Virulence Factors of Meningitis-Causing Bacteria: Enabling Brain Entry across the Blood–Brain Barrier


Abstract
1. Introduction
2. Barriers of the Central Nervous System
2.1. Blood–Brain Barrier
2.2. Blood–Cerebrospinal Fluid Barrier
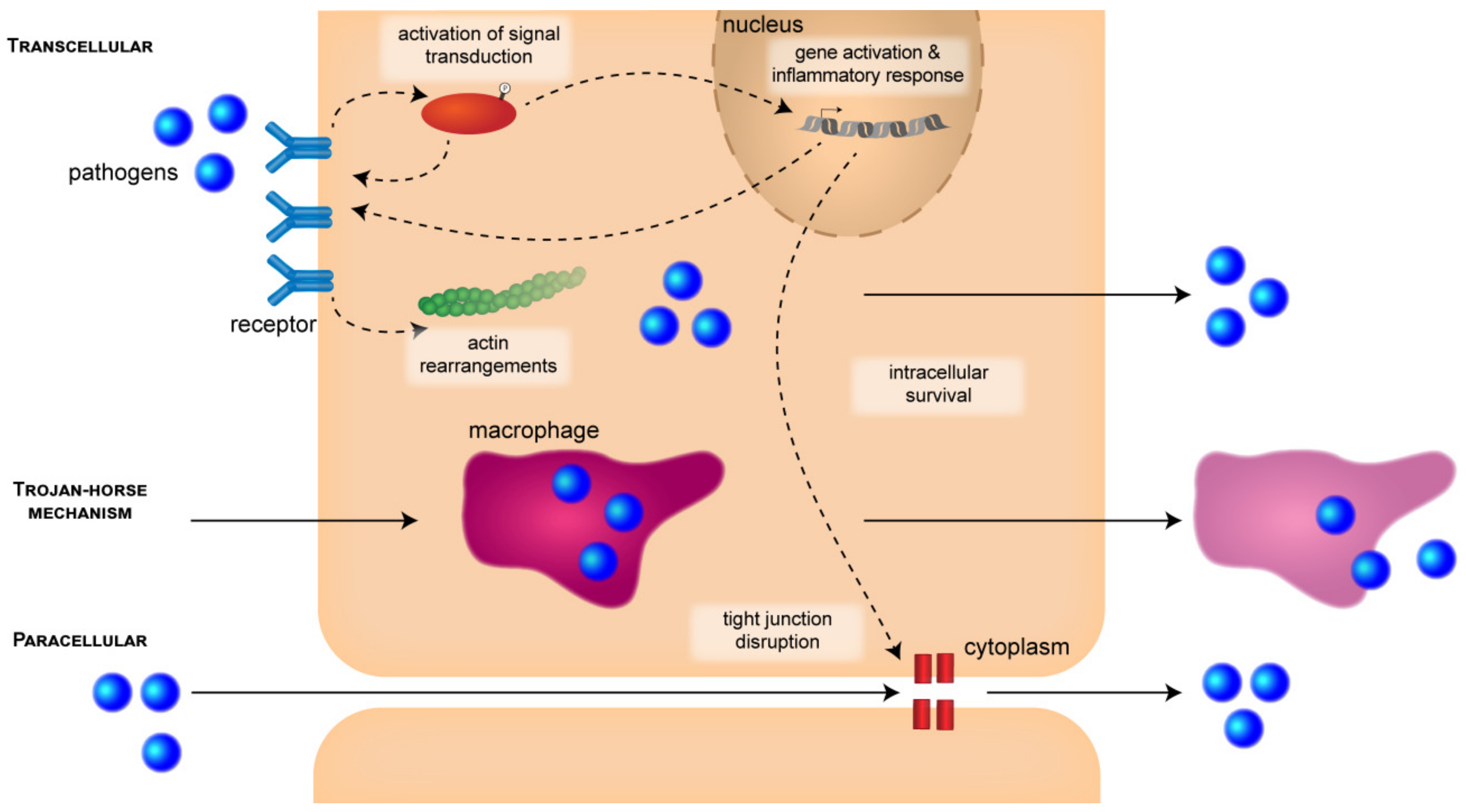
3. Stages during the Pathogenesis of BACTERIAL Meningitis
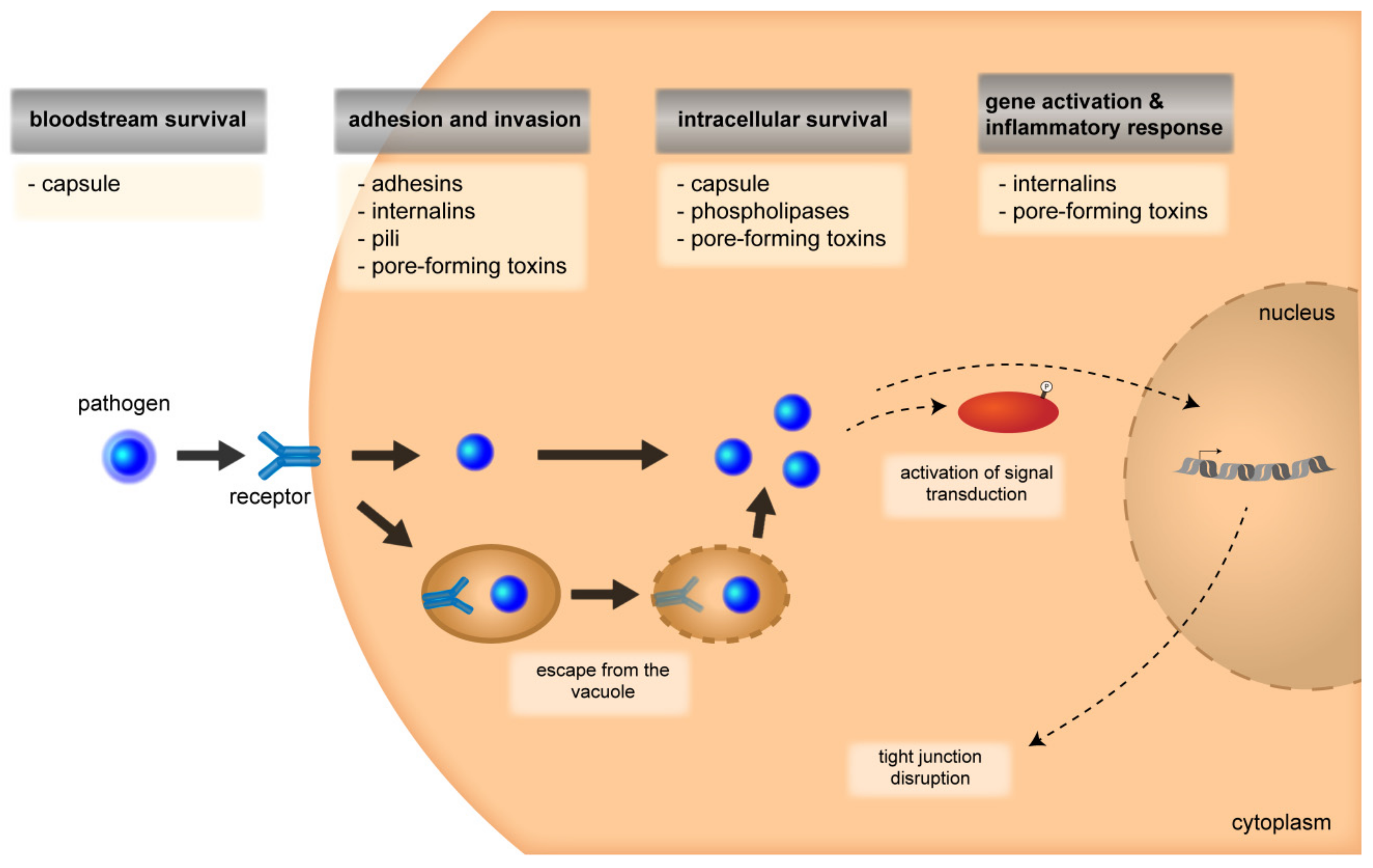
3.1. Attachment and Invasion
3.1.1. CNS Entry Routes
3.1.2. Signal-Transduction Mechanisms and Cytoskeletal Rearrangements
3.2. Intracellular Survival
3.2.1. Multiplication and Intracellular Survival
3.2.2. Disruption of Barrier Integrity and Inflammatory Response
4. Roles of Bacterial Virulence Factors During Invasion Through the Barriers of the CNS
4.1. Gram-Positive Bacteria
4.1.1. Listeria Monocytogenes
4.1.2. Streptococcus suis
4.1.3. Streptococcus Pneumoniae
4.1.4. Group B Streptococcus
4.2. Gram-Negative Bacteria
4.2.1. Escherichia coli
4.2.2. Neisseria Meningitidis
4.2.3. Haemophilus Influenzae
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| BBB | Blood–brain barrier |
| BCSFB | Blood–cerebrospinal fluid barrier |
| BMEC | Brain microvascular endothelial cells |
| CBP | CERB-binding protein |
| CbpA | Choline-binding protein A |
| CECAM | Carcinoembryonic antigen-related cellular adhesion molecule |
| ChoP | Phosphorylcholine |
| CNF1 | Cytotoxic necrotizing factor 1 |
| CNS | Central nervous system |
| CP | Choroid plexus |
| EGFR | Epidermal growth factor receptors |
| ERK | Extracellular signal-regulated kinases |
| FAK | Focal adhesion kinase |
| GCH1 | Guanosine triphosphate cyclohydrolase |
| GlpO | α-glycerophosphate oxidase |
| HBMECs | Human brain microvascular endothelial cells |
| HIBCPP | Human choroid plexus epithelial papilloma |
| HvgA | Hypervirulent GBS adhesin |
| iagA | invasion associated gene A |
| Inl | Internalin |
| JNK | c-JUN N-terminal kinases |
| LLO | Listeriolysin O |
| LOS | Lipooligosaccharide |
| LOX 1 | Lipoprotein receptor 1 |
| LPS | Lipopolysaccharide |
| LTA | Lipoteichoic acid |
| MAPK | Mitogen activated protein kinase |
| MIF | Macrophage migration inhibitory factor |
| MMP | Matrix metalloproteinase |
| Nad A | Neuraminidase A |
| NadA | Neisseria adhesin A |
| NF-κB | Nuclear factor κB |
| NO | Nitric oxide |
| NOD2 | Nucleotide-binding oligomerization domain 2 |
| OMV | Outer membrane vesicles |
| PAFR | Platelet-activating factor receptor |
| PAMPs | Pathogen-associated molecular patterns |
| PCPEC | Primary porcine CP epithelial cells |
| PDGF-B | Platelet-derived growth factor-B |
| PECAM | Platelet endothelial cell adhesion molecule |
| PI3K | Phosphatidylinositol 3-kinase |
| PKC | Protein kinase C |
| plgR | Polymeric immunoglobulin receptor |
| PMN | Polymorphnuclear neutrophils |
| PPRs | Pattern recognition receptors |
| PTX | Pertussis toxin-sensitive |
| Ssr | Serine-rich repeat |
| T3SS | Type three secretion system |
| TF | Transcription factor |
| TGF-β | Transforming growth factor-β |
| TJs | Tight junctions |
| TNF | Tumor necrosis factor |
| VEGF | Vascular Endothelial Growth Factor |
| GBS | Group B streptococcus, Streptococcus agalactiae |
| E. coli | Escherichia coli |
| H. influenzae | Haemophilus influenzae |
| Hia | H. influenzae serotype a |
| Hib | H. influenzae serotype b |
| Hif | H. influenzae serotype f |
| L. monocytogenes | Listeria monocytogenes |
| N. meningitidis | Neisseria meningitidis |
| NTHi | nontypeable H. influenzae |
| S. pneumoniae | Streptococcus pneumoniae |
| S. suis | Streptococcus suis |
References
- Dando, S.J.; Mackay-Sim, A.; Norton, R.; Currie, B.J.; St John, J.A.; Ekberg, J.A.K.; Batzloff, M.; Ulett, G.C.; Beacham, I.R. Pathogens penetrating the central nervous system: Infection pathways and the cellular and molecular mechanisms of invasion. Clin. Microbiol. Rev. 2014, 27, 691–726. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S. Mechanisms of microbial traversal of the blood-brain barrier. Nat. Rev. Microbiol. 2008, 6, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Paulus, W. Choroid plexus: Biology and pathology. Acta Neuropathol. 2010, 119, 75–88. [Google Scholar]
- Doran, K.S.; Fulde, M.; Gratz, N.; Kim, B.J.; Nau, R.; Prasadarao, N.; Schubert-Unkmeir, A.; Tuomanen, E.I.; Valentin-Weigand, P. Host-pathogen interactions in bacterial meningitis. Acta Neuropathol. 2016, 131, 185–209. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.; Sousa, J.C.; Sousa, N.; Palha, J.A. Blood-brain-barriers in aging and in alzheimer‘s disease. Mol. Neurodegener. 2013, 8, 38. [Google Scholar]
- Saunders, N.R.; Habgood, M.D.; Mollgard, K.; Dziegielewska, K.M. The biological significance of brain barrier mechanisms: Help or hindrance in drug delivery to the central nervous system? F1000Res. 2016, 5. [Google Scholar] [CrossRef]
- Lauer, A.N.; Tenenbaum, T.; Schroten, H.; Schwerk, C. The diverse cellular responses of the choroid plexus during infection of the central nervous system. Am. J. Physiol. Cell Physiol. 2018, 314, C152–C165. [Google Scholar]
- Van Sorge, N.M.; Doran, K.S. Defense at the border: The blood-brain barrier versus bacterial foreigners. Future Microbiol. 2012, 7, 383–394. [Google Scholar]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Dias, M.C.; Coisne, C.; Lazarevic, I.; Baden, P.; Hata, M.; Iwamoto, N.; Francisco, D.M.F.; Vanlandewijck, M.; He, L.Q.; Baier, F.A.; et al. Claudin-3-deficient c57bl/6j mice display intact brain barriers. Sci. Rep. 2019, 9, 203. [Google Scholar] [CrossRef] [PubMed]
- Tietz, S.; Engelhardt, B. Brain barriers: Crosstalk between complex tight junctions and adherens junctions. J. Cell Biol. 2015, 209, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.A.; O’Kane, R.L.; Simpson, I.A.; Vina, J.R. Structure of the blood-brain barrier and its role in the transport of amino acids. J. Nutr. 2006, 136, 218s–226s. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-brain barrier: From physiology to disease and back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Weller, R.O.; Sharp, M.M.; Christodoulides, M.; Carare, R.O.; Mollgard, K. The meninges as barriers and facilitators for the movement of fluid, cells and pathogens related to the rodent and human cns. Acta Neuropathol. 2018, 135, 363–385. [Google Scholar] [CrossRef]
- Liddelow, S.A. Development of the choroid plexus and blood-csf barrier. Front. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Schwerk, C.; Tenenbaum, T.; Kim, K.S.; Schroten, H. The choroid plexus-a multi-role player during infectious diseases of the cns. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Bernd, A.; Ott, M.; Ishikawa, H.; Schroten, H.; Schwerk, C.; Fricker, G. Characterization of efflux transport proteins of the human choroid plexus papilloma cell line hibcpp, a functional in vitro model of the blood-cerebrospinal fluid barrier. Pharm. Res. 2015, 32, 2973–2982. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef]
