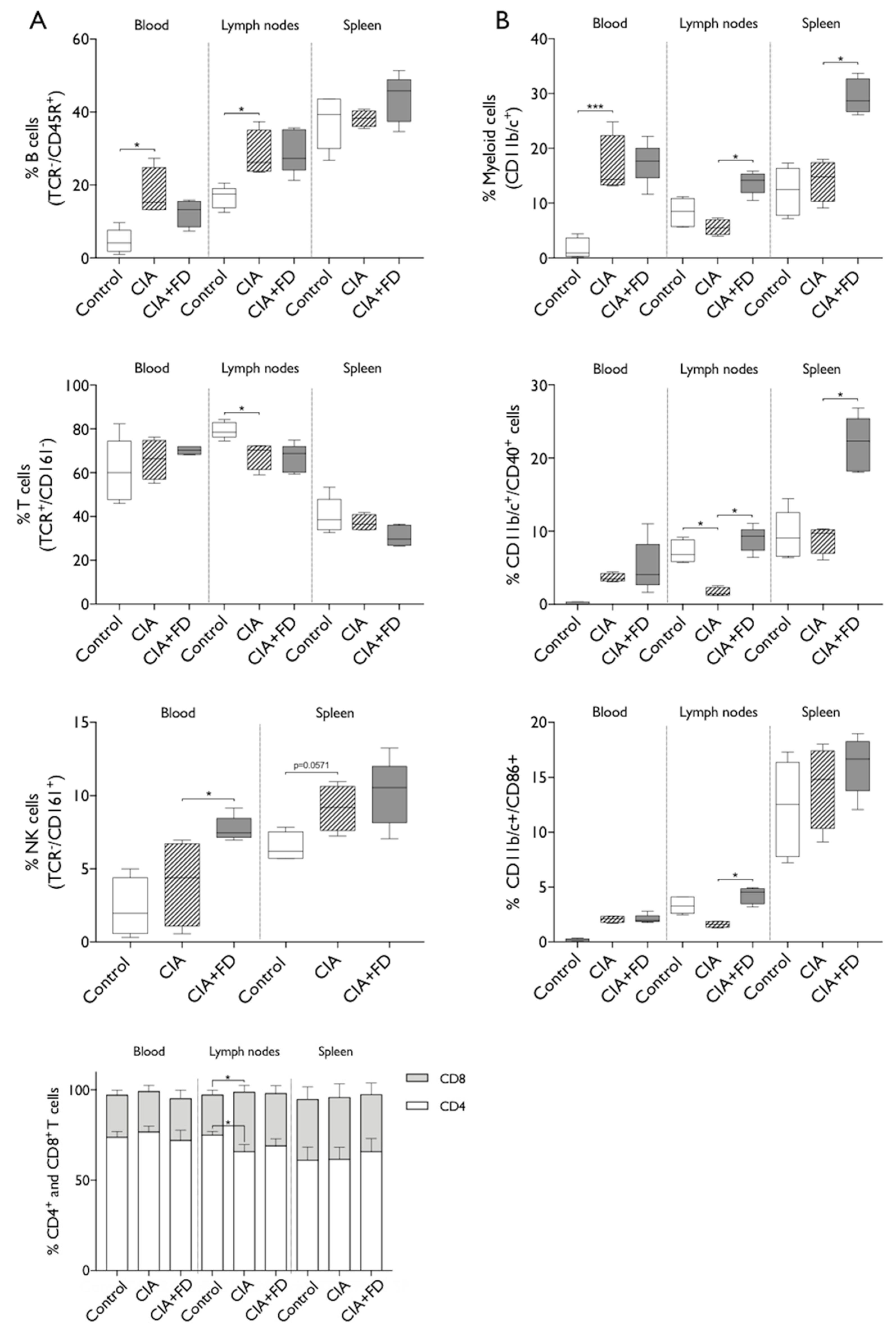
2.2. The Impact of CIA and Bone Injury in Secondary Lymphoid Organs
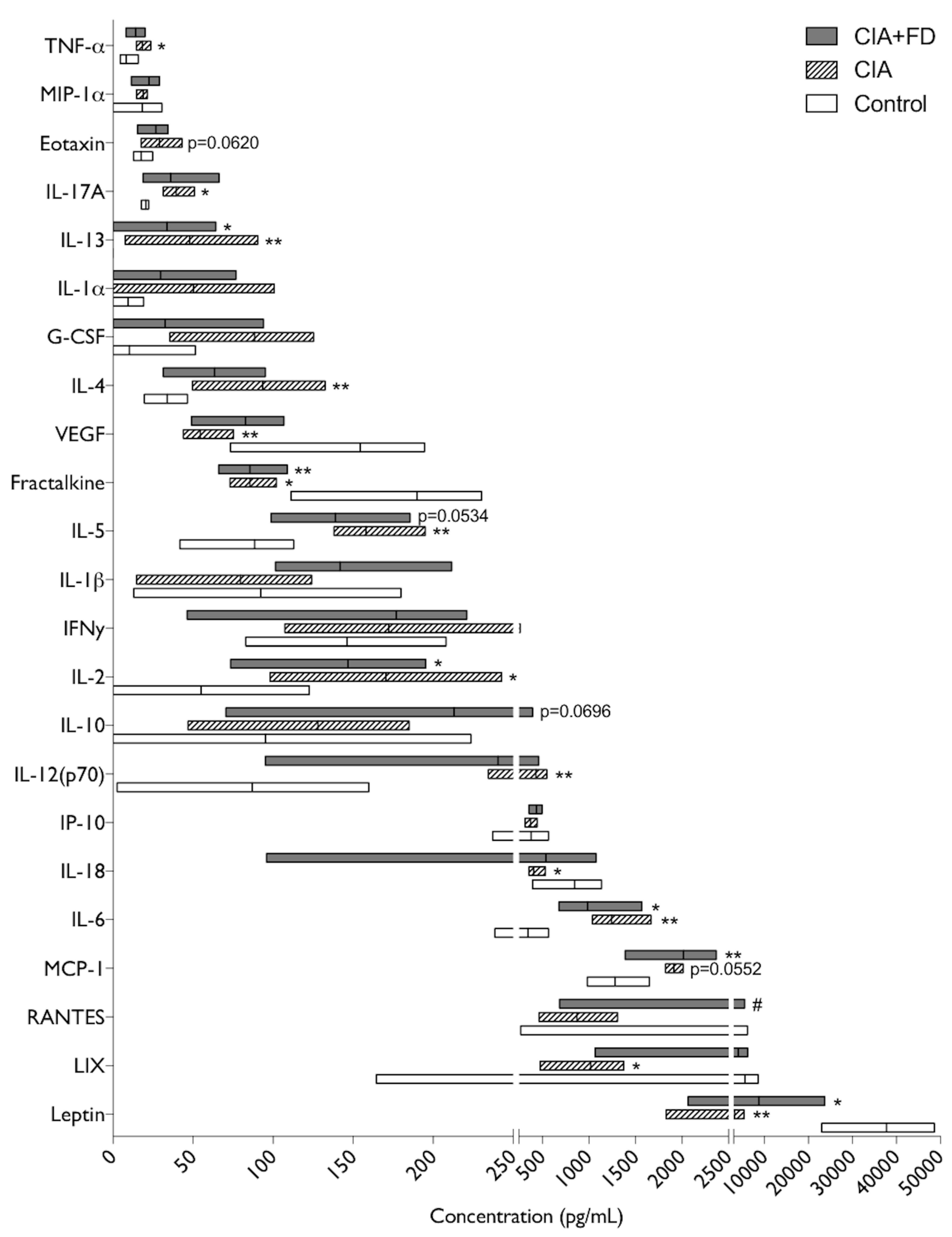
2.3. The Impact of CIA and Bone Defect in Circulating Inflammatory Mediators
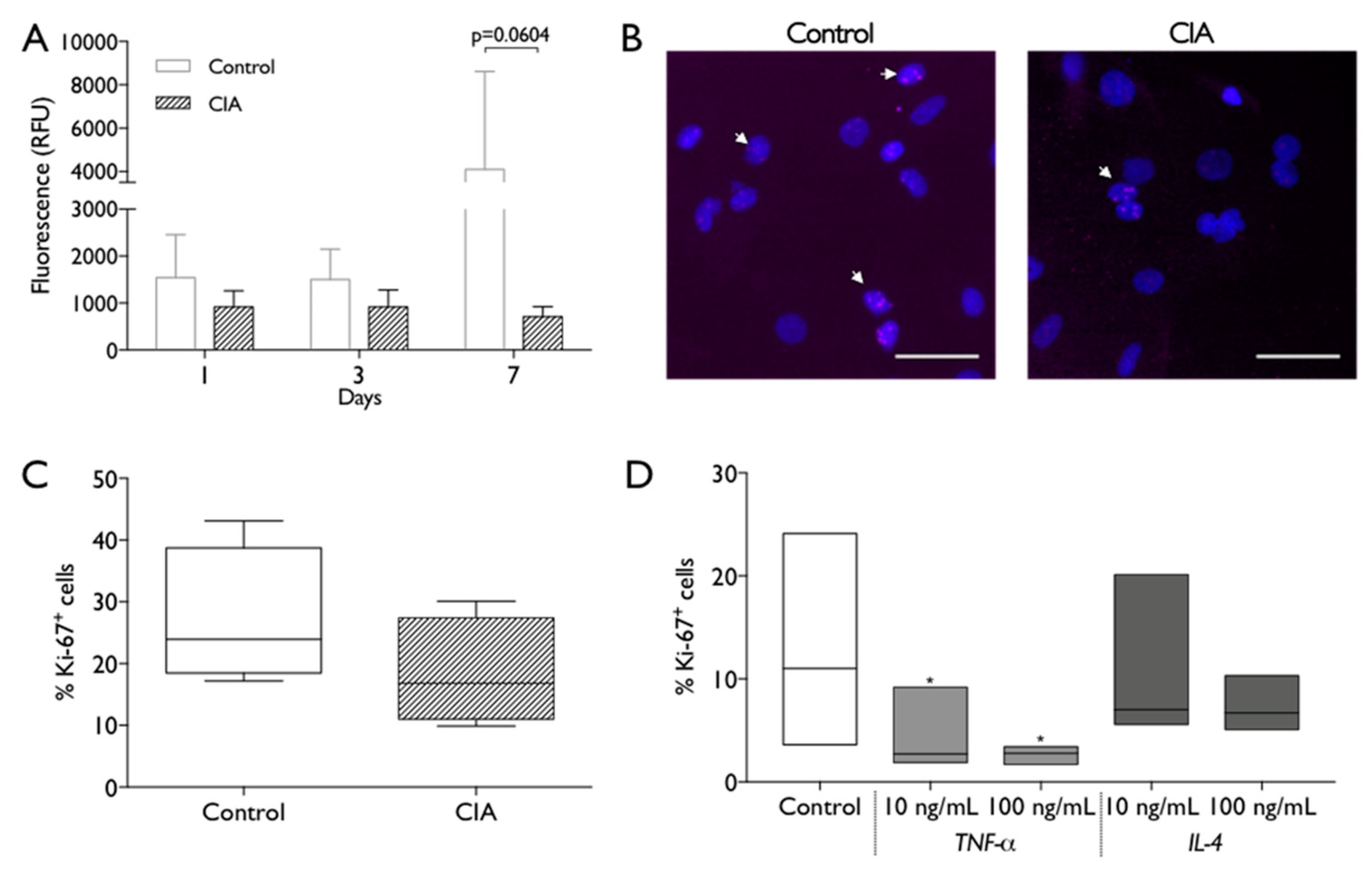
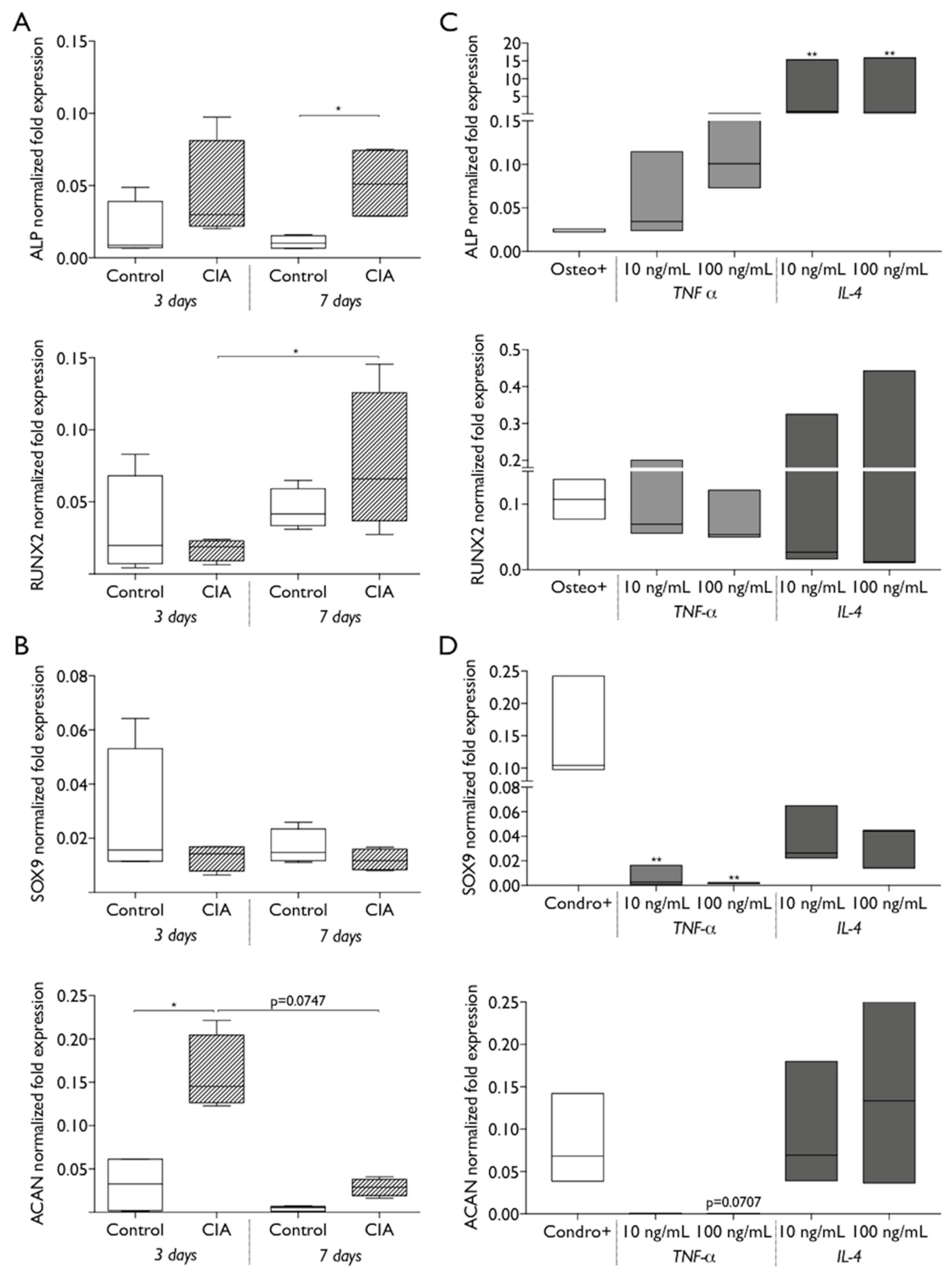
2.4. CIA Induction Impacts Endogenous MSC Biologic Behavior
3. Discussion
4. Materials and Methods
4.1. Bovine Collagen Type II Emulsion Preparation
4.2. Collagen-Induced Arthritis (CIA) Rat Model
4.3. Monitoring of Clinical Arthritis Development
4.4. CIA Rat Model Combined with Bone Injury Model
4.5. Blood, Spleen and Lymph Nodes Collection and Processing
4.6. Histology and Immunohistochemistry Analysis
4.7. Flow Cytometry Analysis
4.8. Plasma Cytokine Quantification
4.9. Isolation of MSC, Primary Culture and Phenotypic Characterization
4.10. MSC Metabolic Activity and Proliferation Assays
4.11. MSC Differentiation Assay
4.12. RNA Isolation and Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CIA | Collagen-induced arthritis |
| MSC | Mesenchymal stem/stromal cells |
| TNF-α | Tumor necrosis factor-alpha |
| IL | Interleukin |
| RA | Rheumatoid Arthritis |
| FD | Femoral defect |
| CIA+FD | Collagen-induced arthritis with femoral defect |
| RT-qPCR | Reverse transcription-quantitative polymerase chain reaction |
| CII-IFA | Collagen type II-Incomplete Freund’s adjuvant |
References
- Van der Woude, D.; van der Helm-van Mil, A.H.M. Update on the epidemiology, risk factors, and disease outcomes of rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2018, 32, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, M.; Cojocaru, I.M.; Silosi, I.; Vrabie, C.D.; Tanasescu, R. Extra-articular manifestations in rheumatoid arthritis. Maedica (Buchar) 2010, 5, 286–291. [Google Scholar] [PubMed]
- Barreira, S.C.; Fonseca, J.E. The impact of conventional and biological disease modifying antirheumatic drugs on bone biology. Rheumatoid arthritis as a case study. Clin. Rev. Allergy Immunol. 2016, 51, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Heinlen, L.; Humphrey, M.B. Skeletal complications of rheumatoid arthritis. Osteoporos. Int. 2017, 28, 2801–2812. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Hsieh, E.; Peng, L.; Yu, C.; Wang, Y.; Wu, C.; Wang, Q.; Li, M.; Zeng, X. Incidence of fractures among patients with rheumatoid arthritis: A systematic review and meta-analysis. Osteoporos. Int. 2018, 29, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Ghazi, M.; Kolta, S.; Briot, K.; Fechtenbaum, J.; Paternotte, S.; Roux, C. Prevalence of vertebral fractures in patients with rheumatoid arthritis: Revisiting the role of glucocorticoids. Osteoporos. Int. 2012, 23, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Claes, L.; Recknagel, S.; Ignatius, A. Fracture healing under healthy and inflammatory conditions. Nat. Rev. Rheumatol. 2012, 8, 133–143. [Google Scholar] [CrossRef]
