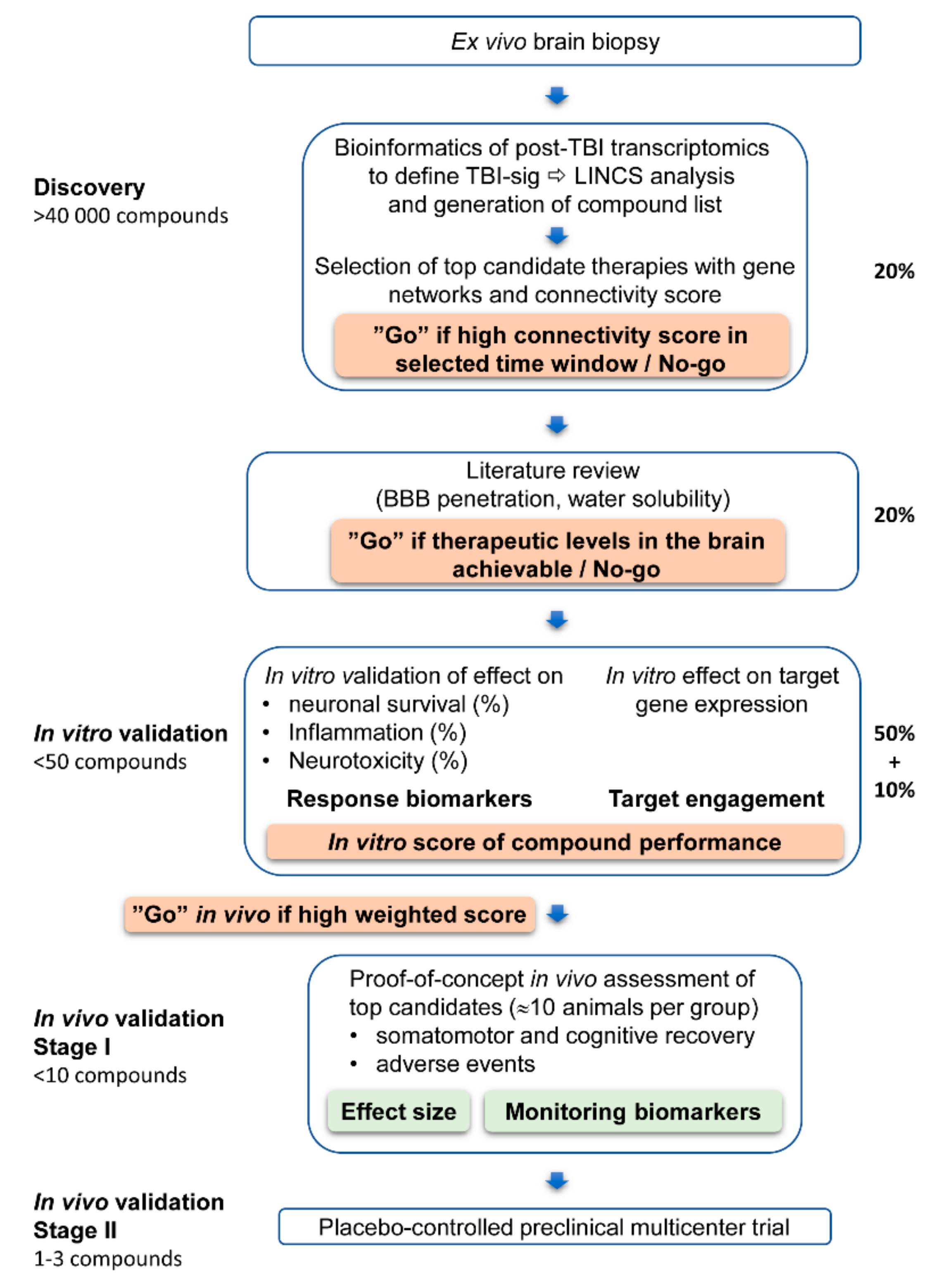
In Vitro and In Vivo Pipeline for Validation of Disease-Modifying Effects of Systems Biology-Derived Network Treatments for Traumatic Brain Injury—Lessons Learned

 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Results
2.1. In Silico Screening of Compounds that Modify TBI-Induced Gene Expression at Both Acute and Chronic Time Points
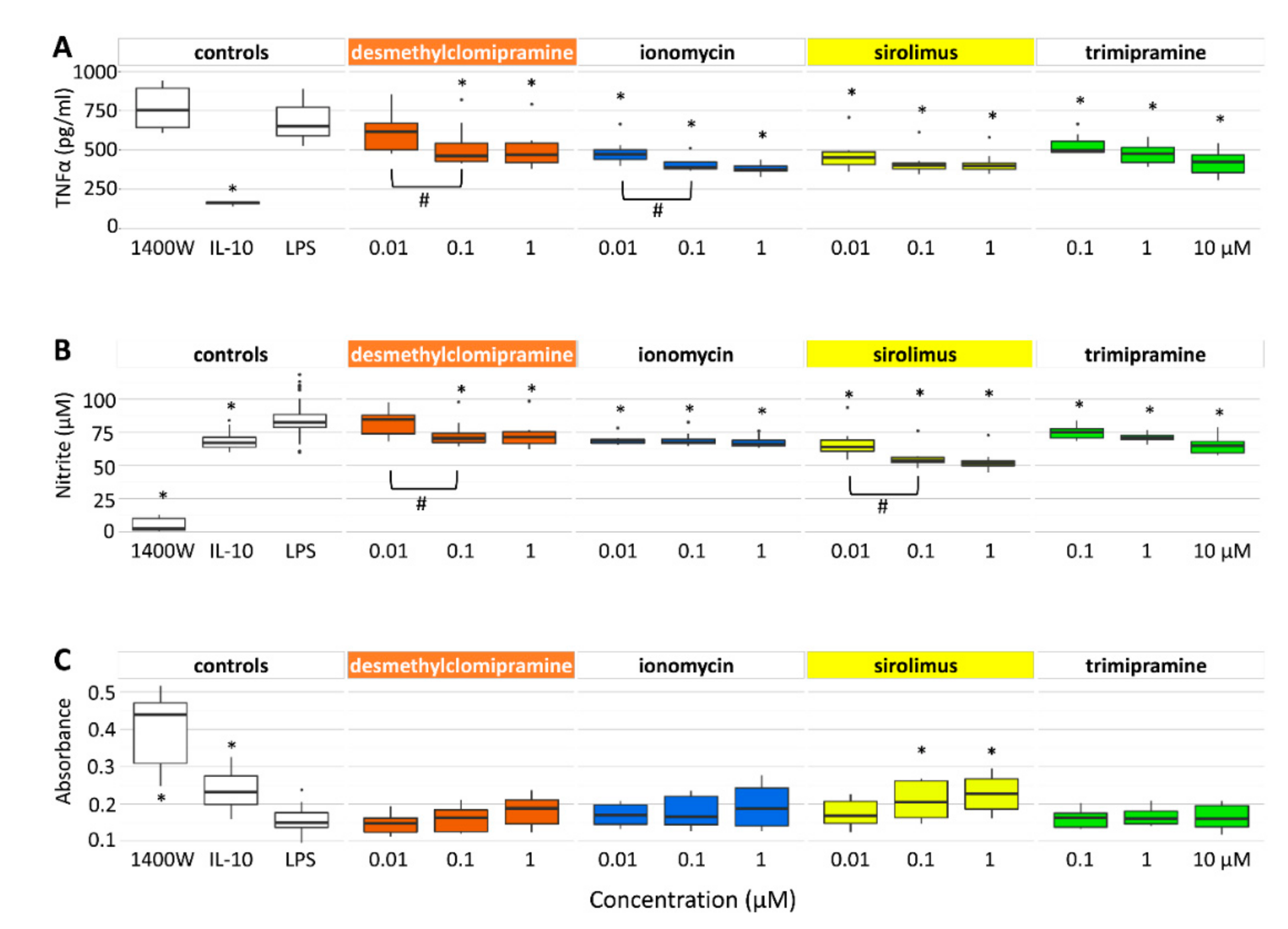
2.2. Desmethylclomipramine, Ionomycin, Trimipramine and Sirolimus Inhibit TNFα and Nitrite Production in Cortical Neuron-BV2 Microglia Co-Culture
2.2.1. TNFα Secretion
2.2.2. Nitrite Levels
2.2.3. Neuronal Viability
2.3. Target Gene Expression After TBI in Vivo and Effect of Candidate Compounds on Target Gene Expression In Vitro
2.3.1. Target Gene Expression after TBI In Vivo
2.3.2. Inflammatory Condition
2.3.3. Non-inflammatory Condition
2.4. Scoring of Candidate Compounds to Proceed to In Vivo Proof-of-Concept Validation
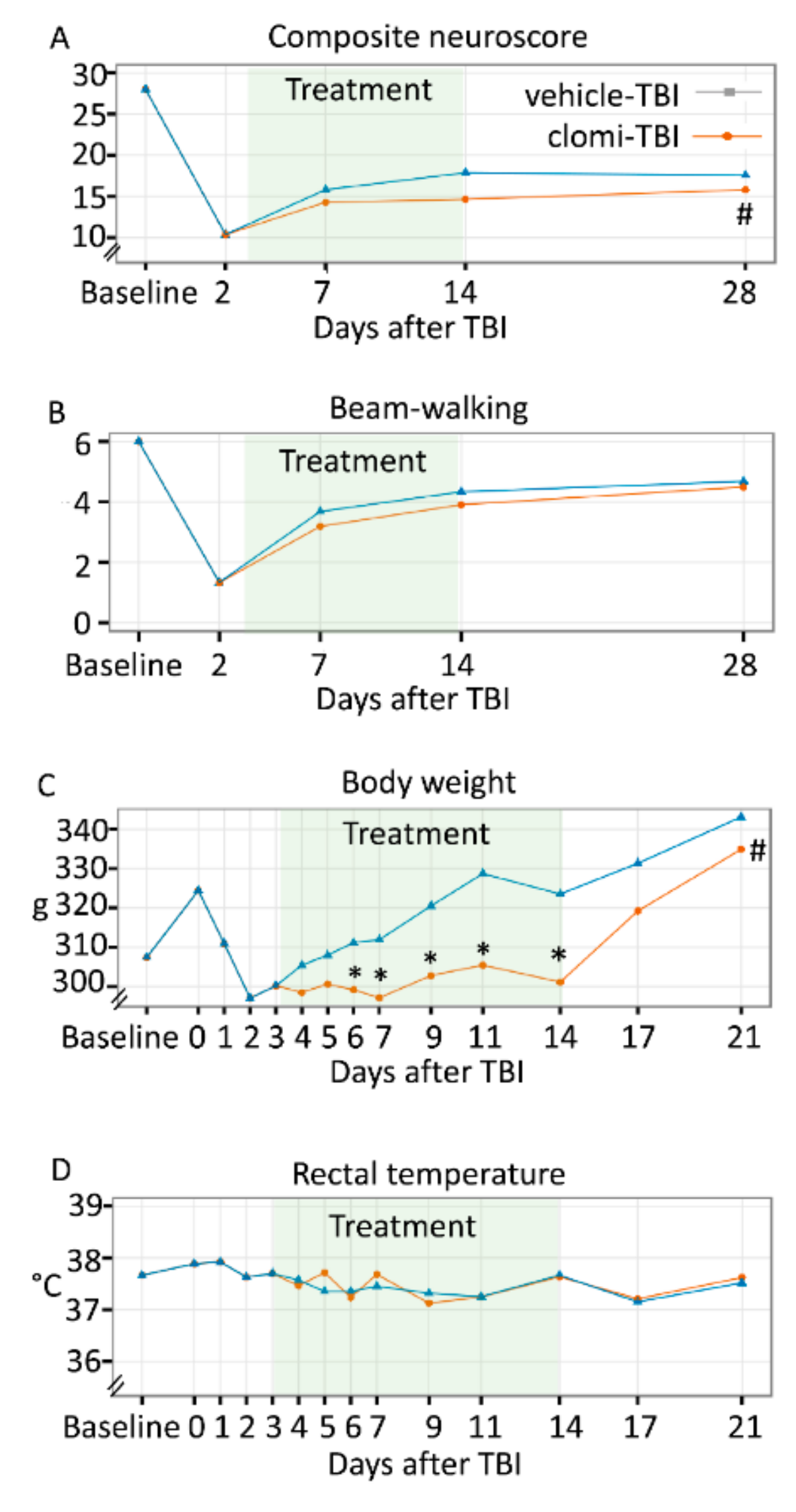
2.5. Clomipramine Treatment in the Acute Post-TBI Phase Had a Negative Effect on Somatomotor Recovery
2.5.1. Acute Mortality, Impact Pressure, Post-impact Apnoea and Acute Post-impact Seizure-like Behaviour
