Modelling Neurotropic Flavivirus Infection in Human Induced Pluripotent Stem Cell-Derived Systems



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Infection and Replication Kinetics of Flaviviruses on hiPSCs, NSCs, and Neurons
2.2. Cytopatic Effects and Cell Death by Apoptosis in Infected Cells
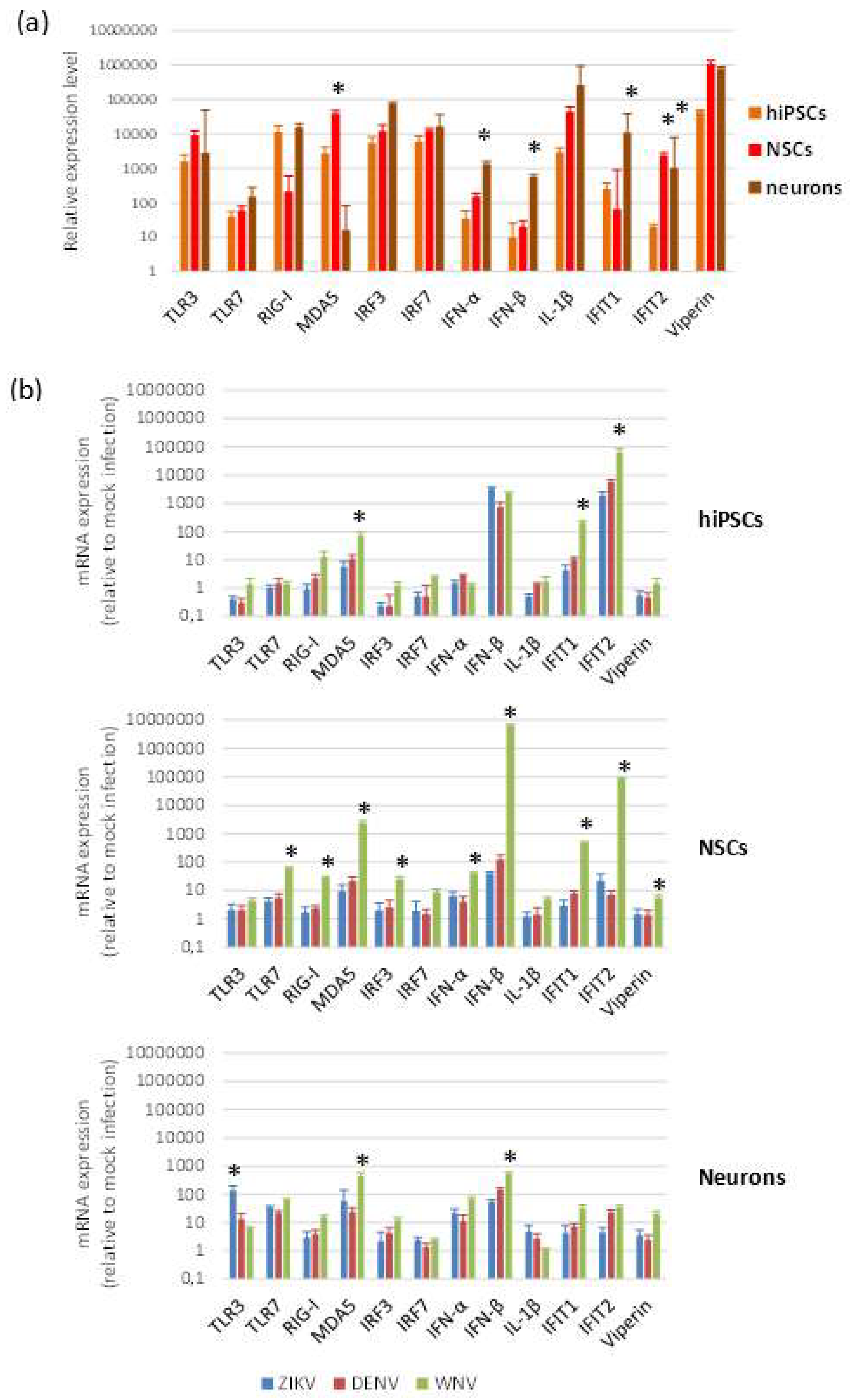
2.3. Expression of Innate Antiviral Immune Response Genes in Infected Cells
2.4. ZIKV Infection and Replication during Neurogenesis
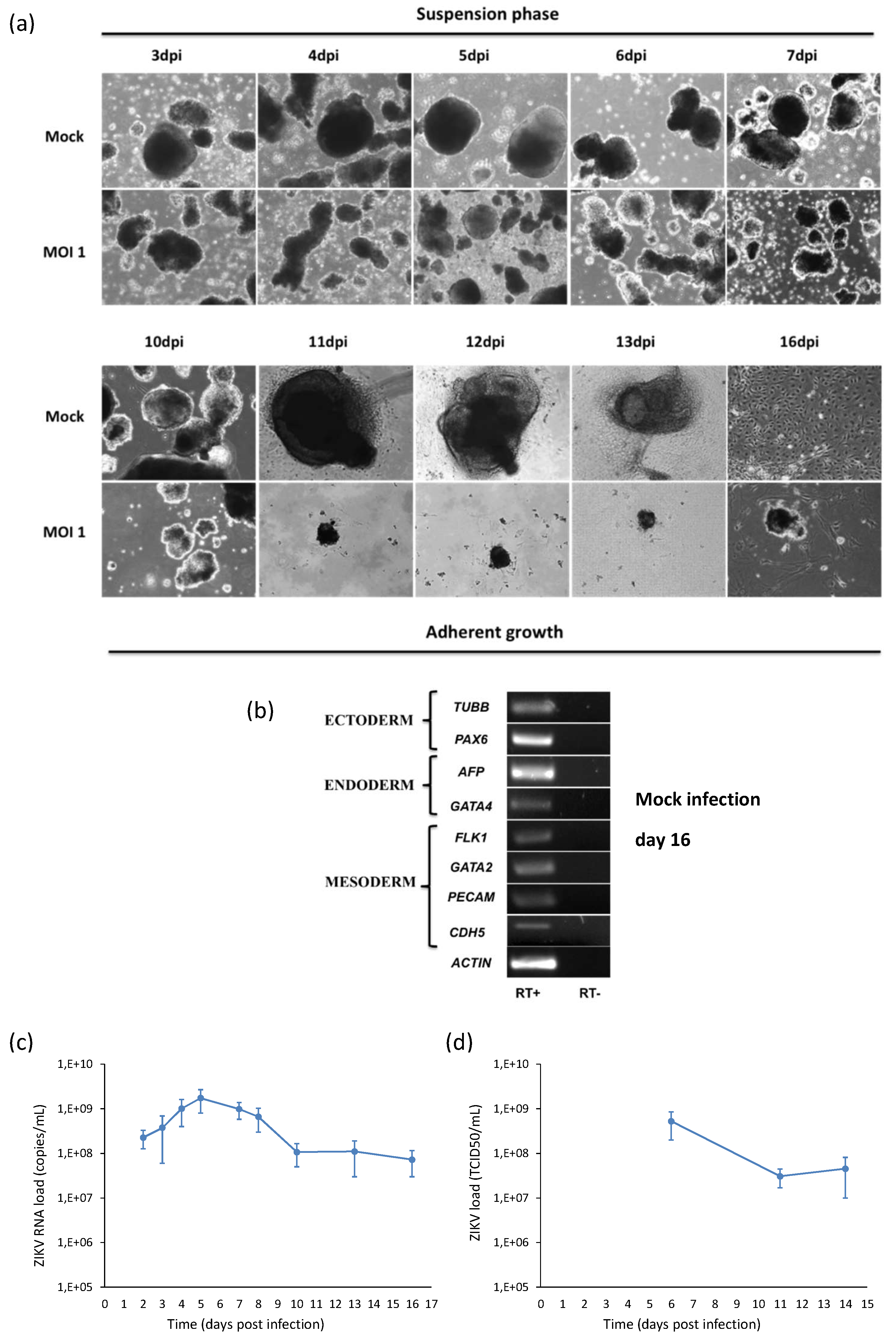
2.5. ZIKV Infection during Embryogenesis
3. Discussion
4. Materials and Methods
4.1. Cells and Culture Protocols
4.2. Embryoid Bodies (EBs) Test
4.3. Viral Strains and Infections
4.4. Infections with ZIKV, WNV, and DENV-2
4.5. Analysis of Virus Replication Kinetics
4.6. Analysis of Gene Expression by RT-PCR and qRT-PCR
4.7. Immunofluorescence Assays
4.8. Apoptosis Assay
4.9. Cell Viability Assay
4.10. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Barzon, L. Ongoing and emerging arbovirus threats in Europe. J. Clin. Virol. 2018, 107, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Barzon, L.; Trevisan, M.; Sinigaglia, A.; Lavezzo, E.; Palù, G. Zika virus: From pathogenesis to disease control. FEMS Microbiol. Lett. 2016, 363, fnw202. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Diamond, M.S. The emergence of Zika virus and its new clinical syndromes. Nature 2018, 560, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Barzon, L.; Percivalle, E.; Pacenti, M.; Rovida, F.; Zavattoni, M.; del Bravo, P.; Cattelan, A.M.; Palù, G.; Baldanti, F. Virus and antibody dynamics in travelers with acute Zika virus infection. Clin. Infect. Dis. 2018, 66, 1173–1180. [Google Scholar] [CrossRef]
- Kurscheidt, F.A.; Mesquita, C.S.S.; Damke, G.M.Z.F.; Damke, E.; Carvalho, A.R.B.A.; Suehiro, T.T.; Teixeira, J.J.V.; da Silva, V.R.S.; Souza, R.P.; Consolaro, M.E.L. Persistence and clinical relevance of Zika virus in the male genital tract. Nat. Rev. Urol. 2019, 16, 211–230. [Google Scholar] [CrossRef]
