β-Cell Maturation and Identity in Health and Disease

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Embryonic Development: Rise of β-Cells
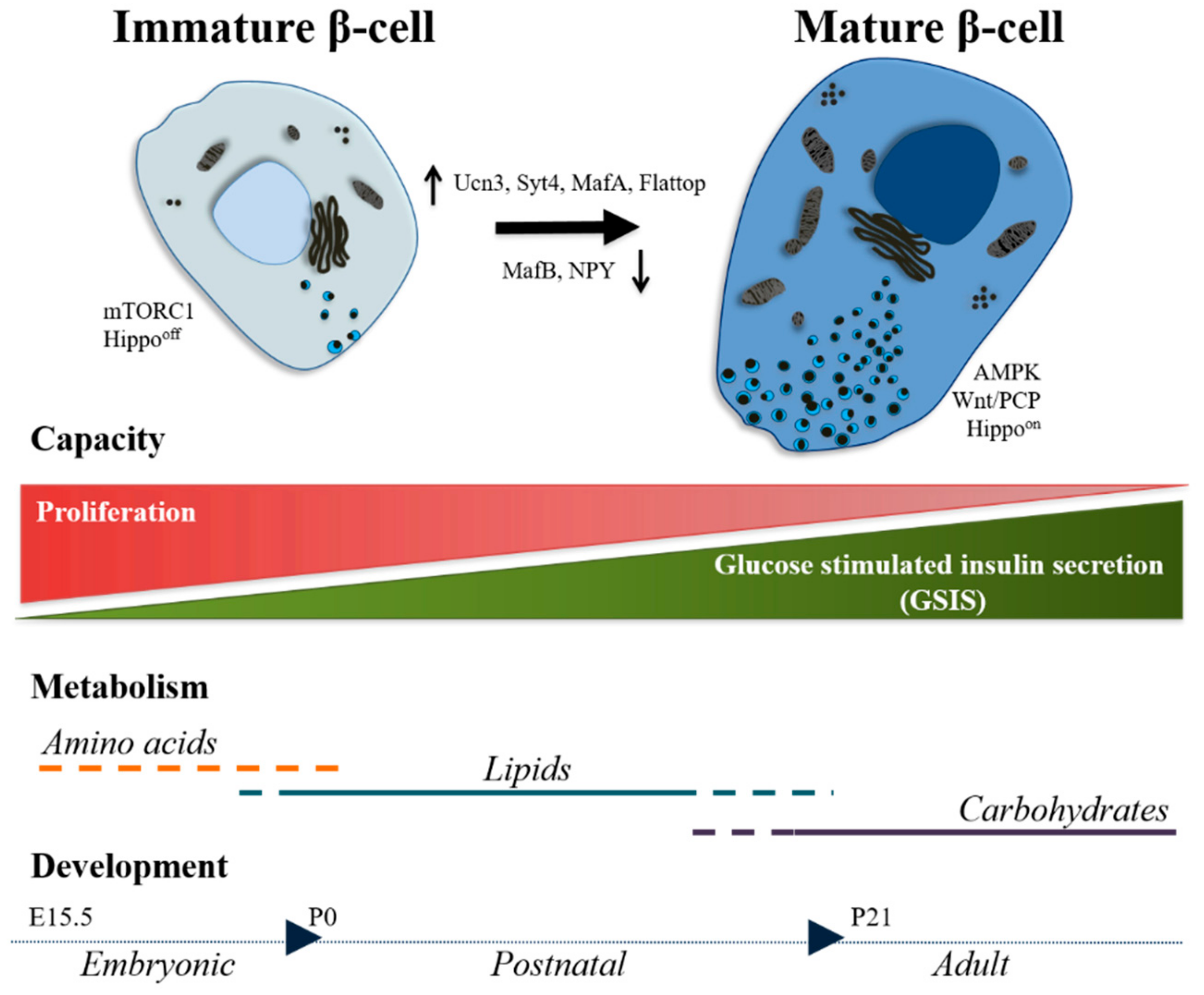
3. Postnatal Development: Road to Maturity
3.1. Markers
3.2. Functionality
3.3. Signaling Pathways
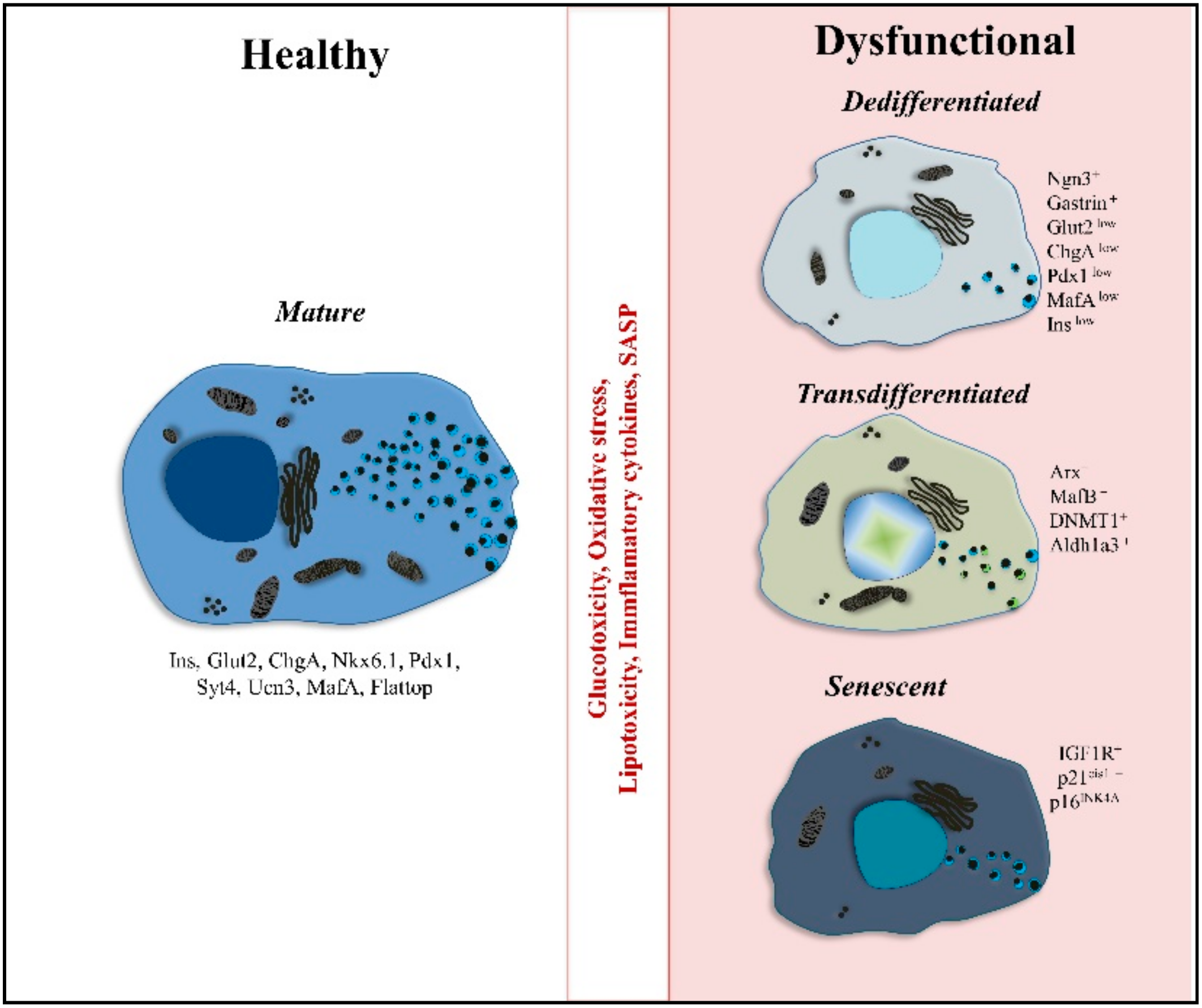
4. Diabetic Conditions: β-Cells Derail
4.1. Dedifferentiated β-Cells
4.2. Polyhormonal Cells and Endcorine Cell Transdifferentiation
4.3. Senescence
5. β-Cell Differentiation Protocols: How to Make “Real” β-Cells
6. Concluding Remarks
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Leslie, R.D.; Palmer, J.; Schloot, N.C.; Lernmark, A. Diabetes at the crossroads: Relevance of disease classification to pathophysiology and treatment. Diabetologia 2016, 59, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.J.; Prentki, M. Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: Time for a conceptual framework shift. Diabetes Vasc. Dis. Res. 2019, 16, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, E. Insulin Therapy and Cancer in Type 2 Diabetes. ISRN Endocrinol. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Liatis, S.; le Roux, C.W.; Kokkinos, A. The role of bariatric surgery to treat diabetes: Current challenges and perspectives. BMC Endocr. Disord. 2017, 17, 50. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.M.J.; Lakey, J.R.T.; Ryan, E.A.; Korbutt, G.S.; Toth, E.; Warnock, G.L.; Kneteman, N.M.; Rajotte, R.V. Islet Transplantation in Seven Patients with Type 1 Diabetes Mellitus Using a Glucocorticoid-Free Immunosuppressive Regimen. N. Engl. J. Med. 2002, 343, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Rother, K.I.; Harlan, D.M. Challenges facing islet transplantation for the treatment of type 1 diabetes mellitus. J. Clin. Investig. 2004, 114, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Swisa, A.; Glaser, B.; Dor, Y. Metabolic stress and compromised identity of pancreatic beta cells. Front. Genet. 2017, 1–11. [Google Scholar] [CrossRef]
- Jiang, W.J.; Peng, Y.C.; Yang, K.M. Cellular signaling pathways regulating β-cell proliferation as a promising therapeutic target in the treatment of diabetes (Review). Exp. Ther. Med. 2018, 16, 3275–3285. [Google Scholar] [CrossRef]
- Ardestani, A.; Paroni, F.; Azizi, Z.; Kaur, S.; Khobragade, V.; Yuan, T.; Frogne, T.; Tao, W.; Oberholzer, J.; Pattou, F.; et al. MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat. Med. 2014, 20, 385–397. [Google Scholar] [CrossRef]
- Muhammad, S.A.; Raza, W.; Nguyen, T.; Bai, B.; Wu, X.; Chen, J. Cellular signaling pathways in insulin resistance-systems biology analyses of microarray dataset reveals new drug target gene signatures of type 2 diabetes mellitus. Front. Physiol. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Jaafar, R.; Tran, S.; Shah, A.; Sun, G.; Valdearcos, M.; Marchetti, P.; Masini, M.; Swisa, A.; Giacometti, S.; Bernal-Mizrachi, E.; et al. mTORC1 to AMPK switching underlies β-cell metabolic plasticity during maturation and diabetes. J. Clin. Investig. 2019, 129, 4124–4137. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 30, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Alismail, H.; Jin, S. Microenvironmental stimuli for proliferation of functional islet β-cells. Cell Biosci. 2014, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Tanaka, Y.; Takahashi, H. Glucose toxicity in β-cells: Type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 2003, 52, 581–587. [Google Scholar] [CrossRef]
- Friedman, R.S.; Lindsay, R.S.; Lilly, J.K.; Nguyen, V.; Sorensen, C.M.; Jacobelli, J.; Krummel, M.F. An evolving autoimmune microenvironment regulates the quality of effector T cell restimulation and function. Proc. Natl. Acad. Sci. USA 2014, 111, 9223–9228. [Google Scholar] [CrossRef] [PubMed]
- Kracht, M.J.L.; Zaldumbide, A.; Roep, B.O. Neoantigens and Microenvironment in Type 1 Diabetes: Lessons from Antitumor Immunity. Trends Endocrinol. Metab. 2016, 27, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Bonner-Weir, S. Pancreatic β Cell Regeneration as a Possible Therapy for Diabetes. Cell Metab. 2018, 27, 57–67. [Google Scholar] [CrossRef]
- Zhou, Q.; Melton, D.A. Pancreas regeneration. Nature 2018, 557, 351–358. [Google Scholar] [CrossRef]
- Tritschler, S.; Theis, F.J.; Lickert, H.; Böttcher, A. Systematic single-cell analysis provides new insights into heterogeneity and plasticity of the pancreas. Mol. Metab. 2017, 6, 974–990. [Google Scholar] [CrossRef]