- Kannan, K.; Ortmann, R.A.; Kimpel, D. Animal models of rheumatoid arthritis and their relevance to human disease. Pathophysiology 2005, 12, 167–181. [Google Scholar] [CrossRef]
- Trentham, D.E.; Townes, A.S.; Kang, A.H. Autoimmunity to type ii collagen an experimental model of arthritis. J. Exp. Med. 1977, 146, 857–868. [Google Scholar] [CrossRef]
- Bevaart, L.; Vervoordeldonk, M.J.; Tak, P.P. Evaluation of therapeutic targets in animal models of arthritis: How does it relate to rheumatoid arthritis? Arthritis Rheum. 2010, 62, 2192–2205. [Google Scholar] [CrossRef]
- Bendele, A. Animal models of rheumatoid arthritis. J. Musculoskelet Neuronal Interact 2001, 1, 377–385. [Google Scholar] [PubMed]
- Mussener, A.; Litton, M.J.; Lindroos, E.; Klareskog, L. Cytokine production in synovial tissue of mice with collagen-induced arthritis (cia). Clin. Exp. Immunol. 1997, 107, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Marinova-Mutafchieva, L.; Williams, R.O.; Mason, L.J.; Mauri, C.; Feldmann, M.; Maini, R.N. Dynamics of proinflammatory cytokine expression in the joints of mice with collagen-induced arthritis (cia). Clin. Exp. Immunol. 1997, 107, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Qiao, H.; Zhai, Z.; Zhang, J.; Tu, J.; Zheng, X.; Qian, N.; Zhou, H.; Lu, E.; Tang, T. Plumbagin ameliorates collagen-induced arthritis by regulating treg/th17 cell imbalances and suppressing osteoclastogenesis. Front. Immunol. 2018, 9, 3102. [Google Scholar] [CrossRef]
- Rosloniec, E.F.; Cremer, M.; Kang, A.; Myers, L.K. Collagen-induced arthritis. Curr. Protoc. Immunol. 2001, 20, 15.5.1–15.5.24. [Google Scholar]
- Endale, M.; Lee, W.M.; Kwak, Y.S.; Kim, N.M.; Kim, B.K.; Kim, S.H.; Cho, J.; Kim, S.; Park, S.C.; Yun, B.S.; et al. Torilin ameliorates type ii collagen-induced arthritis in mouse model of rheumatoid arthritis. Int. Immunopharmacol. 2013, 16, 232–242. [Google Scholar] [CrossRef]
- Luross, J.A.; Williams, N.A. The genetic and immunopathological processes underlying collagen-induced arthritis. Immunology 2001, 103, 407–416. [Google Scholar] [CrossRef]
- Alvaro-Gracia, J.M.; Jover, J.A.; Garcia-Vicuna, R.; Carreno, L.; Alonso, A.; Marsal, S.; Blanco, F.; Martinez-Taboada, V.M.; Taylor, P.; Martin-Martin, C.; et al. Intravenous administration of expanded allogeneic adipose-derived mesenchymal stem cells in refractory rheumatoid arthritis (cx611): Results of a multicentre, dose escalation, randomised, single-blind, placebo-controlled phase ib/iia clinical trial. Ann. Rheum. Dis. 2017, 76, 196–202. [Google Scholar] [CrossRef]