- Carod-Artal, F.J.; Wichmann, O.; Farrar, J.; Gascón, J. Neurological complications of dengue virus infection. Lancet Neurol. 2013, 12, 906–919. [Google Scholar] [CrossRef]
- Marinho, P.E.S.; Alvarenga, P.P.M.; Crispim, A.P.C.; Candiani, T.M.S.; Alvarenga, A.M.; Bechler, I.M.; Alves, P.A.; Dornas, F.P.; de Oliveira, D.B.; Bentes, A.A.; et al. Wild-type yellow fever virus RNA in cerebrospinal fluid of child. Emerg. Infect. Dis. 2019, 25, 1567–1570. [Google Scholar] [CrossRef]
- Suthar, M.S.; Diamond, M.S.; Gale, M.J. West Nile virus infection and immunity. Nat. Rev. Microbiol. 2013, 11, 115–128. [Google Scholar] [CrossRef]
- Ferreira, R.O.; Garcez, P.P. Dissecting the toxic effects of Zika virus proteins on neural progenitor cells. Neuron 2019, 101, 989–991. [Google Scholar] [CrossRef]
- Nazerai, L.; Pravsgaard Christensen, J.; Randrup Thomsen, A. A ‘furry-tale’ of Zika virus infection: What have we learned from animal models? Viruses 2019, 11, 29. [Google Scholar] [CrossRef]
- Alves Dos Santos, E.; Fink, K. Animal models for dengue and Zika vaccine development. Adv. Exp. Med. Biol. 2018, 1062, 215–239. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, M.; Sinigaglia, A.; Desole, G.; Berto, A.; Pacenti, M.; Palù, G.; Barzon, L. Modeling viral infectious diseases and development of antiviral therapies using human induced pluripotent stem cell-derived systems. Viruses. 2015, 7, 3835–3856. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.L.; Tang, H.; Song, H. Advances in Zika virus research: Stem cell models, challenges, and opportunities. Cell Stem Cell 2016, 19, 690–702. [Google Scholar] [CrossRef]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef]
- Garcez, P.P.; Loiola, E.C.; Madeiro da Costa, R.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika virus impairs growth in human neurospheres and brain organoids. Science. 2016, 352, 816–818. [Google Scholar] [CrossRef]
- Dang, J.; Tiwari, S.K.; Lichinchi, G.; Qin, Y.; Patil, V.S.; Eroshkin, A.M.; Rana, T.M. Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell 2016, 19, 258–265. [Google Scholar] [CrossRef]