2.5.2. Pathological Outcome
2.5.3. Composite Neuroscore
2.5.4. Beam-walking
2.6. Clomipramine Treatment Did Not Affect Spatial Learning or Spatial Memory
Morris Water-maze
2.7. Clomipramine Treatment Reduced Weight Gain
2.7.1. Body Weight
2.7.2. Rectal Temperature
2.7.3. General Health
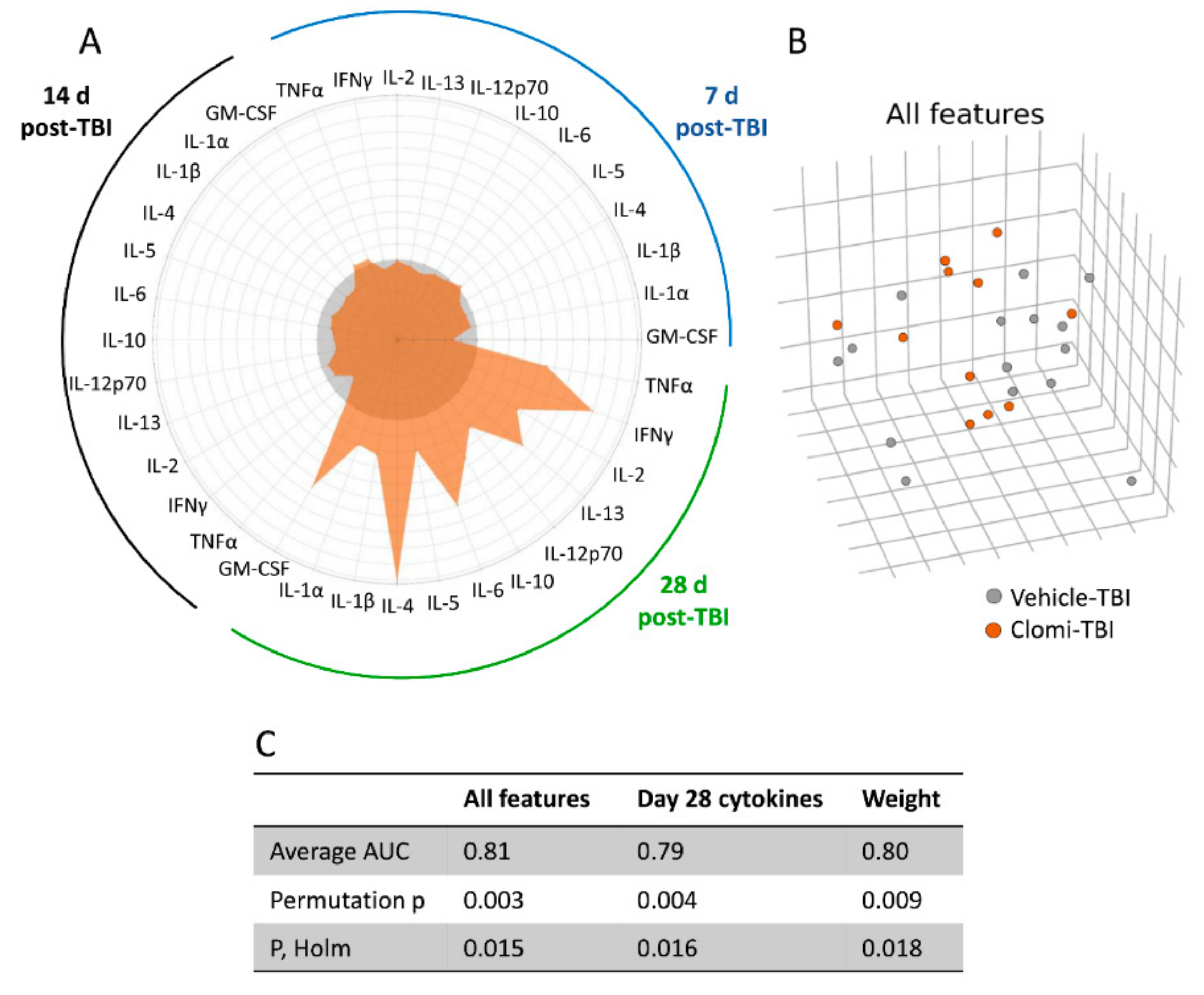
2.8. Clomipramine Treatment Delayed the Reduction of the TBI-Induced Plasma Cytokine Response
2.8.1. Plasma Cytokine Response to Injury and Clomipramine
2.8.2. Machine Learning Separated Clomipramine- and Saline-treated Animals at 28 days post-TBI
3. Discussion
3.1. In Silico Analysis Predicted a Wide Therapeutic Time-Window for Desmethylclomipramine and Ionomycin
3.2. In Vitro Analysis Revealed that Desmethylclomipramine, Ionomycin, Trimipramine and Sirolimus Reduce Neuroinflammation and Nitric Oxide—Mediated Neurotoxicity
3.3. Target Expression and Target Engagement
3.4. Go/No-Go Analysis for Selection of a Compound for In Vivo Testing
3.5. In Vivo Assessment Failed to Demonstrate Favorable Disease-Modifying Effects of Clomipramine on TBI Functional Outcome and Plasma Cytokines