- Park, E.H.; Lim, H.S.; Lee, S.; Roh, K.; Seo, K.W.; Kang, K.S.; Shin, K. Intravenous infusion of umbilical cord blood-derived mesenchymal stem cells in rheumatoid arthritis: A phase ia clinical trial. Stem Cells Transl. Med. 2018, 7, 636–642. [Google Scholar] [CrossRef]
- Gao, F.; Chiu, S.M.; Motan, D.A.; Zhang, Z.; Chen, L.; Ji, H.L.; Tse, H.F.; Fu, Q.L.; Lian, Q. Mesenchymal stem cells and immunomodulation: Current status and future prospects. Cell Death Dis. 2016, 7, e2062. [Google Scholar] [CrossRef]
- Ansboro, S.; Roelofs, A.J.; De Bari, C. Mesenchymal stem cells for the management of rheumatoid arthritis: Immune modulation, repair or both? Curr. Opin. Rheumatol. 2017, 29, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Su, W.; Lin, X.; Guo, Z.; Wang, J.; Zhang, Q.; Brand, D.; Ryffel, B.; Huang, J.; Liu, Z.; et al. Adoptive transfer of human gingiva-derived mesenchymal stem cells ameliorates collagen-induced arthritis via suppression of th1 and th17 cells and enhancement of regulatory t cell differentiation. Arthritis Rheum. 2013, 65, 1181–1193. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Cen, Y.; Wang, Q. Mesenchymal stem cells alleviate experimental rheumatoid arthritis through microrna-regulated ikappab expression. Sci. Rep. 2016, 6, 28915. [Google Scholar] [CrossRef] [PubMed]
- Augello, A.; Tasso, R.; Negrini, S.M.; Cancedda, R.; Pennesi, G. Cell therapy using allogeneic bone marrow mesenchymal stem cells prevents tissue damage in collagen-induced arthritis. Arthritis Rheum. 2007, 56, 1175–1186. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Zhang, Z.; Zhou, M.; Sun, Y.; Su, D.; Feng, X.; Gao, X.; Shi, S.; Chen, W.; et al. Allogeneic mesenchymal stem cells inhibited t follicular helper cell generation in rheumatoid arthritis. Sci. Rep. 2015, 5, 12777. [Google Scholar] [CrossRef]
- Palmblad, K.; Erlandsson-Harris, H.; Tracey, K.J.; Andersson, U. Dynamics of early synovial cytokine expression in rodent collagen-induced arthritis: A therapeutic study using a macrophage-deactivating compound. Am. J. Pathol. 2001, 158, 491–500. [Google Scholar] [CrossRef]
- Komatsu, N.; Takayanagi, H. Inflammation and bone destruction in arthritis: Synergistic activity of immune and mesenchymal cells in joints. Front. Immunol. 2012, 3, 77. [Google Scholar] [CrossRef]
- Asquith, D.L.; Miller, A.M.; McInnes, I.B.; Liew, F.Y. Animal models of rheumatoid arthritis. Eur. J. Immunol. 2009, 39, 2040–2044. [Google Scholar] [CrossRef]
- Trentham, D.E.; Dynesius, R.A.; David, J.R. Passive transfer by cells of type ii collagen-induced arthritis in rats. J. Clin. Investig. 1978, 62, 359–366. [Google Scholar] [CrossRef]