- Zhong, F.; Jiang, Y. Endogenous Pancreatic β Cell Regeneration: A Potential Strategy for the Recovery of β Cell Deficiency in Diabetes. Front. Endocrinol. (Lausanne) 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Kieffer, T.J.; Woltjen, K.; Osafune, K.; Yabe, D.; Inagaki, N. Beta-cell replacement strategies for diabetes. J. Diabetes Investig. 2018, 9, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Sneddon, J.B.; Tang, Q.; Stock, P.; Bluestone, J.A.; Roy, S.; Desai, T.; Hebrok, M. Stem Cell Therapies for Treating Diabetes: Progress and Remaining Challenges. Cell Stem Cell 2018, 22, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Peloso, A.; Citro, A.; Zoro, T.; Cobianchi, L.; Kahler-Quesada, A.; Bianchi, C.M.; Andres, A.; Berishvili, E.; Piemonti, L.; Berney, T.; et al. Regenerative medicine and diabetes: Targeting the extracellular matrix beyond the stem cell approach and encapsulation technology. Front. Endocrinol. (Lausanne) 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nostro, M.C.; Sarangi, F.; Yang, C.; Holland, A.; Elefanty, A.G.; Stanley, E.G.; Greiner, D.L.; Keller, G. Efficient generation of NKX6-1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Rep. 2015, 4, 591–604. [Google Scholar] [CrossRef]
- Pagliuca, F.W.; Millman, J.R.; Gürtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic β cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef]
- Rezania, A.; Bruin, J.E.; Xu, J.; Narayan, K.; Fox, J.K.; O’Neil, J.J.; Kieffer, T.J. Enrichment of human embryonic stem cell-derived NKX6.1-Expressing pancreatic progenitor cells accelerates the maturation of insulin-secreting cells in vivo. Stem Cells 2013, 31, 2432–2442. [Google Scholar] [CrossRef]
- Russ, H.A.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015, 34, 1759–1772. [Google Scholar] [CrossRef]
- Bastidas-Ponce, A.; Tritschler, S.; Dony, L.; Scheibner, K.; Tarquis-Medina, M.; Salinno, C.; Schirge, S.; Burtscher, I.; Böttcher, A.; Theis, F.J.; et al. Comprehensive single cell mRNA profiling reveals a detailed roadmap for pancreatic endocrinogenesis. Development 2019, 146, 1–16. [Google Scholar] [CrossRef]
- Mamidi, A.; Prawiro, C.; Seymour, P.A.; de Lichtenberg, K.H.; Jackson, A.; Serup, P.; Semb, H. Mechanosignalling via integrins directs fate decisions of pancreatic progenitors. Nature 2018, 564, 114–118. [Google Scholar] [CrossRef]
- Bakhti, M.; Böttcher, A.; Lickert, H. Modelling the endocrine pancreas in health and disease. Nat. Rev. Endocrinol. 2019, 15, 155–171. [Google Scholar] [CrossRef]
- Blum, B.; Hrvatin, S.; Schuetz, C.; Bonal, C.; Rezania, A.; Melton, D.A. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat. Biotechnol. 2012, 30, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.C.; Wright, C. Pancreas organogenesis: From bud to plexus to gland. Dev. Dyn. 2011, 240, 530–565. [Google Scholar] [CrossRef] [PubMed]
- Bastidas-Ponce, A.; Scheibner, K.; Lickert, H.; Bakhti, M. Cellular and molecular mechanisms coordinating pancreas development. Development 2017, 144, 2873–2888. [Google Scholar] [CrossRef] [PubMed]
- Brereton, M.F.; Iberl, M.; Shimomura, K.; Zhang, Q.; Adriaenssens, A.E.; Proks, P.; Spiliotis, I.I.; Dace, W.; Mattis, K.K.; Ramracheya, R.; et al. Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, H.; Gu, X.; Enge, M.; Dai, X.; Wang, Y.; Damond, N.; Downie, C.; Liu, K.; Wang, J.; Xing, Y.; et al. Converting Adult Pancreatic Islet α Cells into β Cells by Targeting Both Dnmt1 and Arx. Cell Metab. 2017, 25, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C.; Bonner-Weir, S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 2004, 53 (Suppl. 3), S16–S21. [Google Scholar] [CrossRef]
- Liu, J.S.E.; Hebrok, M. All mixed up: Defining roles for β-cell subtypes in mature islets. Genes Dev. 2017, 31, 228–240. [Google Scholar] [CrossRef]
- Gutierrez, G.D.; Gromada, J.; Sussel, L. Heterogeneity of the pancreatic beta cell. Front. Genet. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Romer, A.I.; Sussel, L. Pancreatic islet cell development and regeneration. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 255–264. [Google Scholar] [CrossRef]
- Nasteska, D.; Hodson, D.J. The role of beta cell heterogeneity in islet function and insulin release. J. Mol. Endocrinol. 2018, 61, R43–R60. [Google Scholar] [CrossRef]
- Bonner-weir, S. Pancreatic β-cell heterogeneity revisited. Nature 1916, 535, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Roscioni, S.S.; Migliorini, A.; Gegg, M.; Lickert, H. Impact of islet architecture on β-cell heterogeneity, plasticity and function. Nat. Publ. Gr. 2016, 12, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Nair, G.; Hebrok, M. Islet formation in mice and men: Lessons for the generation of functional insulin-producing β-cells from human pluripotent stem cells. Curr. Opin. Genet. Dev. 2015, 32, 171–180. [Google Scholar] [CrossRef]