4. Materials and Methods
4.1. In Silico Analysis of Transcriptomic Modifying Compounds
4.1.1. The Effect of Test Compounds on Acute and Chronic TBI Transcriptomic
4.1.2. In Silico Identification of Monitoring Biomarkers for in Vitro and in Vivo Analyses.
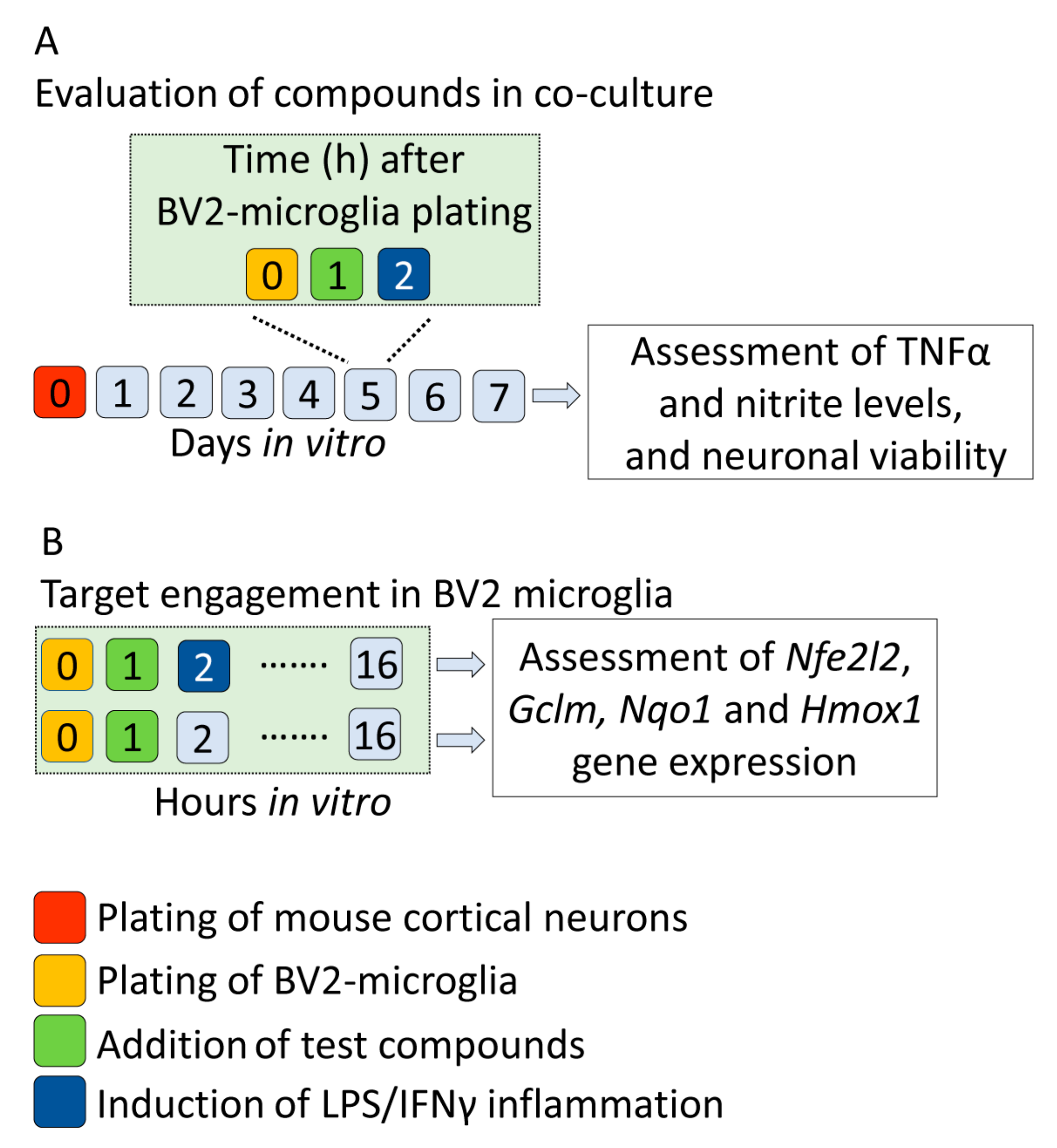
4.2. In Vitro Validation of in Silico Discovered Compounds
4.2.1. Preparation of Neuron-BV2 Co-Cultures
4.2.2. Treatment of Co-Cultures with Test Compounds
4.2.3. Effect of Test Compounds on In Vitro Monitoring Biomarkers with Focus on Inflammation, Nitric Oxide—Mediated Neurotoxicity and Neuronal Viability
4.2.4. Nfe2l2 Upregulation and Activation of Nrf2 Transcription Factor as Indicators of Target Engagement
4.2.5. Scoring of Candidate Compounds for In Vivo Validation
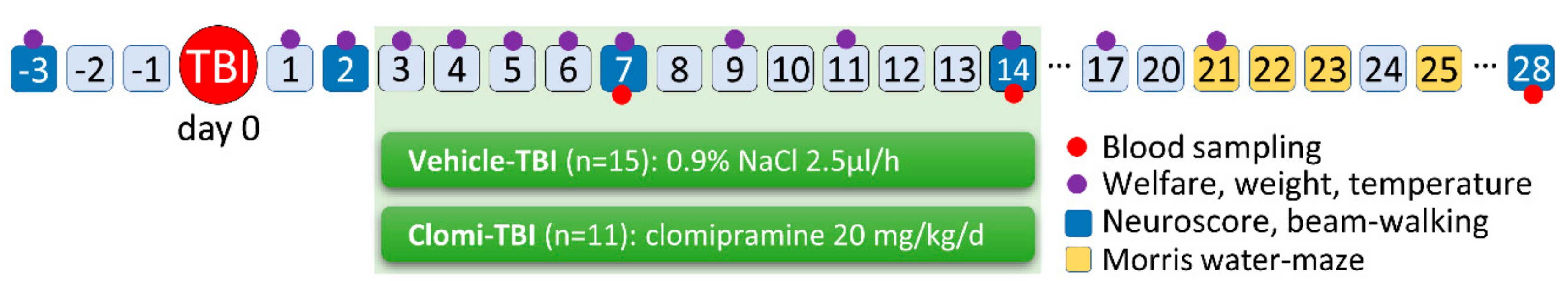
4.3. In Vivo Validation of Treatment on Post-TBI Functional Recovery
4.3.1. Animal Model and In Vivo Treatment Trial
4.3.2. Behavioural Analysis
4.3.3. Monitoring of Adverse Events
4.3.4. Histology
4.3.5. Analysis of Plasma-Monitored Biomarkers
4.3.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | area under the curve |
| CMap | Connectivity Map |
| DMEM | Dulbecco’s modified Eagles medium |
| DMSO | dimethyl sulfoxide |
| ELISA | enzyme-linked immunosorbent assay |
| FBS | foetal bovine serum |
| FDR | false discovery rate |
| FPI | fluid-percussion injury |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| IFNy | interferon gamma |
| IL | interleukin |
| LINCS | Library of Integrated Network-Based Cellular Signatures |
| LPS | lipopolysaccharide |
| MAP2 | microtubule-associated protein 2 |
| ML | machine learning |
| NO | nitric oxide |
| NO2- | nitrite |
| PBS | phosphate-buffered saline |
| STAR | Spliced Transcripts Alignment to Reference |
| TBI | traumatic brain injury |
| TNFα | tumour necrosis factor alpha |
| 1400W | N-(3-(aminomethyl)benzyl)acetamidine |
References
- Corrigan, J.D.; Selassie, A.W.; Orman, J.A.L. The epidemiology of traumatic brain injury. J. Head Trauma Rehabil. 2010, 25, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet. Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef]
- Saatman, K.E.; Duhaime, A.-C.; Bullock, R.; Maas, A.I.R.; Valadka, A.; Manley, G.T. Classification of traumatic brain injury for targeted therapies. J. Neurotrauma 2008, 25, 719–738. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.; Engelhard, K. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Mejia, R.O.S.; Ona, V.O.; Li, M.; Friedlander, R.M. Minocycline Reduces Traumatic Brain Injury-mediated Caspase-1 Activation, Tissue Damage, and Neurological Dysfunction. Neurosurgery 2001, 48, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Loane, D.J. Neuroinflammation after traumatic brain injury: Opportunities for therapeutic intervention. Brain. Behav. Immun. 2012, 26, 1191–1201. [Google Scholar] [CrossRef]
- Marklund, N.; Clausen, F.; Lewén, A.; Hovda, D.A.; Olsson, Y.; Hillered, L. alpha-Phenyl-tert-N-butyl nitrone (PBN) improves functional and morphological outcome after cortical contusion injury in the rat. Acta Neurochir. 2001, 143, 73–81. [Google Scholar] [CrossRef]
- Hall, E.D.; Vaishnav, R.A.; Mustafa, A.G. Antioxidant therapies for traumatic brain injury. Neurotherapeutics 2010, 7, 51–61. [Google Scholar] [CrossRef]
- Erlich, S.; Alexandrovich, A.; Shohami, E.; Pinkas-Kramarski, R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol. Dis. 2007, 26, 86–93. [Google Scholar] [CrossRef]
- Siavelis, J.C.; Bourdakou, M.M.; Athanasiadis, E.I.; Spyrou, G.M.; Nikita, K.S. Bioinformatics methods in drug repurposing for Alzheimer’s disease. Brief. Bioinform. 2016, 17, 322–335. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]
- Gao, L.; Zhao, G.; Fang, J.-S.; Yuan, T.-Y.; Liu, A.-L.; Du, G.-H. Discovery of the neuroprotective effects of alvespimycin by computational prioritization of potential anti-parkinson agents. FEBS J. 2014, 281, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Smalley, J.L.; Breda, C.; Mason, R.P.; Kooner, G.; Luthi-Carter, R.; Gant, T.W.; Giorgini, F. Connectivity mapping uncovers small molecules that modulate neurodegeneration in Huntington’s disease models. J. Mol. Med. 2016, 94, 235–245. [Google Scholar] [CrossRef]
- So, H.-C.; Chau, C.K.-L.; Chiu, W.-T.; Ho, K.-S.; Lo, C.-P.; Yim, S.H.-Y.; Sham, P.-C. Analysis of genome-wide association data highlights candidates for drug repositioning in psychiatry. Nat. Neurosci. 2017, 20, 1342–1349. [Google Scholar] [CrossRef]
- Delahaye-Duriez, A.; Srivastava, P.; Shkura, K.; Langley, S.R.; Laaniste, L.; Moreno-Moral, A.; Danis, B.; Mazzuferi, M.; Foerch, P.; Gazina, E.V.; et al. Rare and common epilepsies converge on a shared gene regulatory network providing opportunities for novel antiepileptic drug discovery. Genome Biol. 2016, 17, 245. [Google Scholar] [CrossRef] [PubMed]