- Sullivan, T.D.; Lascolea, L.J.; Neter, E. Relationship between the magnitude of bacteremia in children and the clinical-disease. Pediatrics 1982, 69, 699–702. [Google Scholar]
- Dietzman, D.E.; Fischer, G.W.; Schoenknecht, F.D. Neonatal escherichia coli septicemia--bacterial counts in blood. J. Pediatr. 1974, 85, 128–130. [Google Scholar] [CrossRef]
- Kim, K.S. Human meningitis-associated escherichia coli. Ecosal Plus 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.M.; Alpert, G.; Campos, J.M.; Plotkin, S.A. Routine quantitative blood cultures in children with haemophilus influenzae or streptococcus pneumoniae bacteremia. Pediatrics 1985, 76, 901–904. [Google Scholar] [PubMed]
- Virji, M. Ins and outs of microbial adhesion. Top. Curr. Chem. 2009, 288, 139–156. [Google Scholar] [PubMed]
- Cossart, P.; Helenius, A. Endocytosis of viruses and bacteria. Cold Spring Harb. Perspect. Biol. 2014, 6, a016972. [Google Scholar] [CrossRef] [PubMed]
- Cossart, P.; Sansonetti, P.J. Bacterial invasion: The paradigms of enteroinvasive pathogens. Science 2004, 304, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Mota, L.J.; Cornelis, G.R. The bacterial injection kit: Type iii secretion systems. Ann. Med. 2005, 37, 234–249. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Tirado, F.H.; Doering, T.L. False friends: Phagocytes as trojan horses in microbial brain infections. PLoS Path. 2017, 13, e1006680. [Google Scholar] [CrossRef]
- Selbach, M.; Backert, S. Cortactin: An achilles’ heel of the actin cytoskeleton targeted by pathogens. Trends Microbiol. 2005, 13, 181–189. [Google Scholar] [CrossRef]
- Stradal, T.E.B.; Schelhaas, M. Actin dynamics in host-pathogen interaction. Febs Lett. 2018, 592, 3658–3669. [Google Scholar] [CrossRef]
- Frischknecht, F.; Way, M. Surfing pathogens and the lessons learned for actin polymerization. Trends Cell Biol. 2001, 11, 30–38. [Google Scholar] [CrossRef]
- Lamason, R.L.; Welch, M.D. Actin-based motility and cell-to-cell spread of bacterial pathogens. Curr. Opin. Microbiol. 2017, 35, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Borkowski, J.; Li, L.; Steinmann, U.; Quednau, N.; Stump-Guthier, C.; Weiss, C.; Findeisen, P.; Gretz, N.; Ishikawa, H.; Tenenbaum, T.; et al. Neisseria meningitidis elicits a pro-inflammatory response involving ikappabzeta in a human blood-cerebrospinal fluid barrier model. J. Neuroinflammation 2014, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Dinner, S.; Kaltschmidt, J.; Stump-Guthier, C.; Hetjens, S.; Ishikawa, H.; Tenenbaum, T.; Schroten, H.; Schwerk, C. Mitogen-activated protein kinases are required for effective infection of human choroid plexus epithelial cells by listeria monocytogenes. Microbes Infect. 2017, 19, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Krachler, A.M.; Woolery, A.R.; Orth, K. Manipulation of kinase signaling by bacterial pathogens. J. Cell Biol. 2011, 195, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Pizarro-Cerda, J.; Cossart, P. Bacterial adhesion and entry into host cells. Cell 2006, 124, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Peng, L.; Gai, Z.; Zhang, L.; Jong, A.; Cao, H.; Huang, S.H. Pathogenic triad in bacterial meningitis: Pathogen invasion, nf-kappab activation, and leukocyte transmigration that occur at the blood-brain barrier. Front. Microbiol 2016, 7, 148. [Google Scholar] [CrossRef]
- Banerjee, A.; Kim, B.J.; Carmona, E.M.; Cutting, A.S.; Gurney, M.A.; Carlos, C.; Feuer, R.; Prasadarao, N.V.; Doran, K.S. Bacterial pili exploit integrin machinery to promote immune activation and efficient blood-brain barrier penetration. Nat. Commun. 2011, 2, 462. [Google Scholar] [CrossRef]
- Koedel, U.; Scheld, W.M.; Pfister, H.W. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect. Dis. 2002, 2, 721–736. [Google Scholar] [CrossRef]
- Kim, S.Y.; Buckwalter, M.; Soreq, H.; Vezzani, A.; Kaufer, D. Blood-brain barrier dysfunction-induced inflammatory signaling in brain pathology and epileptogenesis. Epilepsia 2012, 53, 37–44. [Google Scholar] [CrossRef]
- Haberl, R.L.; Anneser, F.; Kodel, U.; Pfister, H.W. Is nitric-oxide involved as a mediator of cerebrovascular changes in the early phase of experimental pneumococcal meningitis. Neurol. Res. 1994, 16, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Leppert, D.; Leib, S.L.; Grygar, C.; Miller, K.M.; Schaad, U.B.; Hollander, G.A. Matrix metalloproteinase (mmp)-8 and mmp-9 in cerebrospinal fluid during bacterial meningitis: Association with blood-brain barrier damage and neurological sequelae. Clin. Infect. Dis. 2000, 31, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Zwijnenburg, P.J.; de Bie, H.M.; Roord, J.J.; van der Poll, T.; van Furth, A.M. Chemotactic activity of cxcl5 in cerebrospinal fluid of children with bacterial meningitis. J. Neuroimmunol. 2003, 145, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Azuma, H.; Tsuda, N.; Sasaki, K.; Okuno, A. Clinical significance of cytokine measurement for detection of meningitis. J. Pediatr 1997, 131, 463–465. [Google Scholar] [CrossRef]
- Coutinho, L.G.; Grandgirard, D.; Leib, S.L.; Agnez-Lima, L.F. Cerebrospinal-fluid cytokine and chemokine profile in patients with pneumococcal and meningococcal meningitis. BMC Infect. Dis. 2013, 13, 326. [Google Scholar] [CrossRef]
- Cress, B.F.; Englaender, J.A.; He, W.Q.; Kasper, D.; Linhardt, R.J.; Koffas, M.A.G. Masquerading microbial pathogens: Capsular polysaccharides mimic host-tissue molecules. Fems Microbiol. Rev. 2014, 38, 660–697. [Google Scholar] [CrossRef]
- Hauser, S.; Wegele, C.; Stump-Guthier, C.; Borkowski, J.; Weiss, C.; Rohde, M.; Ishikawa, H.; Schroten, H.; Schwerk, C.; Adam, R. Capsule and fimbriae modulate the invasion of haemophilus influenzae in a human blood-cerebrospinal fluid barrier model. Int. J. Med. Microbiol. 2018, 308, 829–839. [Google Scholar] [CrossRef]
- Schwerk, C.; Papandreou, T.; Schuhmann, D.; Nickol, L.; Borkowski, J.; Steinmann, U.; Quednau, N.; Stump, C.; Weiss, C.; Berger, J.; et al. Polar invasion and translocation of neisseria meningitidis and streptococcus suis in a novel human model of the blood-cerebrospinal fluid barrier. PLoS ONE 2012, 7, e30069. [Google Scholar] [CrossRef]
- Gendrin, C.; Merillat, S.; Vornhagen, J.; Coleman, M.; Armistead, B.; Ngo, L.; Aggarwal, A.; Quach, P.; Berrigan, J.; Rajagopal, L. Diminished capsule exacerbates virulence, blood-brain barrier penetration, intracellular persistence, and antibiotic evasion of hyperhemolytic group b streptococci. J. Infect. Dis. 2018, 217, 1128–1138. [Google Scholar] [CrossRef]
- Wu, Z.F.; Wu, C.Y.; Shao, J.; Zhu, Z.Z.; Wang, W.X.; Zhang, W.W.; Tang, M.; Pei, N.; Fan, H.J.; Li, J.G.; et al. The streptococcus suis transcriptional landscape reveals adaptation mechanisms in pig blood and cerebrospinal fluid. RNA 2014, 20, 882–898. [Google Scholar] [CrossRef]