- Shih, H.P.; Wang, A.; Sander, M. Pancreas organogenesis: From lineage determination to morphogenesis. Annu. Rev. Cell Dev. Biol. 2013, 29, 81–105. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.E.; Berry, A.A.; Kirkwood-Wilson, R.; Roberts, N.A.; Hearn, T.; Salisbury, R.J.; Blaylock, J.; Hanley, K.P.; Hanley, N.A. Development of the human pancreas from foregut to endocrine commitment. Diabetes 2013, 62, 3514–3522. [Google Scholar] [CrossRef]
- Bechard, M.E.; Bankaitis, E.D.; Hipkens, S.B.; Ustione, A.; Piston, D.W.; Yang, Y.P.; Magnuson, M.A.; Wright, C.V.E. Precommitment low-level neurog3 expression defines a long-lived mitotic endocrine-biased progenitor pool that drives production of endocrine-committed cells. Genes Dev. 2016, 30, 1852–1865. [Google Scholar] [CrossRef]
- Wang, S.; Yan, J.; Anderson, D.A.; Xu, Y.; Kanal, M.C.; Cao, Z.; Wright, C.V.E.; Gu, G. Neurog3 gene dosage regulates allocation of endocrine and exocrine cell fates in the developing mouse pancreas. Dev. Biol. 2010, 339, 26–37. [Google Scholar] [CrossRef]
- Byrnes, L.E.; Wong, D.M.; Subramaniam, M.; Meyer, N.P.; Gilchrist, C.L.; Knox, S.M.; Tward, A.D.; Ye, C.J.; Sneddon, J.B. Lineage dynamics of murine pancreatic development at single-cell resolution. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Johansson, K.A.; Dursun, U.; Jordan, N.; Gu, G.; Beermann, F.; Gradwohl, G.; Grapin-Botton, A. Temporal Control of Neurogenin3 Activity in Pancreas Progenitors Reveals Competence Windows for the Generation of Different Endocrine Cell Types. Dev. Cell 2007, 12, 457–465. [Google Scholar] [CrossRef]
- Bankaitis, E.D.; Bechard, M.E.; Wright, C.V.E. Feedback control of growth, differentiation, and morphogenesis of pancreatic endocrine progenitors in an epithelial plexus niche. Genes Dev. 2015, 29, 2203–2216. [Google Scholar] [CrossRef]
- Desgraz, R.; Herrera, P.L. Pancreatic neurogenin 3-expressing cells are unipotent islet precursors. Development 2009, 136, 3567–3574. [Google Scholar] [CrossRef] [PubMed]
- Larsen, H.L.; Grapin-Botton, A. The molecular and morphogenetic basis of pancreas organogenesis. Semin. Cell Dev. Biol. 2017. [CrossRef] [PubMed]
- Jennings, R.E.; Berry, A.A.; Gerrard, D.T.; Wearne, S.J.; Strutt, J.; Withey, S.; Chhatriwala, M.; Piper Hanley, K.; Vallier, L.; Bobola, N.; et al. Laser Capture and Deep Sequencing Reveals the Transcriptomic Programmes Regulating the Onset of Pancreas and Liver Differentiation in Human Embryos. Stem Cell Rep. 2017, 9, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar] [PubMed]
- Gradwohl, G.; Dierich, A.; LeMeur, M.; Guillemot, F. Neurogenin3 Is Required for the Development of the Four Endocrine Cell Lineages of the Pancreas. Proc. Natl. Acad. Sci. USA 2000, 97, 1607–1611. [Google Scholar] [CrossRef] [PubMed]
- Collombat, P.; Mansouri, A.; Hecksher-sørensen, J.; Serup, P.; Krull, J.; Gradwohl, G.; Gruss, P. Opposing actions of Arx and Pax4 in endocrine pancreas development Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003, 17, 2591–2603. [Google Scholar] [CrossRef] [PubMed]
- Arda, H.E.; Benitez, C.M.; Kim, S.K. Gene regulatory networks governing pancreas development. Dev. Cell 2013, 25, 5–13. [Google Scholar] [CrossRef]
- Bramswig, N.C.; Kaestner, K.H. Transcriptional regulation of α-cell differentiation. Diabetes Obes. Metab. 2011, 13, 13–20. [Google Scholar] [CrossRef]
- Dassaye, R.; Naidoo, S.; Cerf, M.E. Transcription factor regulation of pancreatic organogenesis, differentiation and maturation. Islets 2016, 8, 13–34. [Google Scholar] [CrossRef]
- Jensen, J. Gene Regulatory Factors in Pancreatic Development. Dev. Dyn. 2004, 229, 176–200. [Google Scholar] [CrossRef]
- Jennings, R.E.; Berry, A.A.; Strutt, J.P.; Gerrard, D.T.; Hanley, N.A. Human pancreas development. Development 2015, 142, 3126–3137. [Google Scholar] [CrossRef] [PubMed]
- Lango Allen, H.; Flanagan, S.E.; Shaw-Smith, C.; De Franco, E.; Akerman, I.; Caswell, R.; Ferrer, J.; Hattersley, A.T.; Ellard, S. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat. Genet. 2012, 44, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Velazco-Cruz, L.; Song, J.; Maxwell, K.G.; Goedegebuure, M.M.; Augsornworawat, P.; Hogrebe, N.J.; Millman, J.R. Acquisition of Dynamic Function in Human Stem Cell-Derived β Cells. Stem Cell Rep. 2019, 12, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Bakhti, M.; Scheibner, K.; Tritschler, S.; Bastidas-ponce, A. Establishment of a high-resolution 3D modeling system for studying pancreatic epithelial cell biology in vitro. Mol. Metab. 2019, 30. [Google Scholar] [CrossRef]