- Santos, S.G.; Lamghari, M.; Almeida, C.R.; Oliveira, M.I.; Neves, N.; Ribeiro, A.C.; Barbosa, J.N.; Barros, R.; Maciel, J.; Martins, M.C.; et al. Adsorbed fibrinogen leads to improved bone regeneration and correlates with differences in the systemic immune response. Acta Biomater. 2013, 9, 7209–7217. [Google Scholar] [CrossRef]
- Ibraheem, A.S.; El-Sayed, M.F.; Ahmed, R.A. Lymph node histopathological studies in a combined adjuvant–collagen induced arthritis model in albino rat rattus rattus. J. Basic Appl. Zool. 2013, 66, 195–205. [Google Scholar] [CrossRef][Green Version]
- Hawkins, P.; Armstrong, R.; Boden, T.; Garside, P.; Knight, K.; Lilley, E.; Seed, M.; Wilkinson, M.; Williams, R.O. Applying refinement to the use of mice and rats in rheumatoid arthritis research. Inflammopharmacology 2015, 23, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, T.; Huang, H.; Cheng, W.; Lai, Y.; Bai, X.; Chen, J.; Yue, Y.; Zheng, Z.; Guo, C.; et al. Fracture healing in a collagen-induced arthritis rat model: Radiology and histology evidence. J. Orthop. Res. 2018, 36, 2876–2885. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Capkova, K.; Hribalova, V.; Vannucci, L.; Danyi, I.; Maly, M.; Fiserova, A. Collagen-induced arthritis: Severity and immune response attenuation using multivalent n-acetyl glucosamine. Clin. Exp. Immunol. 2014, 177, 121–133. [Google Scholar] [CrossRef]
- Alivernini, S.; Tolusso, B.; Ferraccioli, G.; Gremese, E.; Kurowska-Stolarska, M.; McInnes, I.B. Driving chronicity in rheumatoid arthritis: Perpetuating role of myeloid cells. Clin. Exp. Immunol. 2018, 193, 13–23. [Google Scholar] [CrossRef]
- Benson, R.A.; Patakas, A.; Conigliaro, P.; Rush, C.M.; Garside, P.; McInnes, I.B.; Brewer, J.M. Identifying the cells breaching self-tolerance in autoimmunity. J. Immunol. 2010, 184, 6378–6385. [Google Scholar] [CrossRef]
- Dalbeth, N.; Callan, M.F. A subset of natural killer cells is greatly expanded within inflamed joints. Arthritis Rheum. 2002, 46, 1763–1772. [Google Scholar] [CrossRef]
- De Matos, C.T.; Berg, L.; Michaelsson, J.; Fellander-Tsai, L.; Karre, K.; Soderstrom, K. Activating and inhibitory receptors on synovial fluid natural killer cells of arthritis patients: Role of cd94/nkg2a in control of cytokine secretion. Immunology 2007, 122, 291–301. [Google Scholar] [CrossRef]
- Lo, C.K.; Lam, Q.L.; Sun, L.; Wang, S.; Ko, K.H.; Xu, H.; Wu, C.Y.; Zheng, B.J.; Lu, L. Natural killer cell degeneration exacerbates experimental arthritis in mice via enhanced interleukin-17 production. Arthritis Rheum. 2008, 58, 2700–2711. [Google Scholar] [CrossRef]