- Al-Obaidi, M.M.J.; Desa, M.N.M. Mechanisms of blood brain barrier disruption by different types of bacteria, and bacterial-host interactions facilitate the bacterial pathogen invading the brain. Cell. Mol. Neurobiol. 2018, 38, 1349–1368. [Google Scholar] [CrossRef] [PubMed]
- Disson, O.; Lecuit, M. Targeting of the central nervous system by listeria monocytogenes. Virulence 2012, 3, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Radoshevich, L.; Cossart, P. Listeria monocytogenes: Towards a complete picture of its physiology and pathogenesis. Nat. Rev. Microbiol. 2018, 16, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Drevets, D.A.; Leenen, P.J.M.; Greenfield, R.A. Invasion of the central nervous system by intracellular bacteria. Clin. Microbiol. Rev. 2004, 17, 323–347. [Google Scholar] [CrossRef] [PubMed]
- Pagelow, D.; Chhatbar, C.; Beineke, A.; Liu, X.K.; Nerlich, A.; van Vorst, K.; Rohde, M.; Kalinke, U.; Forster, R.; Halle, S.; et al. The olfactory epithelium as a port of entry in neonatal neurolisteriosis. Nat. Commun. 2018, 9, 4269. [Google Scholar] [CrossRef] [PubMed]
- Greiffenberg, L.; Goebel, W.; Kim, K.S.; Daniels, J.; Kuhn, M. Interaction of listeria monocytogenes with human brain microvascular endothelial cells: An electron microscopic study. Infect. Immun. 2000, 68, 3275–3279. [Google Scholar] [CrossRef]
- Grundler, T.; Quednau, N.; Stump, C.; Orian-Rousseau, V.; Ishikawa, H.; Wolburg, H.; Schroten, H.; Tenenbaum, T.; Schwerk, C. The surface proteins inla and inlb are interdependently required for polar basolateral invasion by listeria monocytogenes in a human model of the blood-cerebrospinal fluid barrier. Microbes Infect. 2013, 15, 291–301. [Google Scholar] [CrossRef]
- Ghosh, P.; Halvorsen, E.M.; Ammendolia, D.A.; Mor-Vaknin, N.; O’Riordan, M.X.D.; Brumell, J.H.; Markovitz, D.M.; Higgins, D.E. Invasion of the brain by listeria monocytogenes is mediated by inlf and host cell vimentin. MBio 2018, 9. [Google Scholar] [CrossRef]
- Tang, P.; Rosenshine, I.; Finlay, B.B. Listeria-monocytogenes, an invasive bacterium, stimulates map kinase upon attachment to epithelial-cells. Mol. Biol. Cell 1994, 5, 455–464. [Google Scholar] [CrossRef][Green Version]
- Tang, P.; Sutherland, C.L.; Gold, M.R.; Finlay, B.B. Listeria monocytogenes invasion of epithelial cells requires the mek-1/erk-2 mitogen-activated protein kinase pathway. Infect. Immun. 1998, 66, 1106–1112. [Google Scholar]
- Pizarro-Cerda, J.; Kuhbacher, A.; Cossart, P. Entry of listeria monocytogenes in mammalian epithelial cells: An updated view. Cold Spring Harb. Perspect. Med. 2012, 2, a010009. [Google Scholar] [CrossRef] [PubMed]
- Veiga, E.; Cossart, P. Listeria hijacks the clathrin-dependent endocytic machinery to invade mammalian cells. Nat. Cell Biol. 2005, 7. [Google Scholar] [CrossRef] [PubMed]
- Veiga, E.; Guttman, J.A.; Bonazzi, M.; Boucrot, E.; Toledo-Arana, A.; Lin, A.E.; Enninga, J.; Pizarro-Cerda, J.; Finlay, B.B.; Kirchhausen, T.; et al. Invasive and adherent bacterial pathogens co-opt host clathrin for infection. Cell Host Microbe 2007, 2, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Kayal, S.; Lilienbaum, A.; Join-Lambert, O.; Li, X.X.; Israel, A.; Berche, P. Listeriolysin o secreted by listeria monocytogenes induces nf-kappa b signalling by activating the i kappa b kinase complex. Mol. Microbiol. 2002, 44, 1407–1419. [Google Scholar] [CrossRef]
- Tang, P.; Rosenshine, I.; Cossart, P.; Finlay, B.B. Listeriolysin o activates mitogen-activated protein kinase in eucaryotic cells. Infect. Immun. 1996, 64, 2359–2361. [Google Scholar]
- Weiglein, I.; Goebel, W.; Troppmair, J.; Rapp, U.R.; Demuth, A.; Kuhn, M. Listeria monocytogenes infection of hela cells results in listeriolysin o-mediated transient activation of the raf-mek-map kinase pathway. Fems Microbiol. Lett. 1997, 148, 189–195. [Google Scholar] [CrossRef]
- Lambrechts, A.; Gevaert, K.; Cossart, P.; Vandekerckhove, J.; Van Troys, M. Listeria comet tails: The actin-based motility machinery at work. Trends Cell Biol. 2008, 18, 220–227. [Google Scholar] [CrossRef]
- Kocks, C.; Gouin, E.; Tabouret, M.; Berche, P.; Ohayon, H.; Cossart, P. L-monocytogenes-induced actin assembly requires the acta gene-product, a surface protein. Cell 1992, 68, 521–531. [Google Scholar] [CrossRef]
- Gouin, E.; Adib-Conquy, M.; Balestrino, D.; Nahori, M.A.; Villiers, V.; Colland, F.; Dramsi, S.; Dussurget, O.; Cossart, P. The listeria monocytogenes inlc protein interferes with innate immune responses by targeting the i kappa b kinase subunit ikk alpha. Proc. Natl. Acad. Sci. USA 2010, 107, 17333–17338. [Google Scholar] [CrossRef]
- Rajabian, T.; Gavicherla, B.; Heisig, M.; Muller-Altrock, S.; Goebel, W.; Gray-Owen, S.D.; Ireton, K. The bacterial virulence factor inlc perturbs apical cell junctions and promotes cell-to-cell spread of listeria. Nat. Cell Biol. 2009, 11, 1212–1218. [Google Scholar] [CrossRef]
- Gottschalk, M.; Xu, J.G.; Calzas, C.; Segura, M. Streptococcus suis: A new emerging or an old neglected zoonotic pathogen? Future Microbiol. 2010, 5, 371–391. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, T.; Papandreou, T.; Gellrich, D.; Friedrichs, U.; Seibt, A.; Adam, R.; Wewer, C.; Galla, H.J.; Schwerk, C.; Schroten, H. Polar bacterial invasion and translocation of streptococcus suis across the blood-cerebrospinal fluid barrier in vitro. Cell. Microbiol. 2009, 11, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Vanier, G.; Segura, M.; Gottschalk, M. Characterization of the invasion of porcine endothelial cells by streptococcus suis serotype 2. Can. J. Vet. Res. Rev. Can. De Rech. Vet. 2007, 71, 81–89. [Google Scholar]
- Benga, L.; Goethe, R.; Rohde, M.; Valentin-Weigand, P. Non-encapsulated strains reveal novel insights in invasion and survival of streptococcus suis in epithelial cells. Cell. Microbiol. 2004, 6, 867–881. [Google Scholar] [CrossRef]
- Willenborg, J.; Fulde, M.; de Greeff, A.; Rohde, M.; Smith, H.E.; Valentin-Weigand, P.; Goethe, R. Role of glucose and ccpa in capsule expression and virulence of streptococcus suis. Microbiology 2011, 157, 1823–1833. [Google Scholar] [CrossRef]
- Charland, N.; Nizet, V.; Rubens, C.E.; Kim, K.S.; Lacouture, S.; Gottschalk, M. Streptococcus suis serotype 2 interactions with human brain microvascular endothelial cells. Infect. Immun. 2000, 68, 637–643. [Google Scholar] [CrossRef]
- Benga, L.; Friedl, P.; Valentin-Weigand, P. Adherence of streptococcus suis to porcine endothelial cells. J. Vet. Med. Ser. B-Infect. Dis. Vet. Public Health 2005, 52, 392–395. [Google Scholar] [CrossRef]
- Vanier, G.; Segura, M.; Friedl, P.; Lacouture, S.; Gottschalk, M. Invasion of porcine brain microvascular endothelial cells by streptococcus suis serotype 2. Infect. Immun. 2004, 72, 1441–1449. [Google Scholar] [CrossRef]
- Fittipaldi, N.; Segura, M.; Grenier, D.; Gottschalk, M. Virulence factors involved in the pathogenesis of the infection caused by the swine pathogen and zoonotic agent streptococcus suis. Future Microbiol. 2012, 7, 259–279. [Google Scholar] [CrossRef]