- Mirza, N.; Sills, G.J.; Pirmohamed, M.; Marson, A.G. Identifying new antiepileptic drugs through genomics-based drug repurposing. Hum. Mol. Genet. 2017, 26, 527–537. [Google Scholar] [CrossRef]
- Jacunski, A.; Tatonetti, N.P. Connecting the Dots: Applications of Network Medicine in Pharmacology and Disease. Clin. Pharmacol. Ther. 2013, 94, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef]
- Kolodkin, A.; Boogerd, F.C.; Plant, N.; Bruggeman, F.J.; Goncharuk, V.; Lunshof, J.; Moreno-Sanchez, R.; Yilmaz, N.; Bakker, B.M.; Snoep, J.L.; et al. Emergence of the silicon human and network targeting drugs. Eur. J. Pharm. Sci. 2012, 46, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Silverman, E.K.; Loscalzo, J. Developing New Drug Treatments in the Era of Network Medicine. Clin. Pharmacol. Ther. 2013, 93, 26–28. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef] [PubMed]
- Lipponen, A.; El-Osta, A.; Kaspi, A.; Ziemann, M.; Khurana, I.; Harikrishnan, K.N.; Navarro-Ferrandis, V.; Puhakka, N.; Paananen, J.; Pitkänen, A. Transcription factors Tp73, Cebpd, Pax6, and Spi1 rather than DNA methylation regulate chronic transcriptomics changes after experimental traumatic brain injury. Acta Neuropathol. Commun. 2018, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Lipponen, A.; Paananen, J.; Puhakka, N.; Pitkänen, A. Analysis of Post-Traumatic Brain Injury Gene Expression Signature Reveals Tubulins, Nfe2l2, Nfkb, Cd44, and S100a4 as Treatment Targets. Sci. Rep. 2016, 6, 31570. [Google Scholar] [CrossRef] [PubMed]
- Lapinlampi, N.; Melin, E.; Aronica, E.; Bankstahl, J.P.; Becker, A.; Bernard, C.; Gorter, J.A.; Gröhn, O.; Lipsanen, A.; Lukasiuk, K.; et al. Common data elements and data management: Remedy to cure underpowered preclinical studies. Epilepsy Res. 2017, 129, 87–90. [Google Scholar] [CrossRef]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Gallorini, M.; Petzel, C.; Bolay, C.; Hiller, K.-A.; Cataldi, A.; Buchalla, W.; Krifka, S.; Schweikl, H. Activation of the Nrf2-regulated antioxidant cell response inhibits HEMA-induced oxidative stress and supports cell viability. Biomaterials 2015, 56, 114–128. [Google Scholar] [CrossRef]
- Satoh, T.; Okamoto, S.; Cui, J.; Watanabe, Y.; Furuta, K.; Suzuki, M.; Tohyama, K.; Lipton, S.A. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophilic [correction of electrophillic] phase II inducers. Proc. Natl. Acad. Sci. USA 2006, 103, 768–773. [Google Scholar] [CrossRef]
- Liu, C.; Hermann, T.E. Characterization of ionomycin as a calcium ionophore. J. Biol. Chem. 1978, 253, 5892–5894. [Google Scholar]
- Salzmann, M.M. A controlled trial with trimipramine, a new anti-depressant drug. Br. J. Psychiatry 1965, 111, 1105–1106. [Google Scholar] [CrossRef]
- Pozniak, C.D.; Radinovic, S.; Yang, A.; McKeon, F.; Kaplan, D.R.; Miller, F.D. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science 2000, 289, 304–306. [Google Scholar] [CrossRef]
- Lee, A.F.; Ho, D.K.; Zanassi, P.; Walsh, G.S.; Kaplan, D.R.; Miller, F.D. Evidence That Np73 Promotes Neuronal Survival by p53-Dependent and p53-Independent Mechanisms. J. Neurosci. 2004, 24, 9174–9184. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Zeng, L.; Brody, D.L.; Wong, M. Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PLoS ONE 2013, 8, e64078. [Google Scholar] [CrossRef] [PubMed]
- Pauletti, A.; Terrone, G.; Shekh-Ahmad, T.; Salamone, A.; Ravizza, T.; Rizzi, M.; Pastore, A.; Pascente, R.; Liang, L.-P.; Villa, B.R.; et al. Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain 2017, 140, 1885–1899. [Google Scholar] [CrossRef] [PubMed]
- Shekh-Ahmad, T.; Eckel, R.; Dayalan Naidu, S.; Higgins, M.; Yamamoto, M.; Dinkova-Kostova, A.T.; Kovac, S.; Abramov, A.Y.; Walker, M.C. KEAP1 inhibition is neuroprotective and suppresses the development of epilepsy. Brain 2018, 141, 1390–1403. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.-C. The Nrf2-ARE Pathway. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef]
- Balant-Gorgia, A.E.; Gex-Fabry, M.; Balant, L.P. Clinical Pharmacokinetics of Clomipramine. Clin. Pharmacokinet. 1991, 20, 447–462. [Google Scholar] [CrossRef]
- Weigmann, H.; Härtter, S.; Bagli, M.; Hiemke, C. Steady state concentrations of clomipramine and its major metabolite desmethylclomipramine in rat brain and serum after oral administration of clomipramine. Eur. Neuropsychopharmacol. 2000, 10, 401–405. [Google Scholar] [CrossRef]
- Johnstone, A.L.; Reierson, G.W.; Smith, R.P.; Goldberg, J.L.; Lemmon, V.P.; Bixby, J.L. A chemical genetic approach identifies piperazine antipsychotics as promoters of CNS neurite growth on inhibitory substrates. Mol. Cell. Neurosci. 2012, 50, 125–135. [Google Scholar] [CrossRef]
- Usher, L.C.; Johnstone, A.; Erturk, A.; Hu, Y.; Strikis, D.; Wanner, I.B.; Moorman, S.; Lee, J.W.; Min, J.; Ha, H.H.; et al. A Chemical Screen Identifies Novel Compounds That Overcome Glial-Mediated Inhibition of Neuronal Regeneration. J. Neurosci. 2010, 30, 4693–4706. [Google Scholar] [CrossRef]