- Vasconcelos, D.M.; Goncalves, R.M.; Almeida, C.R.; Pereira, I.O.; Oliveira, M.I.; Neves, N.; Silva, A.M.; Ribeiro, A.C.; Cunha, C.; Almeida, A.R.; et al. Fibrinogen scaffolds with immunomodulatory properties promote in vivo bone regeneration. Biomaterials 2016, 111, 163–178. [Google Scholar] [CrossRef]
- Silva, A.M.; Almeida, M.I.; Teixeira, J.H.; Ivan, C.; Oliveira, J.; Vasconcelos, D.; Neves, N.; Ribeiro-Machado, C.; Cunha, C.; Barbosa, M.A.; et al. Profiling the circulating mirnome reveals a temporal regulation of the bone injury response. Theranostics 2018, 8, 3902–3917. [Google Scholar] [CrossRef] [PubMed]
- Choy, E. Understanding the dynamics: Pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 2012, 51 (Suppl. 5), v3–v11. [Google Scholar] [CrossRef]
- Szekanecz, Z.; Halloran, M.M.; Volin, M.V.; Woods, J.M.; Strieter, R.M.; Haines, G.K., 3rd; Kunkel, S.L.; Burdick, M.D.; Koch, A.E. Temporal expression of inflammatory cytokines and chemokines in rat adjuvant-induced arthritis. Arthritis Rheum. 2000, 43, 1266–1277. [Google Scholar] [CrossRef]
- Fuseler, J.W.; Conner, E.M.; Davis, J.M.; Wolf, R.E.; Grisham, M.B. Cytokine and nitric oxide production in the acute phase of bacterial cell wall-induced arthritis. Inflammation 1997, 21, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Timmen, M.; Hidding, H.; Wieskotter, B.; Baum, W.; Pap, T.; Raschke, M.J.; Schett, G.; Zwerina, J.; Stange, R. Influence of antitnf-alpha antibody treatment on fracture healing under chronic inflammation. BMC Musculoskelet Disord 2014, 15, 184. [Google Scholar] [CrossRef] [PubMed]
- Szekanecz, Z.; Vegvari, A.; Szabo, Z.; Koch, A.E. Chemokines and chemokine receptors in arthritis. Front. Biosci (Schol Ed.) 2010, 2, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Akahoshi, T.; Wada, C.; Endo, H.; Hirota, K.; Hosaka, S.; Takagishi, K.; Kondo, H.; Kashiwazaki, S.; Matsushima, K. Expression of monocyte chemotactic and activating factor in rheumatoid arthritis. Regulation of its production in synovial cells by interleukin-1 and tumor necrosis factor. Arthritis Rheum. 1993, 36, 762–771. [Google Scholar] [CrossRef]
- Kinne, R.W.; Brauer, R.; Stuhlmuller, B.; Palombo-Kinne, E.; Burmester, G.R. Macrophages in rheumatoid arthritis. Arthritis Res. 2000, 2, 189–202. [Google Scholar] [CrossRef][Green Version]
- Sarkar, S.; Cooney, L.A.; White, P.; Dunlop, D.B.; Endres, J.; Jorns, J.M.; Wasco, M.J.; Fox, D.A. Regulation of pathogenic il-17 responses in collagen-induced arthritis: Roles of endogenous interferon-gamma and il-4. Arthritis Res. Ther. 2009, 11, R158. [Google Scholar] [CrossRef]
- Dimitrijevic, M.; Arsenovic-Ranin, N.; Kosec, D.; Bufan, B.; Nacka-Aleksic, M.; Pilipovic, I.; Leposavic, G. Sexual dimorphism in th17/treg axis in lymph nodes draining inflamed joints in rats with collagen-induced arthritis. Brain Behav. Immun. 2019, 76, 198–214. [Google Scholar] [CrossRef]