- Xu, M.; Lee, E.M.; Wen, Z.; Cheng, Y.; Huang, W.K.; Qian, X.; Tcw, J.; Kouznetsova, J.; Ogden, S.C.; Hammack, C.; et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016, 22, 1101–1107. [Google Scholar] [CrossRef]
- Gabriel, E.; Ramani, A.; Karow, U.; Gottardo, M.; Natarajan, K.; Gooi, L.M.; Goranci-Buzhala, G.; Krut, O.; Peters, F.; Nikolic, M.; et al. Recent Zika virus isolates induce premature differentiation of neural progenitors in human brain organoids. Cell Stem Cell 2017, 20, 397–406.e5. [Google Scholar] [CrossRef]
- Zhou, T.; Tan, L.; Cederquist, G.Y.; Fan, Y.; Hartley, B.J.; Mukherjee, S.; Tomishima, M.; Brennand, K.J.; Zhang, Q.; Schwartz, R.E.; et al. High-content screening in hPSC-neural progenitors identifies drug candidates that inhibit Zika virus infection in fetal-like organoids and adult brain. Cell Stem Cell 2017, 21, 274–283.e5. [Google Scholar] [CrossRef]
- Mesci, P.; Macia, A.; LaRock, C.N.; Tejwani, L.; Fernandes, I.R.; Suarez, N.A.; de A. Zanotto, P.M.; Beltrão-Braga, P.C.B.; Nizet, V.; Muotri, A.R. Modeling neuro-immune interactions during Zika virus infection. Hum. Mol. Genet. 2018, 27, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Muffat, J.; Omer Javed, A.; Keys, H.R.; Lungjangwa, T.; Bosch, I.; Khan, M.; Virgilio, M.C.; Gehrke, L.; Sabatini, D.M.; et al. Genome-wide CRISPR screen for Zika virus resistance in human neural cells. Proc. Natl. Acad. Sci. USA 2019, 116, 9527–9532. [Google Scholar] [CrossRef] [PubMed]
- Muffat, J.; Li, Y.; Omer, A.; Durbin, A.; Bosch, I.; Bakiasi, G.; Richards, E.; Meyer, A.; Gehrke, L.; Jaenisch, R. Human induced pluripotent stem cell-derived glial cells and neural progenitors display divergent responses to Zika and dengue infections. Proc. Natl. Acad. Sci. USA 2018, 115, 7117–7122. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Dao Thi, V.L.; Huang, Y.; Billerbeck, E.; Saha, D.; Hoffmann, H.H.; Wang, Y.; Silva, L.A.V.; Sarbanes, S.; Sun, T.; et al. Intrinsic immunity shapes viral resistance of stem cells. Cell 2018, 172, 423–438.e25. [Google Scholar] [CrossRef]
- Hughes, B.W.; Addanki, K.C.; Sriskanda, A.N.; McLean, E.; Bagasra, O. Infectivity of immature neurons to Zika Virus: A link to congenital Zika syndrome. EBioMedicine 2016, 10, 65–70. [Google Scholar] [CrossRef]