- Sharon, N.; Chawla, R.; Mueller, J.; Vanderhooft, J.; Whitehorn, L.J.; Rosenthal, B.; Gürtler, M.; Estanboulieh, R.R.; Shvartsman, D.; Gifford, D.K.; et al. A Peninsular Structure Coordinates Asynchronous Differentiation with Morphogenesis to Generate Pancreatic Islets. Cell 2019, 176, 790–804. [Google Scholar] [CrossRef]
- Martens, G.A.; Motté, E.; Kramer, G.; Stangé, G.; Gaarn, L.W.; Hellemans, K.; Nielsen, J.H.; Aerts, J.M.; Ling, Z.; Pipeleers, D. Functional characteristics of neonatal rat β cells with distinct markers. J. Mol. Endocrinol. 2013, 52, 11–28. [Google Scholar] [CrossRef]
- Dore, A.B.; Magde, G.E.; Grogan, W.M.; Webb, S.R. Biphasic Development of Posnatal Mouse Pancreas. Neonatology 1981, 40, 209–217. [Google Scholar] [CrossRef]
- Bonner-Weir, S.; Aguayo-Mazzucato, C.; Weir, G.C. Dynamic development of the pancreas from birth to adulthood. Ups. J. Med. Sci. 2016, 121, 155–158. [Google Scholar] [CrossRef]
- Stolovich-Rain, M.; Enk, J.; Vikesa, J.; Nielsen, F.; Saada, A.; Glaser, B.; Dor, Y. Weaning Triggers a Maturation Step of Pancreatic β Cells. Dev. Cell 2015, 32, 535–545. [Google Scholar] [CrossRef]
- Van Der Meulen, T.; Donaldson, C.J.; Cáceres, E.; Hunter, A.E.; Cowing-Zitron, C.; Pound, L.D.; Adams, M.W.; Zembrzycki, A.; Grove, K.L.; Huising, M.O. Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat. Med. 2015, 21, 769–776. [Google Scholar] [CrossRef] [PubMed]
- van der Meulen, T.; Xie, R.; Kelly, O.G.; Vale, W.W.; Sander, M.; Huising, M.O. Urocortin 3 Marks Mature Human Primary and Embryonic Stem Cell-Derived Pancreatic Alpha and Beta Cells. PLoS ONE 2012, 7, E52181. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Walker, E.M.; Dadi, P.K.; Hu, R.; Xu, Y.; Zhang, W.; Sanavia, T.; Mun, J.; Liu, J.; Nair, G.G.; et al. Synaptotagmin 4 Regulates Pancreatic β Cell Maturation by Modulating the Ca2+ Sensitivity of Insulin Secretion Vesicles. Dev. Cell 2018, 45, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Rodnoi, P.; Rajkumar, M.; Moin, A.S.M.; Georgia, S.K.; Butler, A.E.; Dhawan, S. Neuropeptide Y expression marks partially differentiated β cells in mice and humans. JCI Insight 2017, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Whim, M.D. Pancreatic beta cells synthesize neuropeptide Y and can rapidly release peptide co-transmitters. PLoS ONE 2011, 6, E19478. [Google Scholar] [CrossRef] [PubMed]
- Hang, Y.; Stein, R. MafA and MafB activity in pancreatic β cells. Trends Endocrinol. Metab. 2011, 22, 364–373. [Google Scholar] [CrossRef]
- Artner, I.; Blanchi, B.; Raum, J.C.; Guo, M.; Kaneko, T.; Cordes, S.; Sieweke, M.; Stein, R. MafB is required for islet beta cell maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 3853–3858. [Google Scholar] [CrossRef]
- Artner, I.; Hang, Y.; Mazur, M.; Yamamoto, T.; Guo, M.; Lindner, J.; Magnuson, M.A.; Stein, R. MafA and MafB regulate genes critical to β-cells in a unique temporal manner. Diabetes 2010, 59, 2530–2539. [Google Scholar] [CrossRef]
- Nishimura, W.; Takahashi, S.; Yasuda, K. MafA is critical for maintenance of the mature beta cell phenotype in mice. Diabetologia 2015, 58, 566–574. [Google Scholar] [CrossRef]
- Nishimura, W.; Kondo, T.; Salameh, T.; El Khattabi, I.; Dodge, R.; Bonner-Weir, S.; Sharma, A. A switch from MafB to MafA expression accompanies differentiation to pancreatic β-cells. Dev. Biol. 2006, 293, 526–539. [Google Scholar] [CrossRef]
- Cyphert, H.A.; Walker, E.M.; Hang, Y.; Dhawan, S.; Haliyur, R.; Bonatakis, L.; Avrahami, D.; Brissova, M.; Kaestner, K.H.; Bhushan, A.; et al. Examining how the MAFB transcription factor affects islet β-cell function postnatally. Diabetes 2019, 68, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Morán, I.; Akerman, I.; Van De Bunt, M.; Xie, R.; Benazra, M.; Nammo, T.; Arnes, L.; Nakić, N.; García-Hurtado, J.; Rodríguez-Seguí, S.; et al. Human β cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 2012, 16, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Akerman, I.; Tu, Z.; Beucher, A.; Rolando, D.M.Y.; Sauty-Colace, C.; Benazra, M.; Nakic, N.; Yang, J.; Wang, H.; Pasquali, L.; et al. Human Pancreatic β Cell lncRNAs Control Cell-Specific Regulatory Networks. Cell Metab. 2017, 25, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.; Böttcher, A.; Burtscher, I.; Hasenoeder, S.; Van Campenhout, C.; Aichler, M.; Walch, A.; Grant, S.G.N.; Lickert, H. Flattop regulates basal body docking and positioning in mono- and multiciliated cells. Elife 2014, 3, 1–24. [Google Scholar] [CrossRef]
- Bader, E.; Migliorini, A.; Gegg, M.; Moruzzi, N.; Gerdes, J.; Roscioni, S.S.; Bakhti, M.; Brandl, E.; Irmler, M.; Beckers, J.; et al. Identification of proliferative and mature β-cells in the islets of Langerhans. Nature 2016, 535, 430–434. [Google Scholar] [CrossRef]