- Thornton, S.; Boivin, G.P.; Kim, K.N.; Finkelman, F.D.; Hirsch, R. Heterogeneous effects of il-2 on collagen-induced arthritis. J. Immunol. 2000, 165, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Iwasaki, T.; Kitano, S.; Satake, A.; Nomura, S.; Furukawa, T.; Matsui, K.; Sano, H. Il-2-anti-il-2 monoclonal antibody immune complexes inhibit collagen-induced arthritis by augmenting regulatory t cell functions. J. Immunol. 2018, 201, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Kouro, T.; Takatsu, K. Il-5- and eosinophil-mediated inflammation: From discovery to therapy. Int. Immunol. 2009, 21, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Chalan, P.; Bijzet, J.; van den Berg, A.; Kluiver, J.; Kroesen, B.J.; Boots, A.M.; Brouwer, E. Analysis of serum immune markers in seropositive and seronegative rheumatoid arthritis and in high-risk seropositive arthralgia patients. Sci. Rep. 2016, 6, 26021. [Google Scholar] [CrossRef]
- De Franco, M.; Gille-Perramant, M.F.; Mevel, J.C.; Couderc, J. T helper subset involvement in two high antibody responder lines of mice (biozzi mice): Hi (susceptible) and hii (resistant) to collagen-induced arthritis. Eur. J. Immunol. 1995, 25, 132–136. [Google Scholar] [CrossRef]
- Hoshino-Negishi, K.; Ohkuro, M.; Nakatani, T.; Kuboi, Y.; Nishimura, M.; Ida, Y.; Kakuta, J.; Hamaguchi, A.; Kumai, M.; Kamisako, T.; et al. Role of anti-fractalkine antibody in suppression of joint destruction by inhibiting migration of osteoclast precursors to the synovium in experimental arthritis. Arthritis Rheumatol. 2019, 71, 222–231. [Google Scholar] [CrossRef]
- Nanki, T.; Urasaki, Y.; Imai, T.; Nishimura, M.; Muramoto, K.; Kubota, T.; Miyasaka, N. Inhibition of fractalkine ameliorates murine collagen-induced arthritis. J. Immunol. 2004, 173, 7010–7016. [Google Scholar] [CrossRef]
- Deng, J.; Liu, Y.; Yang, M.; Wang, S.; Zhang, M.; Wang, X.; Ko, K.H.; Hua, Z.; Sun, L.; Cao, X.; et al. Leptin exacerbates collagen-induced arthritis via enhancement of th17 cell response. Arthritis Rheum. 2012, 64, 3564–3573. [Google Scholar] [CrossRef]
- Okano, T.; Koike, T.; Tada, M.; Sugioka, Y.; Mamoto, K.; Inui, K.; Okihana, H.; Ebihara, K.; Nakao, K.; Nakamura, H. Hyperleptinemia suppresses aggravation of arthritis of collagen-antibody-induced arthritis in mice. J. Orthop. Sci. 2015, 20, 1106–1113. [Google Scholar] [CrossRef]
- Tian, G.; Liang, J.N.; Wang, Z.Y.; Zhou, D. Emerging role of leptin in rheumatoid arthritis. Clin. Exp. Immunol. 2014, 177, 557–570. [Google Scholar] [CrossRef]