- Shresta, B.; Gottlieb, D.; Diamond, M.S. Infection and injury of neurons by West Nile encephalitis virus. J. Virol. 2003, 73, 13203–13213. [Google Scholar] [CrossRef]
- Lucas, M.; Mashimo, T.; Frenkiel, M.P.; Simon-Chazottes, D.; Montagutelli, X.; Ceccaldi, P.E.; Guénet, J.L.; Desprès, P. Infection of mouse neurons by West Nile is modulated by the interferon-inducible 2′-5′ oligoadenylate synthetase 1b protein. Immun. Cell Biol. 2013, 81, 230–236. [Google Scholar] [CrossRef]
- Cho, H.; Proll, S.C.; Szretter, K.J.; Katze, M.G.; Gale, M.J.; Diamond, M.S. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med. 2013, 19, 458–464. [Google Scholar] [CrossRef]
- Souza, B.S.; Sampaio, G.L.; Pereira, C.S.; Campos, G.S.; Sardi, S.I.; Freitas, L.A.; Figueira, C.P.; Paredes, B.D.; Nonaka, C.K.; Azevedo, C.M.; et al. Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci. Rep. 2016, 6, 39775. [Google Scholar] [CrossRef]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika virus infection in human skin cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef]
- Frumence, E.; Roche, M.; Krejbich-Trotot, P.; El-Kalamouni, C.; Nativel, B.; Rondeau, P.; Missé, D.; Gadea, G.; Viranaicken, W.; Desprès, P. The South Pacific epidemic strain of Zika virus replicates efficiently in human epithelial A549 cells leading to IFN-β production and apoptosis induction. Virology 2016, 493, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Hanners, N.W.; Eitson, J.L.; Usui, N.; Richardson, R.B.; Wexler, E.M.; Konopka, G.; Schoggins, J.W. Western Zika virus in human fetal neural progenitors persists long term with partial cytopathic and limited immunogenic effects. Cell Rep. 2016, 15, 2315–2322. [Google Scholar] [CrossRef] [PubMed]
- Fensterl, V.; Sen, G.C. Interferon-induced Ifit proteins: Their role in viral pathogenesis. J. Virol. 2015, 89, 2462–2468. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Shrestha, B.; Sen, G.C.; Diamond, M.S. A role for Ifit2 in restricting West Nile virus infection in the brain. J. Virol. 2013, 87, 8363–8371. [Google Scholar] [CrossRef]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010, 468, 452–456. [Google Scholar] [CrossRef]