- Azim, K.; Angonin, D.; Marcy, G.; Pieropan, F.; Rivera, A.; Donega, V.; Cantù, C.; Williams, G.; Berninger, B.; Butt, A.M.; et al. Pharmacogenomic identification of small molecules for lineage specific manipulation of subventricular zone germinal activity. PLoS Biol. 2017, 15, e2000698. [Google Scholar] [CrossRef]
- Gresa-Arribas, N.; Viéitez, C.; Dentesano, G.; Serratosa, J.; Saura, J.; Solà, C.; Glass, C.; Saijo, K.; Winner, B.; Marchetto, M.; et al. Modelling Neuroinflammation In Vitro: A Tool to Test the Potential Neuroprotective Effect of Anti-Inflammatory Agents. PLoS ONE 2012, 7, e45227. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Zheng, L.T.; Ock, J.; Lee, M.G.; Kim, S.-H.; Lee, H.-W.; Lee, W.-H.; Park, H.-C.; Suk, K. Inhibition of glial inflammatory activation and neurotoxicity by tricyclic antidepressants. Neuropharmacology 2008, 55, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Faissner, S.; Mishra, M.; Kaushik, D.K.; Wang, J.; Fan, Y.; Silva, C.; Rauw, G.; Metz, L.; Koch, M.; Yong, V.W. Systematic screening of generic drugs for progressive multiple sclerosis identifies clomipramine as a promising therapeutic. Nat. Commun. 2017, 8, 1990. [Google Scholar] [CrossRef]
- Yau, J.L.W.; Noble, J.; Thomas, S.; Kerwin, R.; Morgan, P.E.; Lightman, S.; Seckl, J.R.; Pariante, C.M. The Antidepressant Desipramine Requires the ABCB1 (Mdr1)-Type p-Glycoprotein to Upregulate the Glucocorticoid Receptor in Mice. Neuropsychopharmacology 2007, 32, 2520–2529. [Google Scholar] [CrossRef] [PubMed]
- Aitchison, K.; Datla, K.; Rooprai, H.; Fernando, J.; Dexter, D. Regional distribution of clomipramine and desmethylclomipramine in rat brain and peripheral organs on chronic clomipramine administration. J. Psychopharmacol. 2010, 24, 1261–1268. [Google Scholar] [CrossRef]
- Gex-Fabry, M.; Haffen, E.; Paintaud, G.; Bizouard, P.; Sechter, D.; Bechtel, P.R.; Balant, L.P. Population pharmacokinetics of clomipramine, desmethylclomipramine, and hydroxylated metabolites in patients with depression receiving chronic treatment: Model evaluation. Ther. Drug Monit. 2000, 22, 701–711. [Google Scholar] [CrossRef]
- Stern, R.S.; Marks, I.M.; Mawson, D.; Luscombe, D.K. Clomipramine and exposure for compulsive rituals: II. Plasma levels, side effects and outcome. Br. J. Psychiatry 1980, 136, 161–166. [Google Scholar] [CrossRef]
- Hutter-Paier, B.; Grygar, E.; Loibner, M.; Skofitsch, G.; Windisch, M. Effects of NaCN and ionomycin on neuronal viability and on the abundance of microtubule-associated proteins MAP1, MAP2, and tau in isolated chick cortical neurons. Cell Tissue Res. 2000, 302, 39–47. [Google Scholar] [CrossRef]
- Regenthal, R.; Krueger, M.; Koeppel, C.; Preiss, R. Drug levels: Therapeutic and toxic serum/plasma concentrations of common drugs. J. Clin. Monit. Comput. 1999, 15, 529–544. [Google Scholar] [CrossRef]
- Dello Russo, C.; Lisi, L.; Tringali, G.; Navarra, P. Involvement of mTOR kinase in cytokine-dependent microglial activation and cell proliferation. Biochem. Pharmacol. 2009, 78, 1242–1251. [Google Scholar] [CrossRef]
- Stenton, S.B.; Partovi, N.; Ensom, M.H.H. Sirolimus: The evidence for clinical pharmacokinetic monitoring. Clin. Pharmacokinet. 2005, 44, 769–786. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef]
- Mazzuferi, M.; Kumar, G.; van Eyll, J.; Danis, B.; Foerch, P.; Kaminski, R.M. Nrf2 defense pathway: Experimental evidence for its protective role in epilepsy. Ann. Neurol. 2013, 74, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Wang, H.; Yan, W.; Zhu, L.; Hu, Z.; Ding, Y.; Tang, K. Role of Nrf2 in protection against traumatic brain injury in mice. J. Neurotrauma 2009, 26, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Moore, A.N.; Redell, J.B.; Dash, P.K. Enhancing Expression of Nrf2-Driven Genes Protects the Blood Brain Barrier after Brain Injury. J. Neurosci. 2007, 27, 10240–10248. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, G.; Zhang, J.; Strong, R.; Dash, P.K.; Kan, Y.W.; Grotta, J.C.; Aronowski, J. Transcription Factor Nrf2 Protects the Brain From Damage Produced by Intracerebral Hemorrhage. Stroke 2007, 38, 3280–3286. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, X.; Fan, H.; Liu, Y. Curcumin upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. 2009, 1282, 133–141. [Google Scholar] [CrossRef]
- Shih, A.Y. A Small-Molecule-Inducible Nrf2-Mediated Antioxidant Response Provides Effective Prophylaxis against Cerebral Ischemia In Vivo. J. Neurosci. 2005, 25, 10321–10335. [Google Scholar] [CrossRef]
- Albrecht, J.S.; Kiptanui, Z.; Tsang, Y.; Khokhar, B.; Smith, G.S.; Zuckerman, I.H.; Simoni-Wastila, L. Patterns of Depression Treatment in Medicare Beneficiaries with Depression after Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1223–1229. [Google Scholar] [CrossRef]
