Neutrophil-Derived Microvesicle Induced Dysfunction of Brain Microvascular Endothelial Cells In Vitro

 ,
,
Abstract
1. Introduction
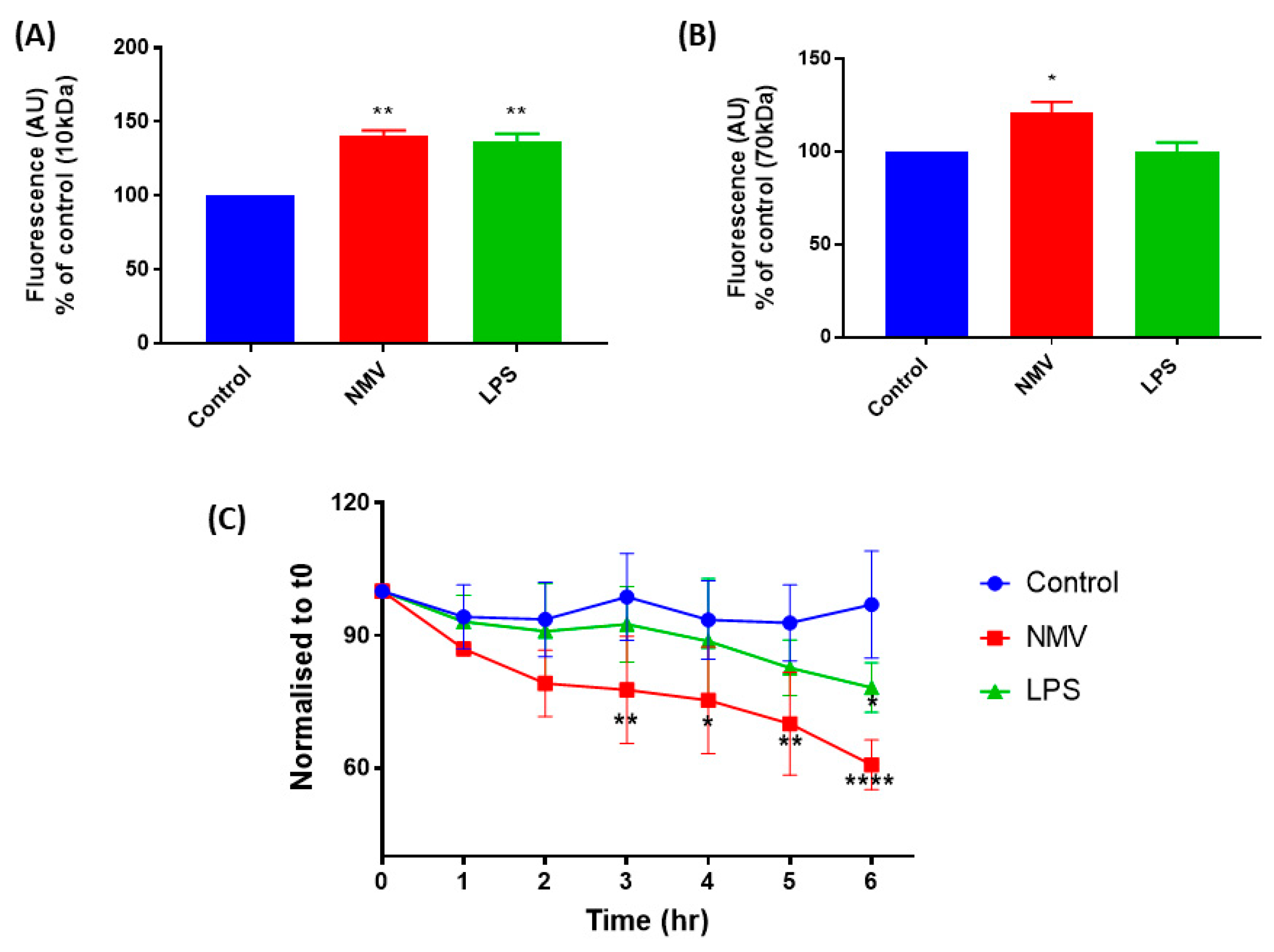
2. Results
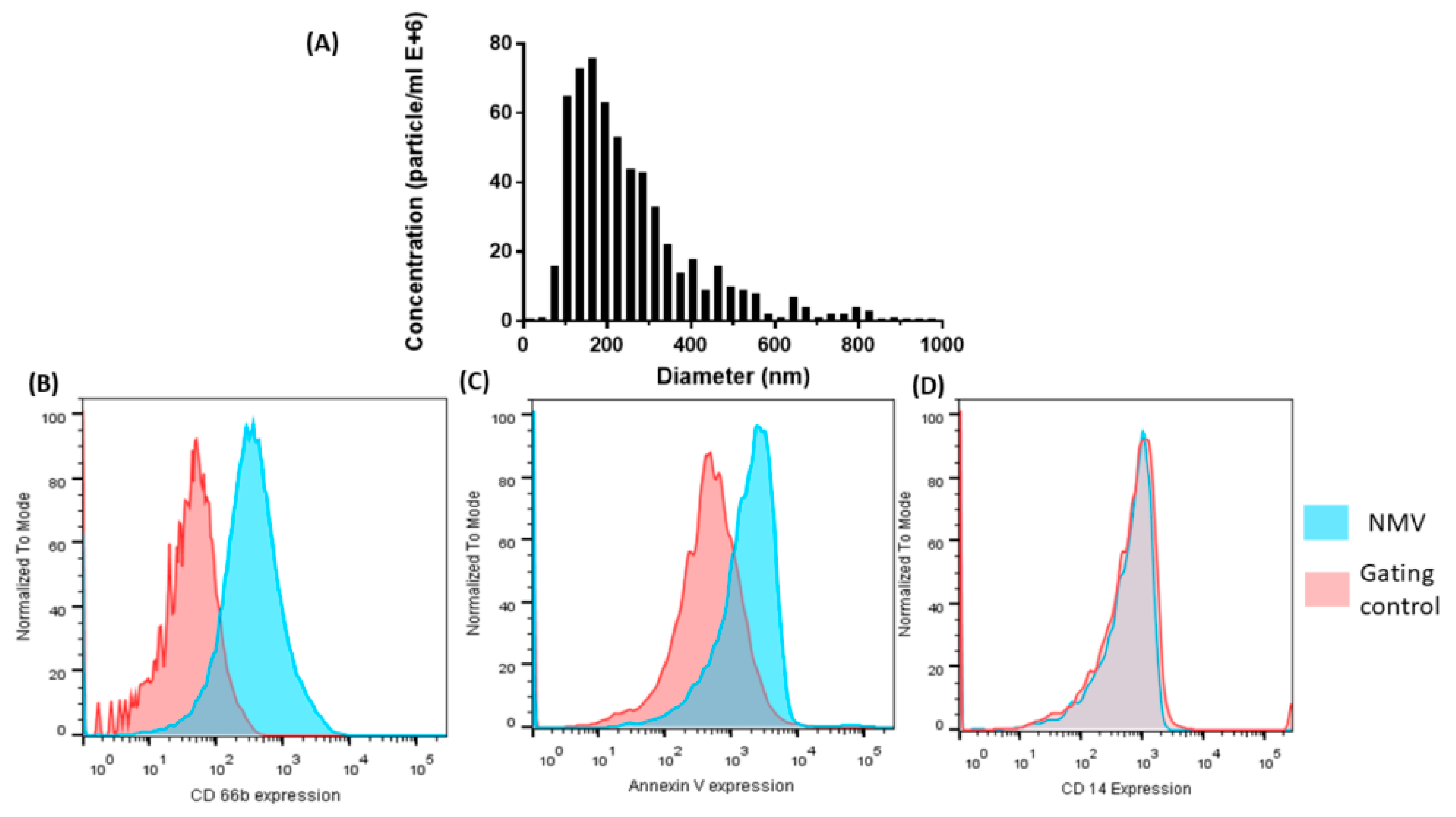
2.1. Characterization of NMV
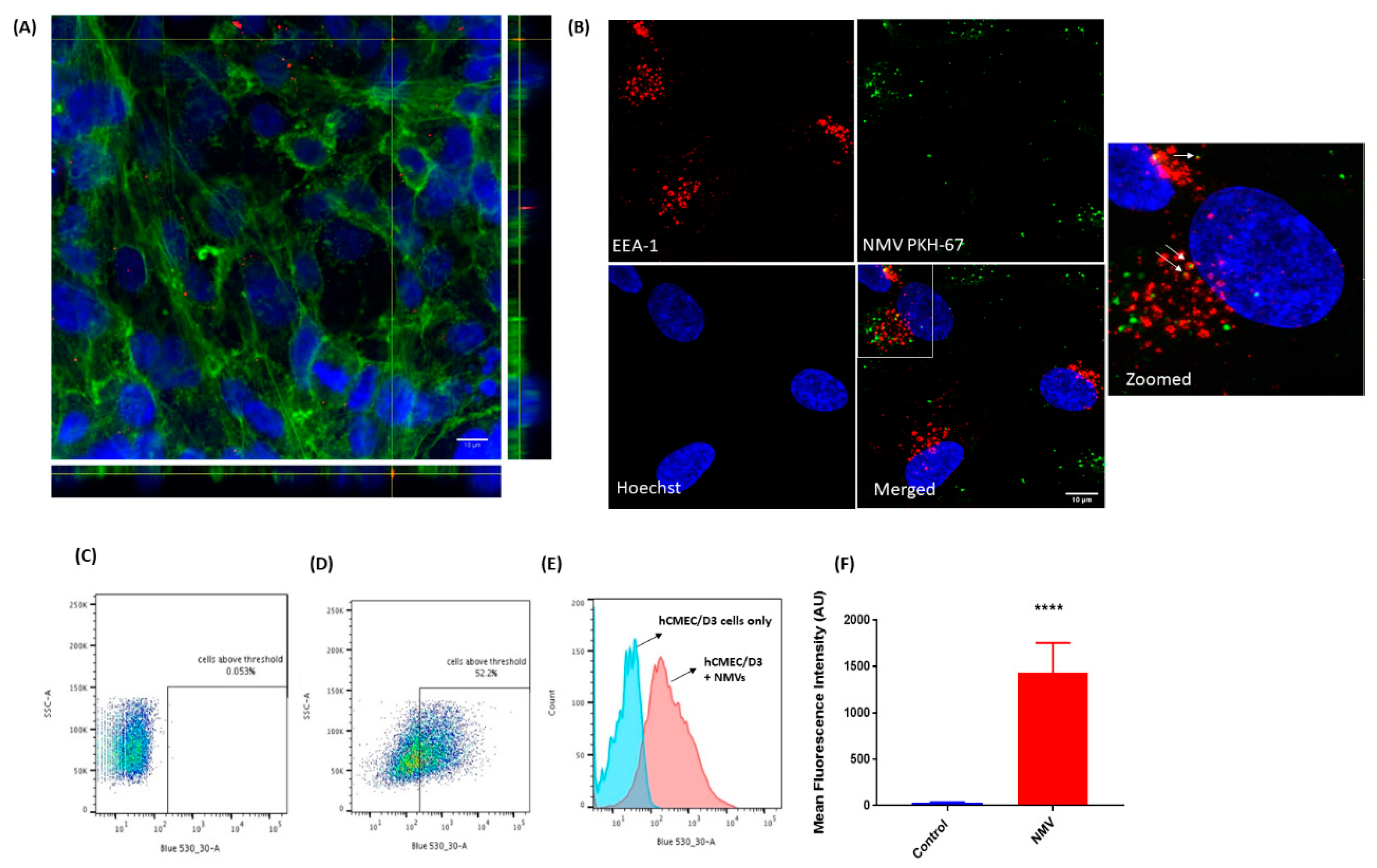
2.2. NMV Are Internalised by Human Brain Endothelial Cells
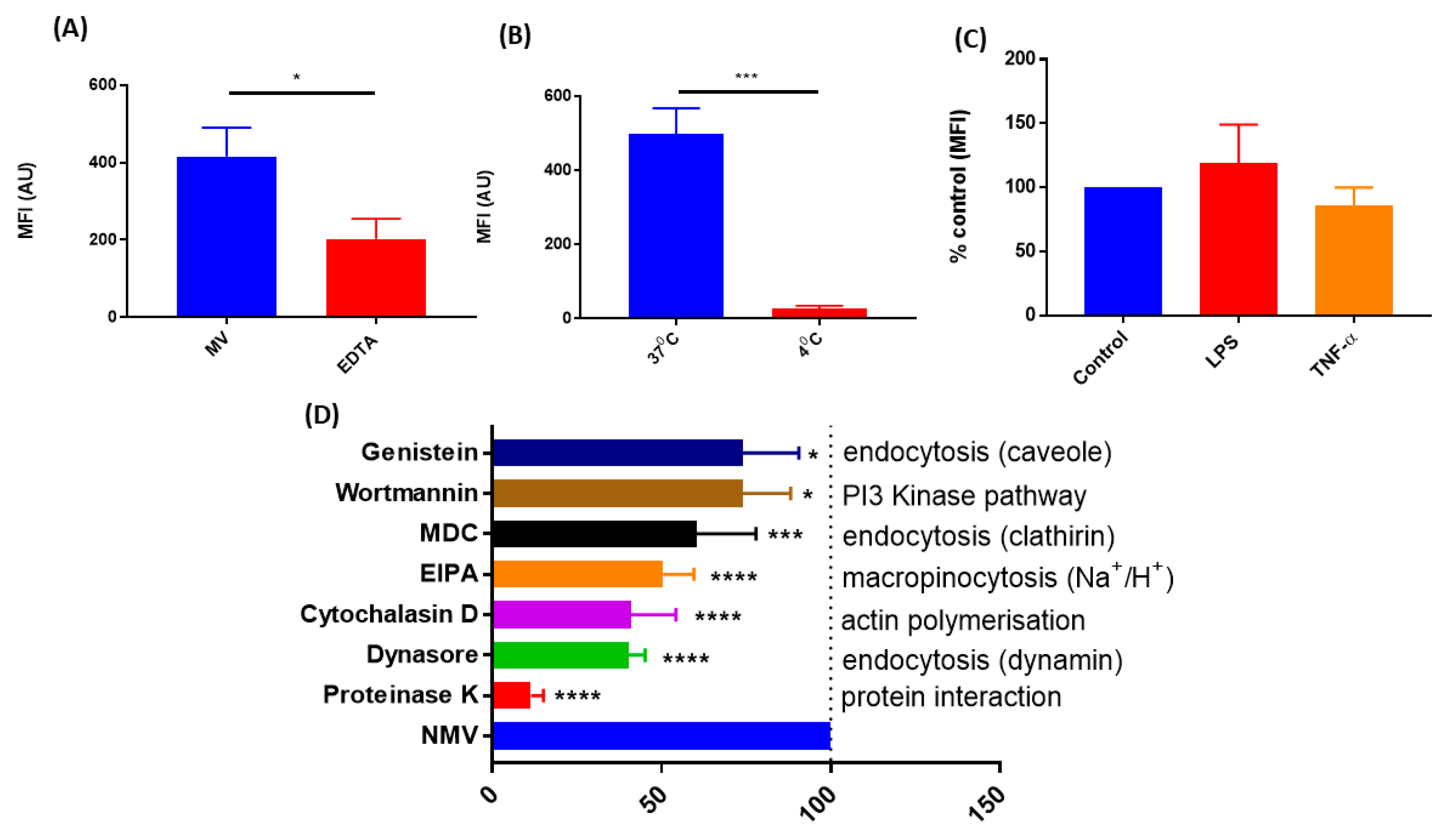
2.3. The Internalization of NMV Occurs via Multiple Pathways
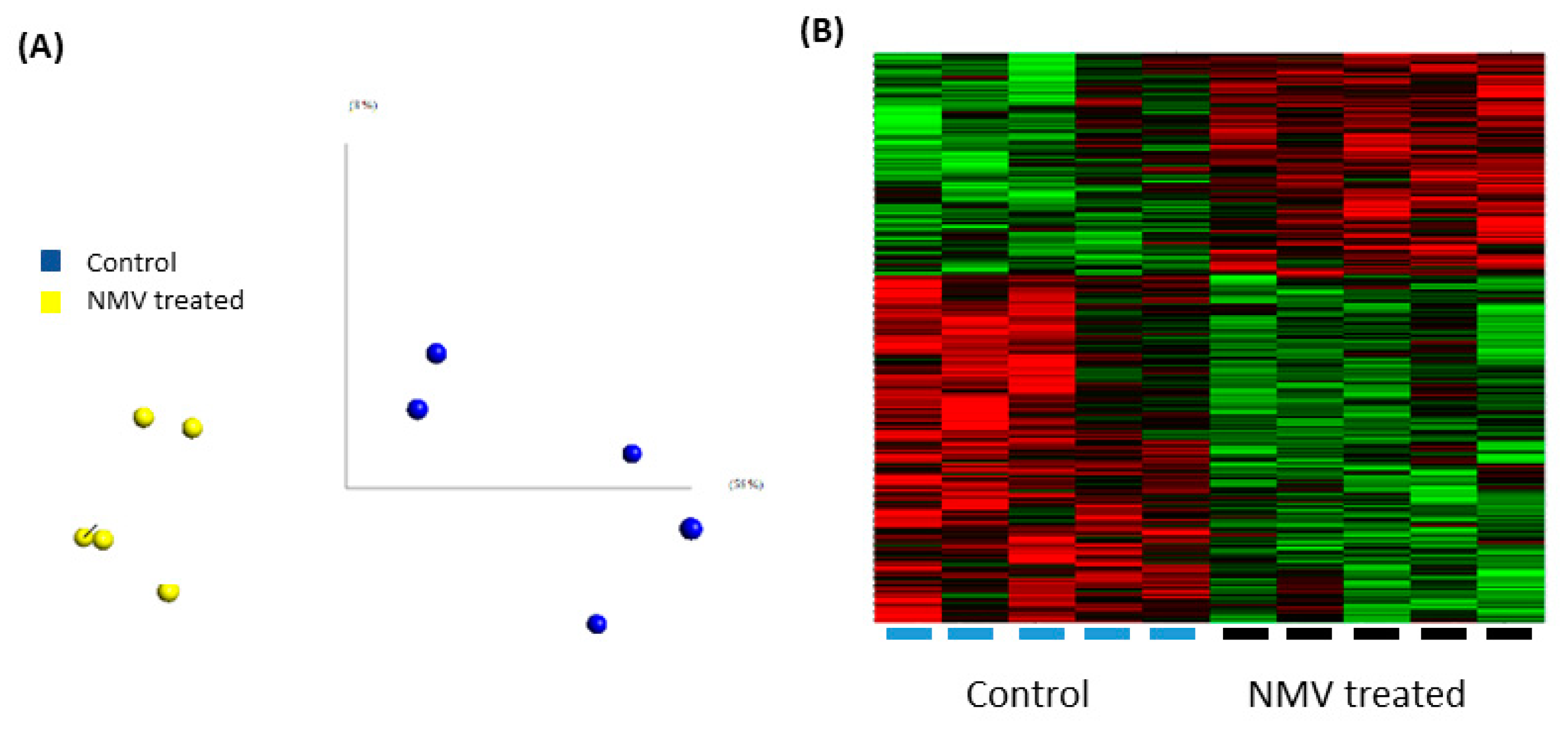
2.4. Internalization of NMV Impacts the Transcriptomic Profile of Brain Endothelial Cells