- Sun, Y.; Li, N.; Zhang, J.; Liu, H.; Liu, J.; Xia, X.; Sun, C.; Feng, X.; Gu, J.; Du, C.; et al. Enolase of streptococcus suis serotype 2 enhances blood-brain barrier permeability by inducing il-8 release. Inflammation 2016, 39, 718–726. [Google Scholar] [CrossRef]
- Lun, S.C.; Perez-Casal, J.; Connor, W.; Willson, P.J. Role of suilysin in pathogenesis of streptococcus suis capsular serotype 2. Microb. Pathog. 2003, 34, 27–37. [Google Scholar] [CrossRef]
- Seitz, M.; Baums, C.G.; Neis, C.; Benga, L.; Fulde, M.; Rohde, M.; Goethe, R.; Valentin-Weigand, P. Subcytolytic effects of suilysin on interaction of streptococcus suis with epithelial cells. Vet. Microbiol. 2013, 167, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Vadeboncoeur, N.; Segura, M.; Al-Numani, D.; Vanier, G.; Gottschalk, M. Pro-inflammatory cytokine and chemokine release by human brain microvascular endothelial cells stimulated by streptococcus suis serotype 2. Fems Immunol. Med. Microbiol. 2003, 35, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Tenenbauma, T.; Matalon, D.; Adam, R.; Seibt, A.; Wewer, C.; Schwerk, C.; Galla, H.J.; Schrotena, H. Dexamethasone prevents alteration of tight junction-associated proteins and barrier function in porcine choroid plexus epithelial cells after infection with streptococcus suis in vitro. Brain Res. 2008, 1229, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wewer, C.; Seibt, A.; Wolburg, H.; Greune, L.; Schmidt, M.A.; Berger, J.; Galla, H.J.; Quitsch, U.; Schwerk, C.; Schroten, H.; et al. Transcellular migration of neutrophil granulocytes through the blood-cerebrospinal fluid barrier after infection with streptococcus suis. J. Neuroinflammation 2011, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, M.; Thwaites, G.E. The diagnosis and management of acute bacterial meningitis in resource-poor settings. Lancet Neurol. 2008, 7, 637–648. [Google Scholar] [CrossRef]
- Iovino, F.; Orihuela, C.J.; Moorlag, H.E.; Molema, G.; Bijlsma, J.J.E. Interactions between blood-borne streptococcus pneumoniae and the blood-brain barrier preceding meningitis. PLoS ONE 2013, 8, e68408. [Google Scholar] [CrossRef]
- Van Ginkel, F.W.; McGhee, J.R.; Watt, J.M.; Campos-Torres, A.; Parish, L.A.; Briles, D.E. Pneumococcal carriage results in ganglioside-mediated olfactory tissue infection (vol 100, pg 14363, 2003). Proc. Natl. Acad. Sci. USA 2004, 101, 6834. [Google Scholar]
- Wartha, F.; Beiter, K.; Albiger, B.; Fernebro, J.; Zychlinsky, A.; Normark, S.; Henriques-Normark, B. Capsule and d-alanylated lipoteichoic acids protect streptococcus pneumoniae against neutrophil extracellular traps. Cell. Microbiol. 2007, 9, 1162–1171. [Google Scholar] [CrossRef]
- Middleton, D.R.; Paschall, A.V.; Duke, J.A.; Avci, F.Y. Enzymatic hydrolysis of pneumococcal capsular polysaccharide renders the bacterium vulnerable to host defense. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef]
- Keller, L.E.; Jones, C.V.; Thornton, J.A.; Sanders, M.E.; Swiatlo, E.; Nahm, M.H.; Park, I.H.; McDaniel, L.S. Pspk of streptococcus pneumoniae increases adherence to epithelial cells and enhances nasopharyngeal colonization. Infect. Immun. 2013, 81, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Li-Korotky, H.S.; Lo, C.Y.; Banks, J.M. Interaction of pneumococcal phase variation, host and pressure/gas composition: Virulence expression of nana, hyla, pspa and cbpa in simulated otitis media. Microb. Pathog. 2010, 49, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Shainheit, M.G.; Muie, M.; Camilli, A. The core promoter of the capsule operon of streptococcus pneumoniae is necessary for colonization and invasive disease. Infect. Immun. 2014, 82, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Iovino, F.; Molema, G.; Bijlsma, J.J.E. Platelet endothelial cell adhesion molecule-1, a putative receptor for the adhesion of streptococcus pneumoniae to the vascular endothelium of the blood-brain barrier. Infect. Immun. 2014, 82, 3555–3566. [Google Scholar] [CrossRef]
- Uchiyama, S.; Carlin, A.F.; Khosravi, A.; Weiman, S.; Banerjee, A.; Quach, D.; Hightower, G.; Mitchell, T.J.; Doran, K.S.; Nizet, V. The surface-anchored nana protein promotes pneumococcal brain endothelial cell invasion. J. Exp. Med. 2009, 206, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Gratz, N.; Loh, L.N.; Mann, B.; Gao, G.; Carter, R.; Rosch, J.; Tuomanen, E.I. Pneumococcal neuraminidase activates tgf-beta signalling. Microbiology 2017, 163, 1198–1207. [Google Scholar]
- Iovino, F.; Engelen-Lee, J.Y.; Brouwer, M.; van de Beek, D.; van der Ende, A.; Seron, M.V.; Mellroth, P.; Muschiol, S.; Bergstrand, J.; Widengren, J.; et al. Pigr and pec am-1 bind to pneumococcal adhesins rrga and pspc mediating bacterial brain invasion. J. Exp. Med. 2017, 214, 1619–1630. [Google Scholar] [CrossRef]
- Bagnoli, F.; Moschioni, M.; Donati, C.; Dimitrovska, V.; Ferlenghi, I.; Facciotti, C.; Muzzi, A.; Giusti, F.; Emolo, C.; Sinisi, A.; et al. A second pilus type in streptococcus pneumoniae is prevalent in emerging serotypes and mediates adhesion to host cells. J. Bacteriol. 2008, 190, 5480–5492. [Google Scholar] [CrossRef]
- Orihuela, C.J.; Mahdavi, J.; Thornton, J.; Mann, B.; Wooldridge, K.G.; Abouseada, N.; Oldfield, N.J.; Self, T.; Ala’Aldeen, D.A.; Tuomanen, E.I. Laminin receptor initiates bacterial contact with the blood brain barrier in experimental meningitis models. J. Clin. Invest. 2009, 119, 1638–1646. [Google Scholar] [CrossRef]
- Schmidt, F.; Kakar, N.; Meyer, T.C.; Depke, M.; Masouris, I.; Burchhardt, G.; Gomez-Mejia, A.; Dhople, V.; Havarstein, L.S.; Sun, Z.; et al. In vivo proteomics identifies the competence regulon and alib oligopeptide transporter as pathogenic factors in pneumococcal meningitis. PLoS Path. 2019, 15, e1007987. [Google Scholar] [CrossRef]
- Chen, J.Q.; Li, N.N.; Wang, B.W.; Liu, X.F.; Liu, J.L.; Chang, Q. Upregulation of cbp by ply can cause permeability of blood-brain barrier to increase meningitis. J. Biochem. Mol. Toxicol. 2019, e22333. [Google Scholar] [CrossRef] [PubMed]
- Zysk, G.; Schneider-Wald, B.K.; Hwang, J.H.; Bejo, L.; Kim, K.S.; Mitchell, T.J.; Hakenbeck, R.; Heinz, H.P. Pneumolysin is the main inducer of cytotoxicity to brain microvascular endothelial cells caused by streptococcus pneumoniae. Infect. Immun. 2001, 69, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Mandi, L.K.; Wang, H.; Van der Hoek, M.B.; Paton, J.C.; Ogunniyi, A.D. Identification of a novel pneumococcal vaccine antigen preferentially expressed during meningitis in mice. J. Clin. Invest. 2012, 122, 2208–2220. [Google Scholar]