- Stawowczyk, M.; Van Scoy, S.; Kumar, K.P.; Reich, N.C. The interferon Stimulated Gene 54 promotes apoptosis. J. Biol. Chem. 2011, 286, 7257–7266. [Google Scholar] [CrossRef]
- Wang, R.; Wang, J.; Paul, A.M.; Acharya, D.; Bai, F.; Huang, F.; Guo, Y.L. Mouse embryonic stem cells are deficient in type I interferon expression in response to viral infections and double-stranded RNA. J. Biol. Chem. 2013, 288, 15926–15936. [Google Scholar] [CrossRef]
- Chen, L.L.; Yang, L.; Carmichael, G.G. Molecular basis for an attenuated cytoplasmic dsRNA response in human embryonic stem cells. Cell Cycle 2010, 9, 3552–3564. [Google Scholar] [CrossRef]
- Wu, K.Y.; Zuo, G.L.; Li, X.F.; Ye, Q.; Deng, Y.Q.; Huang, X.Y.; Cao, W.C.; Qin, C.F.; Luo, Z.G. Vertical transmission of Zika virus targeting the radial glial cells affects cortex development of offspring mice. Cell Res. 2016, 26, 645–654. [Google Scholar] [CrossRef]
- Murry, C.E.; Keller, G. Differentiation of embryonic stem cells to clinically relevant populations: Lessons from embryonic development. Cell 2008, 132, 661–680. [Google Scholar] [CrossRef]
- Richard, A.S.; Shim, B.S.; Kwon, Y.C.; Zhang, R.; Otsuka, Y.; Schmitt, K.; Berri, F.; Diamond, M.S.; Choe, H. AXL-dependent infection of human fetal endothelial cells distinguishes Zika virus from other pathogenic flaviviruses. Proc. Natl. Acad. Sci. USA 2017, 114, 2024–2029. [Google Scholar] [CrossRef] [PubMed]
- Dejarnac, O.; Hafirassou, M.L.; Chazal, M.; Versapuech, M.; Gaillard, J.; Perera-Lecoin, M.; Umana-Diaz, C.; Bonnet-Madin, L.; Carnec, X.; Tinevez, J.Y.; et al. TIM-1 ubiquitination mediates dengue virus entry. Cell Rep. 2018, 23, 1779–1793. [Google Scholar] [CrossRef] [PubMed]
- Puerta-Guardo, H.; Glasner, D.R.; Espinosa, D.A.; Biering, S.B.; Patana, M.; Ratnasiri, K.; Wang, C.; Beatty, P.R.; Harris, E. Flavivirus NS1 triggers tissue-specific vascular endothelial dysfunction reflecting disease tropism. Cell Rep. 2019, 26, 1598–1613.e8. [Google Scholar] [CrossRef]