- Huang, Y.; Zheng, S.; Wang, R.; Tang, C.; Zhu, J.; Li, J. Ccl5 and related genes might be the potential diagnostic biomarkers for the therapeutic strategies of rheumatoid arthritis. Clin. Rheumatol. 2019, 38, 2629–2635. [Google Scholar] [CrossRef] [PubMed]
- Rossato, C.; Albuquerque, L.L.; Katz, I.S.S.; Borrego, A.; Cabrera, W.H.K.; Spadafora-Ferreira, M.; Ribeiro, O.G.; Starobinas, N.; Ibanez, O.M.; De Franco, M.; et al. Early peritoneal cc chemokine production correlates with divergent inflammatory phenotypes and susceptibility to experimental arthritis in mice. J. Immunol. Res. 2019, 2019, 2641098. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Boj, E.; Redlich, K.; Turk, B.; Hanslik-Schnabel, B.; Wanivenhaus, A.; Chott, A.; Smolen, J.S.; Schett, G. Interaction between synovial inflammatory tissue and bone marrow in rheumatoid arthritis. J. Immunol. 2005, 175, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Marinova-Mutafchieva, L.; Williams, R.O.; Funa, K.; Maini, R.N.; Zvaifler, N.J. Inflammation is preceded by tumor necrosis factor-dependent infiltration of mesenchymal cells in experimental arthritis. Arthritis Rheum. 2002, 46, 507–513. [Google Scholar] [CrossRef]
- Santiago-Schwarz, F.; Sullivan, C.; Rappa, D.; Carsons, S.E. Distinct alterations in lineage committed progenitor cells exist in the peripheral blood of patients with rheumatoid arthritis and primary sjogren’s syndrome. J. Rheumatol. 1996, 23, 439–446. [Google Scholar]
- Tomita, T.; Takeuchi, E.; Toyosaki-Maeda, T.; Oku, H.; Kaneko, M.; Takano, H.; Sugamoto, K.; Ohzono, K.; Suzuki, R.; Ochi, T. Establishment of nurse-like stromal cells from bone marrow of patients with rheumatoid arthritis: Indication of characteristic bone marrow microenvironment in patients with rheumatoid arthritis. Rheumatology (Oxford) 1999, 38, 854–863. [Google Scholar] [CrossRef][Green Version]
- Papadaki, H.A.; Kritikos, H.D.; Gemetzi, C.; Koutala, H.; Marsh, J.C.; Boumpas, D.T.; Eliopoulos, G.D. Bone marrow progenitor cell reserve and function and stromal cell function are defective in rheumatoid arthritis: Evidence for a tumor necrosis factor alpha-mediated effect. Blood 2002, 99, 1610–1619. [Google Scholar] [CrossRef]
- Kastrinaki, M.C.; Sidiropoulos, P.; Roche, S.; Ringe, J.; Lehmann, S.; Kritikos, H.; Vlahava, V.M.; Delorme, B.; Eliopoulos, G.D.; Jorgensen, C.; et al. Functional, molecular and proteomic characterisation of bone marrow mesenchymal stem cells in rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 741–749. [Google Scholar] [CrossRef]
- Sun, Y.; Deng, W.; Geng, L.; Zhang, L.; Liu, R.; Chen, W.; Yao, G.; Zhang, H.; Feng, X.; Gao, X.; et al. Mesenchymal stem cells from patients with rheumatoid arthritis display impaired function in inhibiting th17 cells. J. Immunol. Res. 2015, 2015, 284215. [Google Scholar] [CrossRef]