- Jorge, R.E.; Robinson, R.G.; Moser, D.; Tateno, A.; Crespo-Facorro, B.; Arndt, S. Major Depression Following Traumatic Brain Injury. Arch. Gen. Psychiatry 2004, 61, 42. [Google Scholar] [CrossRef]
- Cookson, J. Side-effects of Antidepressants. Br. J. Psychiatry 1993, 163, 20–24. [Google Scholar] [CrossRef]
- Calegari, L.; Gorenstein, C.; Gentil, V.; Planeta, C.S.; Nunes-De-Souza, R.L. Effect of chronic treatment with clomipramine on food intake, macronutrient selection and body weight gain in rats. Biol. Pharm. Bull. 2007, 30, 1541–1546. [Google Scholar] [CrossRef] [PubMed][Green Version]
- García-Marquez, C.; Armario, A. Interaction between chronic stress and clomipramine treatment in rats. Effects on exploratory activity, behavioral despair, and pituitary-adrenal function. Psychopharmacology 1987, 93, 77–81. [Google Scholar] [CrossRef]
- Yang, A.; Daya, T.; Carlton, K.; Yan, J.H.; Schmid, S. Differential effect of clomipramine on habituation and prepulse inhibition in dominant versus subordinate rats. Eur. Neuropsychopharmacol. 2016, 26, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Andrade, P.; Banuelos-Cabrera, I.; Lapinlampi, N.; Paananen, T.; Ciszek, R.; Ndode-Ekane, X.E.; Pitkänen, A. Acute Non-Convulsive Status Epilepticus after Experimental Traumatic Brain Injury in Rats. J. Neurotrauma 2019, 36, 1–18. [Google Scholar] [CrossRef]
- Wroblewski, B.A.; McColgan, K.; Smith, K.; Whyte, J.; Singer, W.D. The incidence of seizures during tricyclic antidepressant drug treatment in a brain-injured population. J. Clin. Psychopharmacol. 1990, 10, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Koella, W.P.; Glatt, A.; Klebs, K.; Dürst, T. Epileptic phenomena induced in the cat by the antidepressants maprotiline, imipramine, clomipramine, and amitriptyline. Biol. Psychiatry 1979, 14, 485–497. [Google Scholar]
- Cardamone, L.; Salzberg, M.R.; O’Brien, T.J.; Jones, N.C. Antidepressant therapy in epilepsy: Can treating the comorbidities affect the underlying disorder? Br. J. Pharmacol. 2013, 168, 1531–1554. [Google Scholar] [CrossRef]
- Woodcock, T.; Morganti-Kossmann, M.C. The Role of Markers of Inflammation in Traumatic Brain Injury. Front. Neurol. 2013, 4, 18. [Google Scholar] [CrossRef]
- Hans, V.H.J.; Kossmann, T.; Lenzlinger, P.M.; Probstmeier, R.; Imhof, H.-G.; Trentz, O.; Morganti-Kossmann, M.C. Experimental Axonal Injury Triggers Interleukin-6 mRNA, Protein Synthesis and Release into Cerebrospinal Fluid. J. Cereb. Blood Flow Metab. 1999, 19, 184–194. [Google Scholar] [CrossRef]
- Kamm, K.; VanderKolk, W.; Lawrence, C.; Jonker, M.; Davis, A.T. The Effect of Traumatic Brain Injury Upon the Concentration and Expression of Interleukin-1?? and Interleukin-10 in the Rat. J. Trauma Inj. Infect. Crit. Care 2006, 60, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.; Robertson, C.; Grossman, R.; Narayan, R. Elevation of tumor necrosis factor in head injury. J. Neuroimmunol. 1990, 30, 213–217. [Google Scholar] [CrossRef]
- Ross, S.A.; Halliday, M.I.; Campbell, G.C.; Byrnes, D.P.; Rowlands, B.J. The presence of tumour necrosis factor in CSF and plasma after severe head injury. Br. J. Neurosurg. 1994, 8, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Woiciechowsky, C.; Schöning, B.; Cobanov, J.; Lanksch, W.R.; Volk, H.-D.; Döcke, W.-D. Early IL-6 Plasma Concentrations Correlate with Severity of Brain Injury and Pneumonia in Brain-Injured Patients. J. Trauma Acute Care Surg. 2002, 52, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.G.; Boles, J.A.; Wagner, A.K. Chronic inflammation after severe traumatic brain injury: Characterization and associations with outcome at 6 and 12 months postinjury. J. Head Trauma Rehabil. 2015, 30, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Buttini, M.; Mir, A.; Appel, K.; Wiederhold, K.H.; Limonta, S.; Gebicke-Haerter, P.J.; Boddeke, H.W.G.M. Lipopolysaccharide induces expression of tumour necrosis factor alpha in rat brain: Inhibition by methylprednisolone and by rolipram. Br. J. Pharmacol. 1997, 122, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Fan, G.; Chen, S.; Wu, Y.; Xu, X.M.; Hsu, C.Y. Methylprednisolone inhibition of TNF-α expression and NF-kB activation after spinal cord injury in rats. Mol. Brain Res. 1998, 59, 135–142. [Google Scholar] [CrossRef]
- Bye, N.; Habgood, M.D.; Callaway, J.K.; Malakooti, N.; Potter, A.; Kossmann, T.; Morganti-Kossmann, M.C. Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp. Neurol. 2007, 204, 220–233. [Google Scholar] [CrossRef]
- Fu, E.S.; Saporta, S. Methylprednisolone inhibits production of interleukin-1beta and interleukin-6 in the spinal cord following compression injury in rats. J. Neurosurg. Anesthesiol. 2005, 17, 82–85. [Google Scholar] [CrossRef]
- iLINCS (Integrative LINCS) Genomics Data Portal. Available online: www.ilincs.org (accessed on 9 April 2018).