- Riedl, W.; Acharya, D.; Lee, J.H.; Liu, G.; Serman, T.; Chiang, C.; Chan, Y.K.; Diamond, M.S.; Gack, M.U. Zika virus NS3 mimics a cellular 14-3-3-binding motif to antagonize RIG-I- and MDA5-mediated innate immunity. Cell Host Microbe 2019, 26, 493–503.e6. [Google Scholar] [CrossRef] [PubMed]
- Esser-Nobis, K.; Aarreberg, L.D.; Roby, J.A.; Fairgrieve, M.R.; Green, R.; Gale, M., Jr. Comparative Analysis of African and Asian lineage-derived Zika virus strains reveals differences in activation of and sensitivity to antiviral innate immunity. J. Virol. 2019, 93, e00640-19. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, M.; Desole, G.; Costanzi, G.; Lavezzo, E.; Palù, G.; Barzon, L. Reprogramming methods do not affect gene expression profile of human induced pluripotent stem cells. Int. J. Mol. Sci. 2017, 18, 206. [Google Scholar] [CrossRef] [PubMed]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desole, G.; Sinigaglia, A.; Riccetti, S.; Masi, G.; Pacenti, M.; Trevisan, M.; Barzon, L. Modelling Neurotropic Flavivirus Infection in Human Induced Pluripotent Stem Cell-Derived Systems. Int. J. Mol. Sci. 2019, 20, 5404. https://doi.org/10.3390/ijms20215404
Desole G, Sinigaglia A, Riccetti S, Masi G, Pacenti M, Trevisan M, Barzon L. Modelling Neurotropic Flavivirus Infection in Human Induced Pluripotent Stem Cell-Derived Systems. International Journal of Molecular Sciences. 2019; 20(21):5404. https://doi.org/10.3390/ijms20215404
Chicago/Turabian StyleDesole, Giovanna, Alessandro Sinigaglia, Silvia Riccetti, Giulia Masi, Monia Pacenti, Marta Trevisan, and Luisa Barzon. 2019. "Modelling Neurotropic Flavivirus Infection in Human Induced Pluripotent Stem Cell-Derived Systems" International Journal of Molecular Sciences 20, no. 21: 5404. https://doi.org/10.3390/ijms20215404
APA StyleDesole, G., Sinigaglia, A., Riccetti, S., Masi, G., Pacenti, M., Trevisan, M., & Barzon, L. (2019). Modelling Neurotropic Flavivirus Infection in Human Induced Pluripotent Stem Cell-Derived Systems. International Journal of Molecular Sciences, 20(21), 5404. https://doi.org/10.3390/ijms20215404