- Dorrell, C.; Schug, J.; Canaday, P.S.; Russ, H.A.; Tarlow, B.D.; Grompe, M.T.; Horton, T.; Hebrok, M.; Streeter, P.R.; Kaestner, K.H.; et al. Human islets contain four distinct subtypes of β cells. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef]
- Johnston, N.R.; Mitchell, R.K.; Haythorne, E.; Pessoa, M.P.; Semplici, F.; Ferrer, J.; Piemonti, L.; Marchetti, P.; Bugliani, M.; Bosco, D.; et al. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab. 2016, 24, 389–401. [Google Scholar] [CrossRef]
- van der Meulen, T.; Mawla, A.M.; DiGruccio, M.R.; Adams, M.W.; Nies, V.; Dólleman, S.; Liu, S.; Ackermann, A.M.; Cáceres, E.; Hunter, A.E.; et al. Virgin Beta Cells Persist throughout Life at a Neogenic Niche within Pancreatic Islets. Cell Metab. 2017, 25, 911–926. [Google Scholar] [CrossRef]
- Kulkarni, R.N.; Mizrachi, E.-B.; Ocana, A.G.; Stewart, A.F. Human β-cell proliferation and intracellular signaling: Driving in the dark without a road map. Diabetes 2012, 61, 2205–2213. [Google Scholar] [CrossRef]
- Bernal-Mizrachi, E.; Kulkarni, R.N.; Scott, D.K.; Mauvais-Jarvis, F.; Stewart, A.F.; Garcia-Ocaña, A. Human β-cell proliferation and intracellular signaling part 2: Still driving in the dark without a road map. Diabetes 2014, 63, 819–831. [Google Scholar] [CrossRef]
- Stewart, A.F.; Hussain, M.A.; García-Ocaña, A.; Vasavada, R.C.; Bhushan, A.; Bernal-Mizrachi, E.; Kulkarni, R.N. Human β-Cell proliferation and intracellular signaling: Part 3. Diabetes 2015, 64, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, L.; Hindi, S.; Sorenson, R.L.; German, M.S. β-Cell adaptation in pregnancy. Diabetes Obes. Metab. 2016, 18, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Arrojo e Drigo, R.; Lev-Ram, V.; Tyagi, S.; Ramachandra, R.; Deerinck, T.; Bushong, E.; Phan, S.; Orphan, V.; Lechene, C.; Ellisman, M.H.; et al. Age Mosaicism across Multiple Scales in Adult Tissues. Cell Metab. 2019, 30, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Linnemann, A.K.; Baan, M.; Davis, D.B. Pancreatic -Cell Proliferation in Obesity. Adv. Nutr. An Int. Rev. J. 2014, 5, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Piccand, J.; Meunier, A.; Merle, C.; Jia, Z.; Barnier, J.V.; Gradwohl, G. Pak3 promotes cell cycle exit and differentiation of β-cells in the embryonic pancreas and is necessary to maintain glucose homeostasis in adult mice. Diabetes 2014, 63, 203–215. [Google Scholar] [CrossRef]
- Puri, S.; Roy, N.; Russ, H.A.; Leonhardt, L.; French, E.K.; Roy, R.; Bengtsson, H.; Scott, D.K.; Stewart, A.F.; Hebrok, M. Replication confers β cell immaturity. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Shirakawa, J.; Kulkarni, R.N. Novel factors modulating human β-cell proliferation. Diabetes Obes. Metab. 2016, 18, 71–77. [Google Scholar] [CrossRef]
- Kang, T.; Jensen, P.; Huang, H.; Lund Christensen, G.; Billestrup, N.; Larsen, M.R. Characterization of the Molecular Mechanisms Underlying Glucose Stimulated Insulin Secretion from Isolated Pancreatic β-cells Using Post-translational Modification Specific Proteomics (PTMomics). Mol. Cell. Proteom. 2018, 17, 95–110. [Google Scholar] [CrossRef]
- Meda, P.; Schuit, F. Glucose-stimulated insulin secretion: The hierarchy of its multiple cellular and subcellular mechanisms. Diabetologia 2013, 56, 2552–2555. [Google Scholar] [CrossRef]
- Jensen, M.V.; Joseph, J.W.; Ronnebaum, S.M.; Burgess, S.C.; Sherry, A.D.; Newgard, C.B. Metabolic cycling in control of glucose-stimulated insulin secretion. Am. J. Physiol. Metab. 2008, 295, E1287–E1297. [Google Scholar] [CrossRef]
- Maechler, P. Mitochondrial function and insulin secretion. Mol. Cell. Endocrinol. 2013, 379, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. The Pancreatic β-Cell: A Bioenergetic Perspective. Physiol. Rev. 2016, 96, 1385–1447. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Ashcroft, F.M. Pancreatic β-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol. Rev. 2017, 98, 117–214. [Google Scholar] [CrossRef] [PubMed]
- Jermendy, A.; Toschi, E.; Aye, T.; Koh, A.; Aguayo-Mazzucato, C.; Sharma, A.; Weir, G.C.; Sgroi, D.; Bonner-Weir, S. Rat neonatal beta cells lack the specialised metabolic phenotype of mature beta cells. Diabetologia 2011, 54, 594–604. [Google Scholar] [CrossRef]
- Wortham, M.; Benthuysen, J.R.; Wallace, M.; Savas, J.N.; Mulas, F.; Divakaruni, A.S.; Liu, F.; Albert, V.; Taylor, B.L.; Sui, Y.; et al. Integrated In Vivo Quantitative Proteomics and Nutrient Tracing Reveals Age-Related Metabolic Rewiring of Pancreatic β Cell Function. Cell Rep. 2018, 25, 2904–2918. [Google Scholar] [CrossRef]