- Rau, T.F.; Kothiwal, A.; Rova, A.; Rhoderick, J.F.; Poulsen, D.J. Phenoxybenzamine is neuroprotective in a rat model of severe traumatic brain injury. Int. J. Mol. Sci. 2014, 15, 1402–1417. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Natunen, T.; Takalo, M.; Kemppainen, S.; Leskelä, S.; Marttinen, M.; Kurkinen, K.M.A.; Pursiheimo, J.-P.; Sarajärvi, T.; Viswanathan, J.; Gabbouj, S.; et al. Relationship between ubiquilin-1 and BACE1 in human Alzheimer’s disease and APdE9 transgenic mouse brain and cell-based models. Neurobiol. Dis. 2016, 85, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Martiskainen, H.; Paldanius, K.M.A.; Natunen, T.; Takalo, M.; Marttinen, M.; Leskelä, S.; Huber, N.; Mäkinen, P.; Bertling, E.; Dhungana, H.; et al. DHCR24 exerts neuroprotection upon inflammation-induced neuronal death. J. Neuroinflamm. 2017, 14, 215. [Google Scholar] [CrossRef] [PubMed]
- Balk, S.D.; Morisi, A.; Gunther, H.S. Phorbol 12-myristate 13-acetate, ionomycin or ouabain, and raised extracellular magnesium induce proliferation of chicken heart mesenchymal cells. Proc. Natl. Acad. Sci. USA 1984, 81, 6418–6421. [Google Scholar] [CrossRef]
- Musa, M.N. Nonlinear kinetics of trimipramine in depressed patients. J. Clin. Pharmacol. 1989, 29, 746–747. [Google Scholar] [CrossRef]
- Kobayashi, M.; Yamamoto, M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid. Redox Signal. 2005, 7, 385–394. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar]
- McIntosh, T.K.K.; Vink, R.; Noble, L.; Yamakami, I.; Fernyak, S.; Soares, H.; Faden, A.L.L. Traumatic brain injury in the rat: Characterization of a lateral fluid-percussion model. Neuroscience 1989, 28, 233–244. [Google Scholar] [CrossRef]
- Kharatishvili, I.; Nissinen, J.P.; Intosh, T.K.M.C.; McIntosh, T.K.; Pitkänen, A. A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience 2006, 140, 685–697. [Google Scholar] [CrossRef]
- Pitkänen, A.; McIntosh, T.K. Animal models of post-traumatic epilepsy. J. Neurotrauma 2006, 23, 241–261. [Google Scholar] [CrossRef] [PubMed]
- Kazuyuki, F.; Akimaro, K.; Shigeru, S.; Kinya, N. Changes of serotonin and catecholamines are related to pharmacokinetic alterations of cloimpramine in rat brain. Eur. J. Pharmacol. 1991, 204, 227–233. [Google Scholar] [CrossRef]
- Friedman, E.; Cooper, T.B. Pharmacokinetics of chlorimipramine and its demethylated metabolite in blood and brain regions of rats treated acutely and chronically with chlorimipramine. J. Pharmacol. Exp. Ther. 1983, 225, 387–390. [Google Scholar] [PubMed]
- Okiyama, K.; Smith, D.H.; Thomas, M.J.; McIntosh, T.K. Evaluation of a novel calcium channel blocker, (S)-emopamil, on regional cerebral edema and neurobehavioral function after experimental brain injury. J. Neurosurg. 1992, 77, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Nissinen, J.; Andrade, P.; Natunen, T.; Hiltunen, M.; Malm, T.; Kanninen, K.; Soares, J.I.; Shatillo, O.; Sallinen, J.; Ndode-Ekane, X.E.; et al. Disease-modifying effect of atipamezole in a model of post-traumatic epilepsy. Epilepsy Res. 2017, 136, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Foltz, C.J.; Ullman-Cullere, M. Guidelines for assessing the health and condition of mice. Lab. Anim. 1999, 28, 28–32. [Google Scholar]
- Ullman-Culleré, M.H.; Foltz, C.J. Body condition scoring: A rapid and accurate method for assessing health status in mice. Lab. Anim. Sci. 1999, 49, 319–323. [Google Scholar]
- Ekolle Ndode-Ekane, X.; Kharatishvili, I.; Pitkänen, A. Unfolded Maps for Quantitative Analysis of Cortical Lesion Location and Extent after Traumatic Brain Injury. J. Neurotrauma 2017, 34, 459–474. [Google Scholar] [CrossRef]
- Van Vliet, E.A.; Puhakka, N.; Mills, J.D.; Srivastava, P.K.; Johnson, M.R.; Roncon, P.; Das Gupta, S.; Karttunen, J.; Simonato, M.; Lukasiuk, K.; et al. Standardization procedure for plasma biomarker analysis in rat models of epileptogenesis: Focus on circulating microRNAs. Epilepsia 2017, 58, 2013–2024. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 6 January 2018).
- Friedman, J.H. Stochastic gradient boosting. Comput. Stat. Data Anal. 2002, 38, 367–378. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Huttunen, H.; Manninen, T.; Kauppi, J.-P.; Tohka, J. Mind reading with regularized multinomial logistic regression. Mach. Vis. Appl. 2013, 24, 1311–1325. [Google Scholar] [CrossRef]
- Ambroise, C.; McLachlan, G.J. Selection bias in gene extraction on the basis of microarray gene-expression data. Proc. Natl. Acad. Sci. USA 2002, 99, 6562–6566. [Google Scholar] [CrossRef] [PubMed]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely randomized trees. Mach. Learn. 2006, 63, 3–42. [Google Scholar] [CrossRef]
- Ojala, M.; Garriga, G.C. Permutation tests for studying classifier performance. J. Mach. Learn. Res. 2010, 11, 1833–1863. [Google Scholar]
- Maas, A.I.R.; Roozenbeek, B.; Manley, G.T. Clinical trials in traumatic brain injury: Past experience and current developments. Neurotherapeutics 2010, 7, 115–126. [Google Scholar] [CrossRef]
- Schouten, J.W. Neuroprotection in traumatic brain injury: A complex struggle against the biology of nature. Curr. Opin. Crit. Care 2007, 13, 134–142. [Google Scholar] [CrossRef]
- Wheaton, P.; Mathias, J.; Vink, R. Impact of pharmacological treatments on outcome in adult rodents after traumatic brain injury: A meta-analysis. J. Psychopharmacol. 2011, 25, 1581–1599. [Google Scholar] [CrossRef]
- Kochanek, P.M.; Bramlett, H.; Dietrich, W.D.; Dixon, C.E.; Hayes, R.L.; Povlishock, J.; Tortella, F.C.; Wang, K.K.W. A novel multicenter preclinical drug screening and biomarker consortium for experimental traumatic brain injury: Operation brain trauma therapy. J. Trauma Inj. Infect. Crit. Care 2011, 71, S15–S24. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | 32 H | 3 Months | ||||
|---|---|---|---|---|---|---|
| Concordance | Cell Line | Signature ID | Concordance | Cell Line | Signature ID | |
| Desmethylclomipramine | 0.123248134 | NEU.KCL | LINCSCP_40633 | 0.239423 | NEU.KCL | LINCSCP_40633 |
| Ionomycin | 0.11614885 | NEU | LINCSCP_41477 | 0.225506 | NEU.KCL | LINCSCP_40564 |
| Trimipramine | 0.106234243 | NEU | LINCSCP_40955 | No score | No score | No score |