- Wang, Y.; Liu, X.; Liu, Q. Nod2 expression in streptococcus pneumoniae meningitis and its influence on the blood-brain barrier. Can. J. Infect. Dis Med. Microbiol 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Barichello, T.; Fagundes, G.D.; Generoso, J.S.; Moreira, A.P.; Costa, C.S.; Zanatta, J.R.; Simoes, L.R.; Petronilho, F.; Dal-Pizzol, F.; Vilela, M.C.; et al. Brain-blood barrier breakdown and pro-inflammatory mediators in neonate rats submitted meningitis by streptococcus pneumoniae. Brain Res. 2012, 1471, 162–168. [Google Scholar] [CrossRef]
- Brouwer, M.C.; Tunkel, A.R.; van de Beek, D. Epidemiology, diagnosis, and antimicrobial treatment of acute bacterial meningitis. Clin. Microbiol. Rev. 2010, 23, 467–492. [Google Scholar] [CrossRef]
- Jones, N.; Bohnsack, J.F.; Takahashi, S.; Oliver, K.A.; Chan, M.S.; Kunst, F.; Glaser, P.; Rusniok, C.; Crook, D.W.M.; Harding, R.M.; et al. Multilocus sequence typing system for group b streptococcus. J. Clin. Microbiol. 2003, 41, 2530–2536. [Google Scholar] [CrossRef]
- Maisey, H.C.; Doran, K.S.; Nizet, V. Recent advances in understanding the molecular basis of group b streptococcus virulence. Expert Rev. Mol. Med. 2008, 10, e27. [Google Scholar] [CrossRef]
- Nizet, V.; Kim, K.S.; Stins, M.; Jonas, M.; Chi, E.Y.; Nguyen, D.; Rubens, C.E. Invasion of brain microvascular endothelial cells by group b streptococci. Infect. Immun. 1997, 65, 5074–5081. [Google Scholar]
- Tazi, A.; Disson, O.; Bellais, S.; Bouaboud, A.; Dmytruk, N.; Dramsi, S.; Mistou, M.Y.; Khun, H.; Mechler, C.; Tardieux, I.; et al. The surface protein hvga mediates group b streptococcus hypervirulence and meningeal tropism in neonates. J. Exp. Med. 2010, 207, 2313–2322. [Google Scholar] [CrossRef]
- Lauer, P.; Rinaudo, C.D.; Soriani, M.; Margarit, I.; Maione, D.; Rosini, R.; Taddei, A.R.; Mora, M.; Rappuoli, R.; Grandi, G.; et al. Genome analysis reveals pili in group b streptococcus. Science 2005, 309, 105. [Google Scholar] [CrossRef] [PubMed]
- Maisey, H.C.; Hensler, M.; Nizet, V.; Doran, K.S. Group b streptococcal pilus proteins contribute to adherence to and invasion of brain microvascular endothelial cells. J. Bacteriol. 2007, 189, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Mu, R.; Kim, B.J.; Paco, C.; Del Rosario, Y.; Courtney, H.S.; Doran, K.S. Identification of a group b streptococcal fibronectin binding protein, sfba, that contributes to invasion of brain endothelium and development of meningitis. Infect. Immun. 2014, 82, 2276–2286. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.S.; Mu, R.; Kim, B.J.; Doran, K.S.; Sullam, P.M. Binding of glycoprotein srr1 of streptococcus agalactiae to fibrinogen promotes attachment to brain endothelium and the development of meningitis. PLoS Path. 2012, 8, e1002947. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.W.; Spencer, B.L.; Holmes, J.A.; Mu, R.; Rego, S.; Weston, T.A.; Hu, Y.; Sanches, G.F.; Yoon, S.; Park, N.; et al. The group b streptococcal surface antigen i/ii protein, bspc, interacts with host vimentin to promote adherence to brain endothelium and inflammation during the pathogenesis of meningitis. PLoS Path. 2019, 15, e1007848. [Google Scholar] [CrossRef]
- Shin, S.; Maneesh, P.S.; Lee, J.S.; Romer, L.H.; Kim, K.S. Focal adhesion kinase is involved in type iii group b streptococcal invasion of human brain microvascular endothelial cells. Microb. Pathog. 2006, 41, 168–173. [Google Scholar] [CrossRef]
- Doran, K.S.; Engelson, E.J.; Khosravi, A.; Maisey, H.C.; Fedtke, I.; Equils, O.; Michelsen, K.S.; Arditi, M.; Peschel, A.; Nizet, V. Blood-brain barrier invasion by group b streptococcus depends upon proper cell-surface anchoring of lipoteichoic acid. J. Clin. Invest. 2005, 115, 2499–2507. [Google Scholar] [CrossRef]
- Luo, S.; Cao, Q.; Ma, K.; Wang, Z.F.; Liu, G.J.; Lu, C.P.; Liu, Y.J. Quantitative assessment of the blood-brain barrier opening caused by streptococcus agalactiae hyaluronidase in a balb/c mouse model. Sci Rep.-Uk 2017, 7, 13529. [Google Scholar] [CrossRef]
- Kim, B.J.; McDonagh, M.A.; Deng, L.; Gastfriend, B.D.; Schubert-Unkmeir, A.; Doran, K.S.; Shusta, E.V. Streptococcus agalactiae disrupts p-glycoprotein function in brain endothelial cells. Fluids Barriers Cns 2019, 16, 1–10. [Google Scholar] [CrossRef]
- Kim, B.J.; Hancock, B.M.; Bermudez, A.; Del Cid, N.; Reyes, E.; van Sorge, N.M.; Lauth, X.; Smurthwaite, C.A.; Hilton, B.J.; Stotland, A.; et al. Bacterial induction of snail1 contributes to blood-brain barrier disruption. J. Clin. Invest. 2015, 125, 2473–2483. [Google Scholar] [CrossRef]
- Rose, R.; Hauser, S.; Stump-Guthier, C.; Weiss, C.; Rohde, M.; Kim, K.S.; Ishikawa, H.; Schroten, H.; Schwerk, C.; Adam, R. Virulence factor-dependent basolateral invasion of choroid plexus epithelial cells by pathogenic Escherichia coli in vitro. Fems Microbiol. Lett. 2018, 365. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Itabashi, H.; Gemski, P.; Sadoff, J.; Warren, R.L.; Cross, A.S. The k1-capsule is the critical determinant in the development of escherichia-coli meningitis in the rat. J. Clin. Invest. 1992, 90, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.S.; Kim, K.S.; Wright, D.C.; Sadoff, J.C.; Gemski, P. Role of lipopolysaccharide and capsule in the serum resistance of bacteremic strains of escherichia-coli. J. Infect. Dis. 1986, 154, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Logue, C.M.; Doetkott, C.; Mangiamele, P.; Wannemuehler, Y.M.; Johnson, T.J.; Tivendale, K.A.; Li, G.W.; Sherwood, J.S.; Nolan, L.K. Genotypic and phenotypic traits that distinguish neonatal meningitis-associated escherichia coli from fecal e. Coli isolates of healthy human hosts. Appl. Environ. Microbiol. 2012, 78, 5824–5830. [Google Scholar] [CrossRef]
- Teng, C.H.; Cai, M.; Shin, S.; Xie, Y.; Kim, K.J.; Khan, N.A.; Di Cello, F.; Kim, K.S. Escherichia coli k1 rs218 interacts with human brain microvascular endothelial cells via type 1 fimbria bacteria in the fimbriated state. Infect. Immun. 2005, 73, 2923–2931. [Google Scholar] [CrossRef]
- Khan, N.A.; Shin, S.; Chung, J.W.; Kim, K.J.; Elliott, S.; Wang, Y.; Kim, K.S. Outer membrane protein a and cytotoxic necrotizing factor-1 use diverse signaling mechanisms for escherichia coli k1 invasion of human brain microvascular endothelial cells. Microb. Pathog. 2003, 35, 35–42. [Google Scholar] [CrossRef]
- Huang, S.H.; Wan, Z.S.; Chen, Y.H.; Jong, A.Y.; Kim, K.S. Further characterization of escherichia coli brain microvascular endothelial cell invasion gene ibea by deletion, complementation, and protein expression. J. Infect. Dis. 2001, 183, 1071–1078. [Google Scholar] [CrossRef]
- Wang, M.H.; Kim, K.S. Cytotoxic necrotizing factor 1 contributes to escherichia coli meningitis. Toxins 2013, 5, 2270–2280. [Google Scholar] [CrossRef]
- Prasadarao, N.V. Identification of escherichia coli outer membrane protein a receptor on human brain microvascular endothelial cells. Infect. Immun. 2002, 70, 4556–4563. [Google Scholar] [CrossRef]
- Reddy, M.A.; Wass, C.A.; Kim, K.S.; Schlaepfer, D.D.; Prasadarao, N.V. Involvement of focal adhesion kinase in escherichia coli invasion of human brain microvascular endothelial cells. Infect. Immun. 2000, 68, 6423–6430. [Google Scholar] [CrossRef]