- Dudics, V.; Kunstar, A.; Kovacs, J.; Lakatos, T.; Geher, P.; Gomor, B.; Monostori, E.; Uher, F. Chondrogenic potential of mesenchymal stem cells from patients with rheumatoid arthritis and osteoarthritis: Measurements in a microculture system. Cells Tissues Organs 2009, 189, 307–316. [Google Scholar] [CrossRef]
- Tong, Y.; Niu, M.; Du, Y.; Mei, W.; Cao, W.; Dou, Y.; Yu, H.; Du, X.; Yuan, H.; Zhao, W. Aryl hydrocarbon receptor suppresses the osteogenesis of mesenchymal stem cells in collagen-induced arthritic mice through the inhibition of beta-catenin. Exp. Cell Res. 2017, 350, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhao, N.; Xu, X.; Xu, Y.; Li, S.; Zhang, J.; Yang, P. Dose-specific effects of tumor necrosis factor alpha on osteogenic differentiation of mesenchymal stem cells. Cell Prolif. 2011, 44, 420–427. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence |
|---|---|
| ALP | Forward 5′-GACAAGAAGCCCTTCACAGC-3′ Reverse 5′-CTGGGCCTGGTAGTTGTTGT-3′ |
| RUNX2 | Forward 5′-CCGATGGGACCGTGGTT-3′ Reverse 5′-CAGCAGAGGCATTTCGTAGCT-3′ |
| SOX9 | Forward 5′-CTGAAGGGCTACGACTGGAC-3′ Reverse 5′-TACTGGTCTGCCAGCTTCCT-3′ |
| ACAN | Forward 5′-CTTGGGCAGAAGAAAGATCG-3′ Reverse 5′-GTGCTTGTAGGTGTTGGGGT-3′ |
| GAPDH | Forward 5′-TGCCACTCAGAAGACTGTGG-3′ Reverse 5′-TTCAGCTCTGGGATGACCTT-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teixeira, J.H.; Silva, A.M.; Almeida, M.I.; Bessa-Gonçalves, M.; Cunha, C.; Barbosa, M.A.; Santos, S.G. The Systemic Immune Response to Collagen-Induced Arthritis and the Impact of Bone Injury in Inflammatory Conditions. Int. J. Mol. Sci. 2019, 20, 5436. https://doi.org/10.3390/ijms20215436
Teixeira JH, Silva AM, Almeida MI, Bessa-Gonçalves M, Cunha C, Barbosa MA, Santos SG. The Systemic Immune Response to Collagen-Induced Arthritis and the Impact of Bone Injury in Inflammatory Conditions. International Journal of Molecular Sciences. 2019; 20(21):5436. https://doi.org/10.3390/ijms20215436
Chicago/Turabian StyleTeixeira, José H., Andreia M. Silva, Maria Inês Almeida, Mafalda Bessa-Gonçalves, Carla Cunha, Mário A. Barbosa, and Susana G. Santos. 2019. "The Systemic Immune Response to Collagen-Induced Arthritis and the Impact of Bone Injury in Inflammatory Conditions" International Journal of Molecular Sciences 20, no. 21: 5436. https://doi.org/10.3390/ijms20215436
APA StyleTeixeira, J. H., Silva, A. M., Almeida, M. I., Bessa-Gonçalves, M., Cunha, C., Barbosa, M. A., & Santos, S. G. (2019). The Systemic Immune Response to Collagen-Induced Arthritis and the Impact of Bone Injury in Inflammatory Conditions. International Journal of Molecular Sciences, 20(21), 5436. https://doi.org/10.3390/ijms20215436