| Sirolimus | No score | No score | No score | −0.18388 | NPC | LINCSCP_42468 |
| Gene | 32 H | 3 Months | ||
|---|---|---|---|---|
| Log2 FC | FDR | Log2 FC | FDR | |
| Nfe2l2 and Nrf2 Target Gene Expression | ||||
| Nfe2l2 | 0.094 | 6.41 × 10−1 | 0.601 | 6.41× 10−5 |
| Gclm | 0.818 | 1.88× 10−8 | 0.016 | 9.51 × 10−1 |
| Hmox1 | 2.860 | 1.69× 10−9 | 1.189 | 2.69× 10−18 |
| Nqo1 | 0.424 | 6.45 × 10−3 | 0.646 | 1.29× 10−7 |
| Nitric Oxide Synthase Gene Expression | ||||
| Nos1 | 0.038 | 6.22 × 10−1 | 0.209 | 3.50 × 10−1 |
| Nos2 | −0.324 | 7.05× 10−7 | 0.587 | 3.32 × 10−2 |
| Nos3 | 0.203 | 1.22 × 10−1 | −0.023 | 9.30 × 10−1 |
| Cytokine Gene Expression | ||||
| Il1a | −0.693 | 1.08 × 10−4 | −0.089 | 8.50 × 10−1 |
| Il1b | 0.478 | 6.15 × 10−2 | 0.598 | NA |
| Il2 | −0.314 | 6.57× 10−6 | −0.009 | NA |
| Il4 | −0.177 | 1.38 × 10−4 | 0.095 | NA |
| Il5 | −0.149 | 9.87× 10−4 | NA | NA |
| Il6 | 0.409 | 2.62 × 10−2 | −0.013 | NA |
| Il10 | −0.300 | 3.81 × 10−3 | 0.161 | NA |
| Il12a | −0.409 | 5.12× 10−5 | −0.002 | 9.96 × 10−1 |
| IL-13 | −0.247 | 1.39 × 10−3 | −0.046 | NA |
| Csf2 (GM-CSF) | −0.211 | 5.91 × 10−3 | NA | NA |
| Ifng | −0.414 | 2.19 × 10−2 | NA | NA |
| Tnf | −0.147 | 5.24 × 10−1 | 0.088 | 8.53 × 10−1 |
| Drug | Inflammatory (LPS/IFNy) | Non-Inflammatory (No LPS/IFNy) | ||
|---|---|---|---|---|
| Log2 FC | p-Value | Log2 FC | p-Value | |
| Desmethylclomipramine | ||||
| Nfe2l2 | 0.282 | 1.87 × 10−2 | 0.200 | 3.83 × 10−2 |
| Gclm | 0.099 | 7.70 × 10−1 | 0.118 | 7.83 × 10−1 |
| Hmox1 | 0.039 | 7.85 × 10−1 | 0.050 | 3.98 × 10−1 |
| Nqo1 | −0.056 | 6.64 × 10−1 | −12.419 | 1.09 × 10−1 |
| Ionomycin | ||||
| Nfe2l2 | −0.052 | 7.87 × 10−1 | 0.125 | 1.65 × 10−1 |
| Gclm | 0.262 | 1.04 × 10−1 | 0.388 | 4.52 × 10−1 |
| Hmox1 | 0.298 | 1.54 × 10-1 | 0.281 | 6.58 × 10-2 |
| Nqo1 | 0.565 | 8.72 × 10-3 | −12.283 | 8.89 × 10-2 |
| Sulforaphane | ||||
| Nfe2l2 | 0.723 | 3.01× 10−7 | −0.382 | 1.26 × 10-2 |
| Gclm | 1.117 | 7.47× 10−8 | 3.305 | 4.61× 10−15 |
| Hmox1 | 0.171 | 1.52 × 10-2 | 1.403 | 3.14× 10−9 |
| Nqo1 | 1.897 | 1.03× 10−12 | −6.055 | 7.23 × 10-2 |
| Scoring Component | Desmethylclomipramine | Ionomycin | Trimipramine | Sirolimus | 1400W | IL-10 |
|---|---|---|---|---|---|---|
| In Silico Analysis of Therapeutic Time Window | ||||||
| Concordance (acute) | 0 | 0.3 | 0 | No score | No score | No score |
| Concordance (chronic) | 0.3 | 0.3 | 0 | 0 | No score | No score |
| Pharmacokinetic Properties | ||||||
| Blood brain barrier penetration | 0.5 | −0.5 | 0.5 | 0.5 | 0.5 | 0 |
| Water solubility | 0.5 | 0 | 0.5 | 0.5 | 0.5 | 0.5 |
| Subtotal | 1.3 | −0.2 | 1 | 1 | 1 | 0.5 |
| Co-culture Outcome | ||||||
| Inflammation (TNFα) | 0.3 | 0.3 | 0.3 | 0.3 | 0.6 | 0.6 |
| Oxidative stress (NO) | 0.15 | 0.15 | 0.15 | 0.3 | 0.15 | 0.15 |
| Neuronal viability (MAP2) | 0 | 0 | 0 | 0.2 | 0.4 | 0.4 |
| Subtotal | 1.75 | 0.25 | 1.45 | 1.8 | 1.15 | 1.65 |
| Target Engagement (Inflammatory Condition) | ||||||
| Nfe2l2 | 0.0125 | 0 | ||||
| Gclm | 0 | 0 | ||||
| Hmox1 | 0 | 0 | ||||
| Nqo1 | 0 | 0.0125 | ||||
| Target Engagement (Non-inflammatory Condition) | ||||||
| Nfe2l2 | 0.0125 | 0 | ||||
| Gclm | 0 | 0 | ||||
| Hmox1 | 0 | 0 | ||||
| Nqo1 | 0 | 0 | ||||
| Total Score | 1.775 | 0.2625 | ||||
| Scoring Component | −5 | −1 | 0 | 1 | 2 | 3 | 4 | 5 | Weight |
|---|---|---|---|---|---|---|---|---|---|
| In Silico Analysis of Therapeutic Time Window | |||||||||
| Concordance (acute) | >Top 60% # | Top 50% # | Top 40% # | Top 30% # | Top 20% # | Top 10% # | 10% | ||
| Concordance (chronic) | >Top 60% # | Top 50% # | Top 40% # | Top 30% # | Top 20% # | Top 10% # | 10% | ||
| Pharmacokinetic Properties | |||||||||
| Blood brain barrier penetration | No | Unknown | Yes | 10% | |||||
| Water solubility | Non soluble | Unknown | Soluble | 10% | |||||
| Co-culture Outcome | |||||||||
| Inflammation (TNFa) | >100%& | 100% & | <90% & | <70% & | <50% & | <30% & | <10% & | 15% | |
| Neurotoxicity (NO) | >100%& | 200% & | <90% & | <70% & | <50% & | <30% & | <10% & | 15% | |
| Neuronal viability (MAP2) | <100%& | >115% & | >100% & | >150% & | >200% & | >250% & | >300% & | 20% | |
| Target Engagement (under Inflammatory Conditions) | |||||||||
| Nfe2l2 | ↓ | - | ↑ | 1.25% | |||||
| Gclm | ↓ | - | ↑ | 1.25% | |||||
| Hmox1 | ↓ | - | ↑ | 1.25% | |||||
| Nqo1 | ↓ | - | ↑ | 1.25% | |||||
| Target Engagement (under Non-inflammatory Condition) | |||||||||
| Nfe2l2 | ↓ | - | ↑ | 1.25% | |||||
| Gclm | ↓ | - | ↑ | 1.25% | |||||
| Hmox1 | ↓ | - | ↑ | 1.25% | |||||
| Nqo1 | ↓ | - | ↑ | 1.25% | |||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lipponen, A.; Natunen, T.; Hujo, M.; Ciszek, R.; Hämäläinen, E.; Tohka, J.; Hiltunen, M.; Paananen, J.; Poulsen, D.; Kansanen, E.; et al. In Vitro and In Vivo Pipeline for Validation of Disease-Modifying Effects of Systems Biology-Derived Network Treatments for Traumatic Brain Injury—Lessons Learned. Int. J. Mol. Sci. 2019, 20, 5395. https://doi.org/10.3390/ijms20215395
Lipponen A, Natunen T, Hujo M, Ciszek R, Hämäläinen E, Tohka J, Hiltunen M, Paananen J, Poulsen D, Kansanen E, et al. In Vitro and In Vivo Pipeline for Validation of Disease-Modifying Effects of Systems Biology-Derived Network Treatments for Traumatic Brain Injury—Lessons Learned. International Journal of Molecular Sciences. 2019; 20(21):5395. https://doi.org/10.3390/ijms20215395
Chicago/Turabian StyleLipponen, Anssi, Teemu Natunen, Mika Hujo, Robert Ciszek, Elina Hämäläinen, Jussi Tohka, Mikko Hiltunen, Jussi Paananen, David Poulsen, Emilia Kansanen, and et al. 2019. "In Vitro and In Vivo Pipeline for Validation of Disease-Modifying Effects of Systems Biology-Derived Network Treatments for Traumatic Brain Injury—Lessons Learned" International Journal of Molecular Sciences 20, no. 21: 5395. https://doi.org/10.3390/ijms20215395
APA StyleLipponen, A., Natunen, T., Hujo, M., Ciszek, R., Hämäläinen, E., Tohka, J., Hiltunen, M., Paananen, J., Poulsen, D., Kansanen, E., Ekolle Ndode-Ekane, X., Levonen, A.-L., & Pitkänen, A. (2019). In Vitro and In Vivo Pipeline for Validation of Disease-Modifying Effects of Systems Biology-Derived Network Treatments for Traumatic Brain Injury—Lessons Learned. International Journal of Molecular Sciences, 20(21), 5395. https://doi.org/10.3390/ijms20215395