- Reddy, M.A.; Prasadarao, N.V.; Wass, C.A.; Kim, K.S. Phosphatidylinositol 3-kinase activation and interaction with focal adhesion kinase in escherichia coli k1 invasion of human brain microvascular endothelial cells. J. Biol. Chem. 2000, 275, 36769–36774. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, S.K.; McNamara, G.; Prasadarao, N.V. Escherichia coli k-1 interaction with human brain micro-vascular endothelial cells triggers phospholipase c-gamma1 activation downstream of phosphatidylinositol 3-kinase. J. Biol. Chem. 2003, 278, 45753–45762. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.K.; Periandythevar, P.; Prasadarao, N.V. Outer membrane protein a of escherichia coli k1 selectively enhances the expression of intercellular adhesion molecule-1 in brain microvascular endothelial cells. Microb. Infect. 2007, 9, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Prasadarao, N.V.; Wass, C.A.; Stins, M.F.; Shimada, H.; Kim, K.S. Outer membrane protein a-promoted actin condensation of brain microvascular endothelial cells is required for escherichia coli invasion. Infect. Immun. 1999, 67, 5775–5783. [Google Scholar]
- Sukumaran, S.K.; Quon, M.J.; Prasadarao, N.V. Escherichia coli k1 internalization via caveolae requires caveolin-1 and protein kinase c alpha interaction in human brain microvascular endothelial cells. J. Biol. Chem. 2002, 277, 50716–50724. [Google Scholar] [CrossRef]
- Kim, K.J.; Elliott, S.J.; Di Cello, F.; Stins, M.F.; Kim, K.S. The k1 capsule modulates trafficking of e-coli-containing vacuoles and enhances intracellular bacterial survival in human brain microvascular endothelial cells. Cell. Microbiol. 2003, 5, 245–252. [Google Scholar] [CrossRef]
- Yang, R.C.; Huang, F.; Fu, J.Y.; Dou, B.B.; Xu, B.J.; Miao, L.; Liu, W.T.; Yang, X.P.; Tan, C.; Chen, H.C.; et al. Differential transcription profiles of long non-coding rnas in primary human brain microvascular endothelial cells in response to meningitic escherichia coli. Sci. Rep. 2016, 6, 38903. [Google Scholar] [CrossRef]
- Yang, R.C.; Qu, X.Y.; Xiao, S.Y.; Li, L.; Xu, B.J.; Fu, J.Y.; Lv, Y.J.; Amjad, N.; Tan, C.; Kim, K.S.; et al. Meningitic escherichia coli-induced upregulation of pdgf-b and icam-1 aggravates blood-brain barrier disruption and neuroinflammatory response. J. Neuroinflamm. 2019, 16, 101. [Google Scholar] [CrossRef]
- Sukumaran, S.K.; Prasadarao, N.V. Escherichia coli k1 invasion increases human brain microvascular endothelial cell monolayer permeability by disassembling vascular-endothelial cadherins at tight junctions. J. Infect. Dis. 2003, 188, 1295–1309. [Google Scholar] [CrossRef]
- Mittal, R.; Gonzalez-Gomez, I.; Goth, K.A.; Prasadarao, N.V. Inhibition of inducible nitric oxide controls pathogen load and brain damage by enhancing phagocytosis of escherichia coli k1 in neonatal meningitis. Am. J. Pathol. 2010, 176, 1292–1305. [Google Scholar] [CrossRef]
- Shanmuganathan, M.V.; Krishnan, S.; Fu, X.W.; Prasadarao, N.V. Attenuation of biopterin synthesis prevents escherichia coli k1 invasion of brain endothelial cells and the development of meningitis in newborn mice. J. Infect. Dis. 2013, 207, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.T.; Lv, Y.J.; Yang, R.C.; Fu, J.Y.; Liu, L.; Wang, H.; Cao, Q.; Tan, C.; Chen, H.C.; Wang, X.R. New insights into meningitic escherichia coli infection of brain microvascular endothelial cells from quantitative proteomics analysis. J. Neuroinflamm. 2018, 15, 291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Shi, M.J.; Niu, Z.; Chen, X.; Wei, J.Y.; Miao, Z.W.; Zhao, W.D.; Chen, Y.H. Activation of brain endothelium by escherichia coli k1 virulence factor cgld promotes polymorphonuclear leukocyte transendothelial migration. Med. Microbiol. Immunol. 2019, 208, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Coureuil, M.; Lecuyer, H.; Bourdoulous, S.; Nassif, X. A journey into the brain: Insight into how bacterial pathogens cross blood-brain barriers. Nat. Rev. Microbiol. 2017, 15, 149–159. [Google Scholar] [CrossRef]
- Schneider, M.C.; Exley, R.M.; Chan, H.; Feavers, I.; Kang, Y.H.; Sim, R.B.; Tang, C.M. Functional significance of factor h binding to neisseria meningitidis. J. Immunol. 2006, 176, 7566–7575. [Google Scholar] [CrossRef]
- Seib, K.L.; Scarselli, M.; Comanducci, M.; Toneatto, D.; Masignani, V. Neisseria meningitidis factor h-binding protein fhbp: A key virulence factor and vaccine antigen. Expert Rev. Vaccines 2015, 14, 841–859. [Google Scholar] [CrossRef]
- Kim, B.J.; Shusta, E.V.; Doran, K.S. Past and current perspectives in modeling bacteria and blood brain barrier interactions. Front. Microbiol. 2019, 10, 1336. [Google Scholar] [CrossRef]
- Stephens, D.S. Biology and pathogenesis of the evolutionarily successful, obligate human bacterium neisseria meningitidis. Vaccine 2009, 27, B71–B77. [Google Scholar] [CrossRef]
- Gomes, S.F.M.; Westermann, A.J.; Sauerwein, T.; Hertlein, T.; Forstner, K.U.; Ohlsen, K.; Metzger, M.; Shusta, E.V.; Kim, B.J.; Appelt-Menzel, A.; et al. Induced pluripotent stem cell-derived brain endothelial cells as a cellular model to study neisseria meningitidis infection. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Nagele, V.; Heesemann, J.; Schielke, S.; Jimenez-Soto, L.F.; Kurzai, O.; Ackermann, N. Neisseria meningitidis adhesin nada targets beta1 integrins: Functional similarity to yersinia invasin. J. Biol. Chem. 2011, 286, 20536–20546. [Google Scholar] [CrossRef]
- Pizza, M.; Rappuoli, R. Neisseria meningitidis: Pathogenesis and immunity. Curr. Opin. Microbiol. 2015, 23, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Unkmeir, A.; Latsch, K.; Dietrich, G.; Wintermeyer, E.; Schinke, B.; Schwender, S.; Kim, K.S.; Eigenthaler, M.; Frosch, M. Fibronectin mediates opc-dependent internalization of neisseria meningitidis in human brain microvascular endothelial cells. Mol. Microbiol. 2002, 46, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Deghmane, A.E.; Giorgini, D.; Larribe, M.; Alonso, J.M.; Taha, M.K. Down-regulation of pili and capsule of neisseria meningitidis upon contact with epithelial cells is mediated by crga regulatory protein. Mol. Microbiol. 2002, 43, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.L.; Pelicic, V. Exceptionally widespread nanomachines composed of type iv pilins: The prokaryotic swiss army knives. Fems Microbiol. Rev. 2015, 39, 134–154. [Google Scholar] [CrossRef]
- Bernard, S.C.; Simpson, N.; Join-Lambert, O.; Federici, C.; Laran-Chich, M.P.; Maissa, N.; Bouzinba-Segard, H.; Morand, P.C.; Chretien, F.; Taouji, S.; et al. Pathogenic neisseria meningitidis utilizes cd147 for vascular colonization. Nat. Med. 2014, 20, 725–731. [Google Scholar] [CrossRef]
- Rudel, T.; Scheurerpflug, I.; Meyer, T.F. Neisseria pilc protein identified as type-4 pilus tip-located adhesin. Nature 1995, 373, 357–359. [Google Scholar] [CrossRef]