- Henquin, J.C.; Nenquin, M. Immaturity of insulin secretion by pancreatic islets isolated from one human neonate. J. Diabetes Investig. 2018, 9, 270–273. [Google Scholar] [CrossRef]
- Wang, Z.; Thurmond, D.C. Mechanisms of biphasic insulin-granule exocytosis—Roles of the cytoskeleton, small GTPases and SNARE proteins. J. Cell Sci. 2009, 122, 893–903. [Google Scholar] [CrossRef]
- Salem, V.; Silva, L.D.; Suba, K.; Georgiadou, E.; Neda Mousavy Gharavy, S.; Akhtar, N.; Martin-Alonso, A.; Gaboriau, D.C.A.; Rothery, S.M.; Stylianides, T.; et al. Leader β-cells coordinate Ca2+ dynamics across pancreatic islets in vivo. Nat. Metab. 2019, 1, 615–629. [Google Scholar] [CrossRef]
- Lei, C.L.; Kellard, J.A.; Hara, M.; Johnson, J.D.; Rodriguez, B.; Briant, L.J.B. Beta-cell hubs maintain Ca2+ oscillations in human and mouse islet simulations. Islets 2018, 10, 151–167. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 361–371. [Google Scholar] [CrossRef]
- Sinagoga, K.L.; Stone, W.J.; Schiesser, J.V.; Schweitzer, J.I.; Sampson, L.; Zheng, Y.; Wells, J.M. Distinct roles for the mTOR pathway in postnatal morphogenesis, maturation and function of pancreatic islets. J. Cell Sci. 2017, 144, 2402–2414. [Google Scholar]
- Ni, Q.; Gu, Y.; Xie, Y.; Yin, Q.; Zhang, H.; Nie, A.; Li, W.; Wang, Y.; Ning, G.; Wang, W.; et al. Raptor regulates functional maturation of murine beta cells. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Eberhard, C.E.; Screaton, R.A. Role of AMPK in pancreatic beta cell function. Mol. Cell. Endocrinol. 2013, 366, 127–134. [Google Scholar] [CrossRef]
- Rourke, J.L.; Hu, Q.; Screaton, R.A. AMPK and Friends: Central Regulators of β Cell Biology. Trends Endocrinol. Metab. 2018, 29, 111–122. [Google Scholar] [CrossRef]
- Yoshihara, E.; Wei, Z.; Lin, C.S.; Fang, S.; Ahmadian, M.; Kida, Y.; Tseng, T.; Dai, Y.; Yu, R.T.; Liddle, C.; et al. ERRγ Is Required for the Metabolic Maturation of Therapeutically Functional Glucose-Responsive β Cells. Cell Metab. 2016, 23, 622–643. [Google Scholar] [CrossRef]
- Sharon, N.; Vanderhooft, J.; Straubhaar, J.; Mueller, J.; Chawla, R.; Zhou, Q.; Engquist, E.N.; Trapnell, C.; Gifford, D.K.; Melton, D.A. Wnt Signaling Separates the Progenitor and Endocrine Compartments during Pancreas Development. Cell Rep. 2019, 27, 2281–2291. [Google Scholar] [CrossRef]
- Cortijo, C.; Gouzi, M.; Tissir, F.; Grapin-Botton, A. Planar Cell Polarity Controls Pancreatic Beta Cell Differentiation and Glucose Homeostasis. Cell Rep. 2012, 2, 1593–1606. [Google Scholar] [CrossRef]
- Scheibner, K.; Bakhti, M.; Bastidas-Ponce, A.; Lickert, H. Wnt signaling: Implications in endoderm development and pancreas organogenesis. Curr. Opin. Cell Biol. 2019, 61, 48–55. [Google Scholar] [CrossRef]
- Liu, X.; Yan, F.; Yao, H.; Chang, M.; Qin, J.; Li, Y.; Wang, Y.; Pei, X. Involvement of RhoA/ROCK in insulin secretion of pancreatic β-cells in 3D culture. Cell Tissue Res. 2014, 358, 359–369. [Google Scholar] [CrossRef]
- Schulze, T.; Morsi, M.; Brüning, D.; Schumacher, K.; Rustenbeck, I. Different responses of mouse islets and MIN6 pseudo-islets to metabolic stimulation: A note of caution. Endocrine 2016, 51, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Carstensen-Kirberg, M.; Röhrig, K.; Niersmann, C.; Margriet Ouwens, D.; Belgardt, B.F.; Roden, M.; Herder, C. Sfrp5 increases glucose-stimulated insulin secretion in the rat pancreatic beta cell line INS-1E. PLoS ONE 2019, 14, e0213650. [Google Scholar] [CrossRef] [PubMed]
- Ardestani, A.; Maedler, K. The Hippo signaling pathway in pancreatic β-cells: Functions and regulations. Endocr. Rev. 2018, 39, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Dai, C.; Guo, M.; Taylor, B.; Harmon, J.S.; Sander, M.; Robertson, R.P.; Powers, A.C.; Stein, R. Inactivation of specific β cell transcription factors in type 2 diabetes. J. Clin. Investig. 2013, 123, 3305–3316. [Google Scholar] [CrossRef]
- Bensellam, M.; Jonas, J.C.; Laybutt, D.R. Mechanisms of β;-cell dedifferentiation in diabetes: Recent findings and future research directions. J. Endocrinol. 2018, 236, R109–R143. [Google Scholar] [CrossRef]
- Dahan, T.; Ziv, O.; Horwitz, E.; Zemmour, H.; Lavi, J.; Swisa, A.; Leibowitz, G.; Ashcroft, F.M.; In’t Veld, P.; Glaser, B.; et al. Pancreatic β-Cells express the fetal islet hormone gastrin in rodent and human diabetes. Diabetes 2017, 66, 426–436. [Google Scholar] [CrossRef]
- Jonas, J.C.; Sharma, A.; Hasenkamp, W.; Ilkova, H.; Patanè, G.; Laybutt, R.; Bonner-Weir, S.; Weir, G.C. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J. Biol. Chem. 1999, 274, 14112–14121. [Google Scholar] [CrossRef]
- Ross Laybutt, D.; Sharma, A.; Sgroi, D.C.; Gaudet, J.; Bonner-Weir, S.; Weir, G.C. Genetic regulation of metabolic pathways in β-cells disrupted by hyperglycemia. J. Biol. Chem. 2002, 277, 10912–10921. [Google Scholar] [CrossRef]