- Nassif, X.; Beretti, J.L.; Lowy, J.; Stenberg, P.; O’Gaora, P.; Pfeifer, J.; Normark, S.; So, M. Roles of pilin and pilc in adhesion of neisseria meningitidis to human epithelial and endothelial cells. Proc. Natl. Acad. Sci. USA 1994, 91, 3769–3773. [Google Scholar] [CrossRef]
- Morand, P.C.; Tattevin, P.; Eugene, E.; Beretti, J.L.; Nassif, X. The adhesive property of the type iv pilus-associated component pilc1 of pathogenic neisseria is supported by the conformational structure of the n-terminal part of the molecule. Mol. Microbiol. 2001, 40, 846–856. [Google Scholar] [CrossRef]
- Virji, M. Pathogenic neisseriae: Surface modulation, pathogenesis and infection control. Nat. Rev. Microbiol. 2009, 7, 274–286. [Google Scholar] [CrossRef]
- Carbonnelle, E.; Hill, D.J.; Morand, P.; Griffiths, N.J.; Bourdoulous, S.; Murillo, I.; Nassif, X.; Virji, M. Meningococcal interactions with the host. Vaccine 2009, 27, B78–B89. [Google Scholar] [CrossRef]
- Virji, M.; Makepeace, K.; Ferguson, D.J.; Achtman, M.; Moxon, E.R. Meningococcal opa and opc proteins: Their role in colonization and invasion of human epithelial and endothelial cells. Mol. Microbiol. 1993, 10, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Gray-Owen, S.D. Neisserial opa proteins: Impact on colonization, dissemination and immunity. Scand. J. Infect. Dis. 2003, 35, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Sa, E.C.C.; Griffiths, N.J.; Virji, M. Neisseria meningitidis opc invasin binds to the sulphated tyrosines of activated vitronectin to attach to and invade human brain endothelial cells. PLoS Pathog 2010, 6, e1000911. [Google Scholar]
- Simonis, A.; Hebling, S.; Gulbins, E.; Schneider-Schaulies, S.; Schubert-Unkmeir, A. Differential activation of acid sphingomyelinase and ceramide release determines invasiveness of neisseria meningitidis into brain endothelial cells. PLoS Path. 2014, 10, e1004160. [Google Scholar] [CrossRef] [PubMed]
- Metruccio, M.M.; Pigozzi, E.; Roncarati, D.; Berlanda Scorza, F.; Norais, N.; Hill, S.A.; Scarlato, V.; Delany, I. A novel phase variation mechanism in the meningococcus driven by a ligand-responsive repressor and differential spacing of distal promoter elements. PLoS Pathog 2009, 5, e1000710. [Google Scholar] [CrossRef] [PubMed]
- Comanducci, M.; Bambini, S.; Caugant, D.A.; Mora, M.; Brunelli, B.; Capecchi, B.; Ciucchi, L.; Rappuoli, R.; Pizza, M. Nada diversity and carriage in neisseria meningitidis. Infect. Immun. 2004, 72, 4217–4223. [Google Scholar] [CrossRef] [PubMed]
- Capecchi, B.; Adu-Bobie, J.; Di Marcello, F.; Ciucchi, L.; Masignani, V.; Taddei, A.; Rappuoli, R.; Pizza, M.; Arico, B. Neisseria meningitidis nada is a new invasin which promotes bacterial adhesion to and penetration into human epithelial cells. Mol. Microbiol. 2005, 55, 687–698. [Google Scholar] [CrossRef]
- Scietti, L.; Sampieri, K.; Pinzuti, I.; Bartolini, E.; Benucci, B.; Liguori, A.; Haag, A.F.; Lo Surdo, P.; Pansegrau, W.; Nardi-Dei, V.; et al. Exploring host-pathogen interactions through genome wide protein microarray analysis. Sci. Rep. 2016, 6, 27996. [Google Scholar] [CrossRef]
- Simonis, A.; Schubert-Unkmeir, A. Interactions of meningococcal virulence factors with endothelial cells at the human blood-cerebrospinal fluid barrier and their role in pathogenicity. Febs Lett. 2016, 590, 3854–3867. [Google Scholar] [CrossRef]
- Lambotin, M.; Hoffmann, I.; Laran-Chich, M.P.; Nassif, X.; Couraud, P.O.; Bourdoulous, S. Invasion of endothelial cells by neisseria meningitidis requires cortactin recruitment by a phosphoinositide-3-kinase/rac1 signalling pathway triggered by the lipo-oligosaccharide. J. Cell Sci. 2005, 118, 3805–3816. [Google Scholar] [CrossRef]
- Soyer, M.; Charles-Orszag, A.; Lagache, T.; Machata, S.; Imhaus, A.F.; Dumont, A.; Millien, C.; Olivo-Marin, J.C.; Dumenil, G. Early sequence of events triggered by the interaction of neisseria meningitidis with endothelial cells. Cell. Microbiol. 2014, 16, 878–895. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Watanabe, H.; Kim, K.S.; Yokoyama, S.; Yanagisawa, T. The meningococcal cysteine transport system plays a crucial role in neisseria meningitidis survival in human brain microvascular endothelial cells. Mbio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Slanina, H.; Hebling, S.; Hauck, C.R.; Schubert-Unkmeir, A. Cell invasion by neisseria meningitidis requires a functional interplay between the focal adhesion kinase, src and cortactin. PLoS ONE 2012, 7, e39613. [Google Scholar] [CrossRef] [PubMed]
- Schubert-Unkmeir, A.; Sokolova, O.; Panzner, U.; Eigenthaler, M.; Frosch, M. Gene expression pattern in human brain endothelial cells in response to neisseria meningitidis. Infect. Immun. 2007, 75, 899–914. [Google Scholar] [CrossRef] [PubMed]
- Sa, E.C.C.; Griffiths, N.J.; Murillo, I.; Virji, M. Neisseria meningitidis opc invasin binds to the cytoskeletal protein alpha-actinin. Cell. Microbiol. 2009, 11, 389–405. [Google Scholar] [CrossRef]
- Coureuil, M.; Lecuyer, H.; Scott, M.G.H.; Boularan, C.; Enslen, H.; Soyer, M.; Mikaty, G.; Bourdoulous, S.; Nassif, X.; Marullo, S. Meningococcus hijacks a beta 2-adrenoceptor/beta-arrestin pathway to cross brain microvasculature endothelium. Cell 2010, 143, 1149–1160. [Google Scholar] [CrossRef]
- Schubert-Unkmeir, A.; Konrad, C.; Slanina, H.; Czapek, F.; Hebling, S.; Frosch, M. Neisseria meningitidis induces brain microvascular endothelial cell detachment from the matrix and cleavage of occludin: A role for mmp-8. PLoS Path. 2010, 6, e1000874. [Google Scholar] [CrossRef]
- Coureuil, M.; Mikaty, G.; Miller, F.; Lecuyer, H.; Bernard, C.; Bourdoulous, S.; Dumenil, G.; Mege, R.M.; Weksler, B.B.; Romero, I.A.; et al. Meningococcal type iv pili recruit the polarity complex to cross the brain endothelium. Science 2009, 325, 83–87. [Google Scholar] [CrossRef]
- Waage, A.; Brandtzaeg, P.; Halstensen, A.; Kierulf, P.; Espevik, T. The complex pattern of cytokines in serum from patients with meningococcal septic shock-association between interleukin-6, interleukin-1, and fatal outcome. J. Exp. Med. 1989, 169, 333–338. [Google Scholar] [CrossRef]
- Sokolova, O.; Heppel, N.; Jagerhuber, R.; Kim, K.S.; Frosch, M.; Eigenthaler, M.; Schubert-Unkmeir, A. Interaction of neisseria meningitidis with human brain microvascular endothelial cells: Role of map- and tyrosine kinases in invasion and inflammatory cytokine release. Cell. Microbiol. 2004, 6, 1153–1166. [Google Scholar] [CrossRef]
- Watt, J.P.; Wolfson, L.J.; O’Brien, K.L.; Henkle, E.; Deloria-Knoll, M.; McCall, N.; Lee, E.; Levine, O.S.; Hajjeh, R.; Mulholland, K.; et al. Burden of disease caused by haemophilus influenzae type b in children younger than 5 years: Global estimates. Lancet 2009, 374, 903–911. [Google Scholar] [CrossRef]