- Laybutt, D.R.; Glandt, M.; Xu, G.; Ahn, Y.B.; Trivedi, N.; Bonner-Weir, S.; Weir, G.C. Critical reduction β-cell mass results in two distinct outcomes over time. Adaptation with impaired glucose tolerance or decompensated diabetes. J. Biol. Chem. 2003, 278, 2997–3005. [Google Scholar] [CrossRef]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef]
- Wang, Z.; York, N.W.; Nichols, C.G.; Remedi, M.S. Pancreatic β cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014, 19, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Diedisheim, M.; Oshima, M.; Albagli, O.; Huldt, C.W.; Ahlstedt, I.; Clausen, M.; Menon, S.; Aivazidis, A.; Andreasson, A.C.; Haynes, W.G.; et al. Modeling human pancreatic beta cell dedifferentiation. Mol. Metab. 2018, 10, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.J.; Chatterjee, A.; Shen, E.; Cox, A.R.; Kushner, J.A. Low-level insulin content within abundant non-b islet endocrine cells in long-standing type 1 diabetes. Diabetes 2019, 68, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of β-cell dedifferentiation in human type 2 diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Rui, J.; Deng, S.; Arazi, A.; Perdigoto, A.L.; Liu, Z.; Herold, K.C. β Cells that Resist Immunological Attack Develop during Progression of Autoimmune Diabetes in NOD Mice. Cell Metab. 2017, 25, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Keenan, H.A.; Sun, J.K.; Levine, J.; Doria, A.; Aiello, L.P.; Eisenbarth, G.; Bonner-Weir, S.; King, G.L. Residual insulin production and pancreatic β-cell turnover after 50 years of diabetes: Joslin medalist study. Diabetes 2010, 59, 2846–2853. [Google Scholar] [CrossRef]
- Spaeth, J.M.; Gupte, M.; Perelis, M.; Yang, Y.P.; Cyphert, H.; Guo, S.; Liu, J.H.; Guo, M.; Bass, J.; Magnuson, M.A.; et al. Defining a Novel Role for the Pdx1 Transcription Factor in Islet β-Cell Maturation and Proliferation During Weaning. Diabetes 2017, 66, 2830–2839. [Google Scholar] [CrossRef]
- Bastidas-Ponce, A.; Roscioni, S.S.; Burtscher, I.; Bader, E.; Sterr, M.; Bakhti, M.; Lickert, H. Foxa2 and Pdx1 cooperatively regulate postnatal maturation of pancreatic β-cells. Mol. Metab. 2017, 6, 524–534. [Google Scholar] [CrossRef]
- Sacco, F.; Seelig, A.; Humphrey, S.J.; Krahmer, N.; Volta, F.; Reggio, A.; Marchetti, P.; Gerdes, J.; Mann, M. Phosphoproteomics Reveals the GSK3-PDX1 Axis as a Key Pathogenic Signaling Node in Diabetic Islets. Cell Metab. 2019, 29, 1422–1432. [Google Scholar] [CrossRef]
- Cheng, C.W.; Villani, V.; Buono, R.; Wei, M.; Kumar, S.; Yilmaz, O.H.; Cohen, P.; Sneddon, J.B.; Perin, L.; Longo, V.D. Fasting-Mimicking Diet Promotes Ngn3-Driven β-Cell Regeneration to Reverse Diabetes. Cell 2017, 168, 775–788. [Google Scholar] [CrossRef]
- Tomita, T. Apoptosis of pancreatic β-cells in Type 1 diabetes. Bosn. J. Basic Med. Sci. 2017, 17, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T. Apoptosis in pancreatic β-islet cells in Type 2 diabetes. Bosn. J. Basic Med. Sci. 2016, 16, 162–179. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Bhushan, A.; Butler, A.E.; Rizza, R.A.; Butler, P.C. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: Indirect evidence for islet regeneration? Diabetologia 2005, 48, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H.E.; McKenzie, M.D.; Angstetra, E.; Campbell, P.D.; Kay, T.W. Beta cell apoptosis in diabetes. Apoptosis 2009, 14, 1389–1404. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C.; Bonner-Weir, S. Islet β cell mass in diabetes and how it relates to function, birth, and death. Ann. N. Y. Acad. Sci. 2013, 1281, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C.; Aguayo-Mazzucato, C.; Bonner-Weir, S. β-cell dedifferentiation in diabetes is important, but what is it? Islets 2013, 5, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Chera, S.; Baronnier, D.; Ghila, L.; Cigliola, V.; Jensen, J.N.; Gu, G.; Furuyama, K.; Thorel, F.; Gribble, F.M.; Reimann, F.; et al. Diabetes recovery by age-dependent conversion of pancreatic δ-cells into insulin producers. Nature 2014, 514, 503–507. [Google Scholar] [CrossRef]
- Moin, A.S.; Butler, A.E. Alterations in Beta Cell Identity in Type 1 and Type 2 Diabetes. Curr. Diab. Rep. 2019, 19, 83. [Google Scholar] [CrossRef]
- Gutiérrez, G.D.; Bender, A.S.; Cirulli, V.; Mastracci, T.L.; Kelly, S.M.; Tsirigos, A.; Kaestner, K.H.; Sussel, L. Pancreatic β cell identity requires continual repression of non-β cell programs. J. Clin. Investig. 2017, 127, 244–259. [Google Scholar] [CrossRef]
- Gao, T.; McKenna, B.; Li, C.; Reichert, M.; Nguyen, J.; Singh, T.; Yang, C.; Pannikar, A.; Doliba, N.; Zhang, T.; et al. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab. 2014, 19, 259–271. [Google Scholar] [CrossRef]
- Seiron, P.; Wiberg, A.; Kuric, E.; Krogvold, L.; Jahnsen, F.L.; Dahl-Jørgensen, K.; Skog, O.; Korsgren, O. Characterisation of the endocrine pancreas in type 1 diabetes: Islet size is maintained but islet number is markedly reduced. J. Pathol. Clin. Res. 2019, 5, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Dirice, E.; Kahraman, S.; De Jesus, D.F.; El Ouaamari, A.; Basile, G.; Baker, R.L.; Yigit, B.; Piehowski, P.D.; Kim, M.-J.; Dwyer, A.J.; et al. Increased β-cell proliferation before immune cell invasion prevents progression of type 1 diabetes. Nat. Metab. 2019, 1, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Thorel, F.; Népote, V.; Avril, I.; Kohno, K.; Desgraz, R.; Chera, S.; Herrera, P.L. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010, 464, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, K.; Chera, S.; van Gurp, L.; Oropeza, D.; Ghila, L.; Damond, N.; Vethe, H.; Paulo, J.A.; Joosten, A.M.; Berney, T.; et al. Diabetes relief in mice by glucose-sensing insulin-secreting human α-cells. Nature 2019, 567, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Bakhti, M.; Lickert, H. Cell makeover for diabetes therapy. Nat. Metab. 2019, 1, 312–313. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C.; van Haaren, M.; Mruk, M.; Lee, T.B.; Crawford, C.; Hollister-Lock, J.; Sullivan, B.A.; Johnson, J.W.; Ebrahimi, A.; Dreyfuss, J.M.; et al. β Cell Aging Markers Have Heterogeneous Distribution and Are Induced by Insulin Resistance. Cell Metab. 2017, 25, 898–910. [Google Scholar] [CrossRef]
- Thompson, P.J.; Shah, A.; Ntranos, V.; Van Gool, F.; Atkinson, M.; Bhushan, A. Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes. Cell Metab. 2019, 29, 1045–1060. [Google Scholar] [CrossRef]
- Mills, J.C.; Stanger, B.Z.; Sander, M. Nomenclature for cellular plasticity: Are the terms as plastic as the cells themselves? EMBO J. 2019, 38, 1–5. [Google Scholar] [CrossRef]
- Shahjalal, H.M.; Abdal Dayem, A.; Lim, K.M.; Jeon, T.-I.; Cho, S.G. Generation of pancreatic β cells for treatment of diabetes: Advances and challenges. Stem Cell Res. Ther. 2018, 9, 1–19. [Google Scholar] [CrossRef]
- Nair, G.G.; Liu, J.S.; Russ, H.A.; Tran, S.; Saxton, M.S.; Chen, R.; Juang, C.; Li, M.-L.; Nguyen, V.Q.; Giacometti, S.; et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat. Cell Biol. 2019, 21, 263–274. [Google Scholar] [CrossRef]
- Alagpulinsa, D.A.; Cao, J.J.L.; Driscoll, R.K.; Sîrbulescu, R.F.; Penson, M.F.E.; Sremac, M.; Engquist, E.N.; Brauns, T.A.; Markmann, J.F.; Melton, D.A.; et al. Alginate-microencapsulation of human stem cell–derived β cells with CXCL12 prolongs their survival and function in immunocompetent mice without systemic immunosuppression. Am. J. Transplant. 2019, 19, 1930–1940. [Google Scholar] [PubMed]
- Veres, A.; Faust, A.L.; Bushnell, H.L.; Engquist, E.N.; Kenty, J.H.R.; Harb, G.; Poh, Y.C.; Sintov, E.; Gürtler, M.; Pagliuca, F.W.; et al. Charting cellular identity during human in vitro β-cell differentiation. Nature 2019, 569, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Theis, F.J.; Lickert, H. A map of β-cell differentiation pathways supports cell therapies for diabetes. Nature 2019, 569, 342–343. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Dominguez, J.R.; Donaghey, J.; Kenty, J.H.R.; Rasouli, N.; Helman, A.; Charlton, J.; Straubhaar, J.R.; Meissner, A.; Melton, D.A. Dissecting mechanisms of human islet differentiation and maturation through epigenome profiling. bioRxiv Genom. 2019, 4, 613026. [Google Scholar]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salinno, C.; Cota, P.; Bastidas-Ponce, A.; Tarquis-Medina, M.; Lickert, H.; Bakhti, M. β-Cell Maturation and Identity in Health and Disease. Int. J. Mol. Sci. 2019, 20, 5417. https://doi.org/10.3390/ijms20215417
Salinno C, Cota P, Bastidas-Ponce A, Tarquis-Medina M, Lickert H, Bakhti M. β-Cell Maturation and Identity in Health and Disease. International Journal of Molecular Sciences. 2019; 20(21):5417. https://doi.org/10.3390/ijms20215417
Chicago/Turabian StyleSalinno, Ciro, Perla Cota, Aimée Bastidas-Ponce, Marta Tarquis-Medina, Heiko Lickert, and Mostafa Bakhti. 2019. "β-Cell Maturation and Identity in Health and Disease" International Journal of Molecular Sciences 20, no. 21: 5417. https://doi.org/10.3390/ijms20215417
APA StyleSalinno, C., Cota, P., Bastidas-Ponce, A., Tarquis-Medina, M., Lickert, H., & Bakhti, M. (2019). β-Cell Maturation and Identity in Health and Disease. International Journal of Molecular Sciences, 20(21), 5417. https://doi.org/10.3390/ijms20215417