- Zarei, A.E.; Almehdar, H.A.; Redwan, E.M. Hib vaccines: Past, present, and future perspectives. J. Immunol. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Bakaletz, L.O.; Novotny, L.A. Nontypeable haemophilus influenzae (nthi). Trends Microbiol. 2018, 26, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Doran, K.S.; Banerjee, A.; Disson, O.; Lecuit, M. Concepts and mechanisms: Crossing host barriers. Cold Spring Harb. Perspect. Med. 2013, 3, a010090. [Google Scholar] [CrossRef]
- Van Ham, S.M.; van Alphen, L.; Mooi, F.R.; van Putten, J.P. Phase variation of h. Influenzae fimbriae: Transcriptional control of two divergent genes through a variable combined promoter region. Cell 1993, 73, 1187–1196. [Google Scholar] [CrossRef]
- Cundell, D.R.; Gerard, N.P.; Gerard, C.; Idanpaan-Heikkila, I.; Tuomanen, E.I. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature 1995, 377, 435–438. [Google Scholar] [CrossRef]
- Swords, W.E.; Ketterer, M.R.; Shao, J.; Campbell, C.A.; Weiser, J.N.; Apicella, M.A. Binding of the non-typeable haemophilus influenzae lipooligosaccharide to the paf receptor initiates host cell signalling. Cell. Microbiol. 2001, 3, 525–536. [Google Scholar] [CrossRef]
- Radin, J.N.; Orihuela, C.J.; Murti, G.; Guglielmo, C.; Murray, P.J.; Tuomanen, E.I. Beta-arrestin 1 participates in platelet-activating factor receptor-mediated endocytosis of streptococcus pneumoniae. Infect. Immun. 2005, 73, 7827–7835. [Google Scholar] [CrossRef]
- Caporarello, N.; Olivieri, M.; Cristaldi, M.; Scalia, M.; Toscano, M.A.; Genovese, C.; Addamo, A.; Salmeri, M.; Lupo, G.; Anfuso, C.D. Blood-brain barrier in a haemophilus influenzae type a in vitro infection: Role of adenosine receptors a(2a) and a(2b). Mol. Neurobiol. 2018, 55, 5321–5336. [Google Scholar] [CrossRef]
- Wispelwey, B.; Lesse, A.J.; Hansen, E.J.; Scheld, W.M. Haemophilus influenzae lipopolysaccharide-induced blood brain barrier permeability during experimental meningitis in the rat. J. Clin. Invest. 1988, 82, 1339–1346. [Google Scholar] [CrossRef]
- Wispelwey, B.; Hansen, E.J.; Scheld, W.M. Haemophilus influenzae outer membrane vesicle-induced blood-brain barrier permeability during experimental meningitis. Infect. Immun. 1989, 57, 2559–2562. [Google Scholar] [PubMed]
- Parisi, D.N.; Martinez, L.R. Intracellular haemophilus influenzae invades the brain: Is zyxin a critical blood brain barrier component regulated by tnf-alpha? Virulence 2014, 5, 645–647. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kostyanev, T.S.; Sechanova, L.P. Virulence factors and mechanisms of antibiotic resistance of haemophilus influenzae. Folia Med. 2012, 54, 19–23. [Google Scholar] [CrossRef]
- Yau, B.; Hunt, N.H.; Mitchell, A.J.; Too, L.K. Bloodbrain barrier pathology and cns outcomes in streptococcus pneumoniae meningitis. Int. J. Mol. Sci. 2018, 19, 3555. [Google Scholar] [CrossRef] [PubMed]
- Ferrieri, P.; Burke, B.; Nelson, J. Production of bacteremia and meningitis in infant rats with group-b streptococcal serotypes. Infect. Immun. 1980, 27, 1023–1032. [Google Scholar]
- Nassif, X.; Bourdoulous, S.; Eugene, E.; Couraud, P.O. How do extracellular pathogens cross the blood-brain barrier? Trends Microbiol. 2002, 10. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Pathogen | Entry Mechanisms | Major Virulence Factors | ||
|---|---|---|---|---|
| Gram-Positive | BBB | BCSFB | BBB | BCSFB |
| L. monocytogenes | Transcellular route [54] “Trojan horse” mechanism within leukocytes [54] Retrograde migration within axons of cranial nerves [54] | Transcellular route [57] | Major invasion protein InlB inducing receptor-mediated endocytosis [25] Bacterial surface protein InlF interacting with surface vimentin [58] Pore-forming cytolysin LLO inducing signaling pathways (NF-κB, MAPK) [64,66] ActA promoting F-actin-based intracellular motility [68] | Major invasion proteins InlA and InlB inducing receptor-mediated endocytosis [34,57] |
| S. suis | Invasion at low rates in porcine models [73,78]. | Invasion demonstrated for porcine and human in vitro models [48,72] Possibly “Trojan-horse” mechanism [85] | Enolase increasing BBB permeability [80] Suilysin inducing pore formation in membranes [81] | Regulation of capsule expression [48,72] |
| S. pneumoniae | Translocation across BBB in vivo and in vitro [87,194] | Only attachment observed in an in vivo mouse model during late stages of infection with high levels of bacteremia [87]. | Altered expression of the capsule for attachment [92,93] Interaction with BBB through NadA [96] Pore-forming toxin pneumolysin [101] | |
| GBS | Traversal of BBB in vivo and in vitro [49,109,195] | Expression of cell-wall anchored pili [111] PilA: promoting attachment of GBS [112] PilB: mediating internalization [112] | ||
| Gram-Negative | BBB | BCSFB | BBB | BCSFB |
| E. coli | Traversal of BBB in vivo and in vitro [22] | Traversal of BCSFB in vitro [121] | Attachent facilitated by type 1 fimbriae and OmpA [125,126] Invasion induced by IbeA [127] and CNF1 [128] Intracellular survival promoted by the E. coli K1 capsule [136] | Role of fimH during adhesion [121] Involvement of OmpA, FimH and IbeA in invasion [121] |
| N. meningitidi | Traversal of BBB in vivo and in vitro [2,196] | Traversal of BCSFB in vitro of choroid plexus epithelial cells [48] Invasion of outer BCSFB in induced pluripotent stem cell-derived brain endothelial cells [149] | Protective function of the polysaccharide capsule during bloodstream survival but attenuated tissue invasion [151] Adherence through pili and surface exposed proteins [154] Invasion is facilitated by Opa and Opc [160] | Capsule attenuates invasion in vitro [48] |
| H. influenzae | Traversal of BBB in vitro [51] | Traversal of BCSFB in vitro [47] | Entry via binding of PAFR [186,187] Attachment facilitated by binding of the laminin receptor [99] | Capsule and fimbriae attenuate invasion [47] Invasion if H. influenzae was observed as intracellular bacterium [47] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herold, R.; Schroten, H.; Schwerk, C. Virulence Factors of Meningitis-Causing Bacteria: Enabling Brain Entry across the Blood–Brain Barrier. Int. J. Mol. Sci. 2019, 20, 5393. https://doi.org/10.3390/ijms20215393
Herold R, Schroten H, Schwerk C. Virulence Factors of Meningitis-Causing Bacteria: Enabling Brain Entry across the Blood–Brain Barrier. International Journal of Molecular Sciences. 2019; 20(21):5393. https://doi.org/10.3390/ijms20215393
Chicago/Turabian StyleHerold, Rosanna, Horst Schroten, and Christian Schwerk. 2019. "Virulence Factors of Meningitis-Causing Bacteria: Enabling Brain Entry across the Blood–Brain Barrier" International Journal of Molecular Sciences 20, no. 21: 5393. https://doi.org/10.3390/ijms20215393
APA StyleHerold, R., Schroten, H., & Schwerk, C. (2019). Virulence Factors of Meningitis-Causing Bacteria: Enabling Brain Entry across the Blood–Brain Barrier. International Journal of Molecular Sciences, 20(21), 5393. https://doi.org/10.3390/ijms20215393