2.5. NMV Increase the Permeability and Decrease the Transendothelial Electrical Resistance of hCMEC/D3
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Neutrophil-Derived Microvesicle Isolation
4.3. Flow Cytometric Analysis
4.4. Nanoparticle Tracking Analysis
4.5. Neutrophil Microvesicle Internalization by Endothelial Cells
4.6. Analysis of Internalization Pathways
4.7. RNA Extraction and Amplification
4.8. Microarray Amplification and Microarray Hybridization
4.9. Microarray Analysis
4.10. Permeability Assay
4.11. Transendothelial Electrical Resistance (TEER)
4.12. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| BBB | Blood brain barrier |
| BMEC | Brain microvascular endothelial cells |
| CNS | Central nervous system |
| EDTA | Ethylenediaminetetraacetic Acid |
| EEA-1 | Early endosomal antigen-1 |
| EIPA | 5-(N-Ethyl-N-isopropyl)amiloride |
| fMLP | N-formylmethionine-leucyl-phenylalanine |
| HCAEC | Human coronary arterial endothelial cells |
| HUVEC | Human umbilical vein endothelial cells |
| ICAM-1 | Intercellular adhesion molecule-1 |
| LPS | Lipopolysachharide |
| MDC | Monodansylcadaverine |
| MFI | Mean Fluorescent Intensity |
| MV | Microvesicles |
| NMV | Neutrophil derived microvesicles |
| NVU | Neuro vascular unit |
| TEER | Tranendothelial electrical resistance |
| TJ | Tight junction |
| VaD | Vascular Dementia |
References
- Cai, Z.; Qiao, P.-F.; Wan, C.-Q.; Cai, M.; Zhou, N.-K.; Li, Q. Role of Blood-Brain Barrier in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 63, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Cao, Y.; Ma, L.; Pei, H.; Rausch, W.D.; Li, H. Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia. Front. Aging Neurosci. 2018, 10, 376. [Google Scholar] [CrossRef] [PubMed]
- Minagar, A.; Alexander, J.S. Blood-brain barrier disruption in multiple sclerosis. Mult. Scler. 2003, 9, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef]
- Cunningham, C.; Hennessy, E. Co-morbidity and systemic inflammation as drivers of cognitive decline: New experimental models adopting a broader paradigm in dementia research. Alzheimers Res. Ther. 2015. [Google Scholar] [CrossRef]
- Lim, A.; Krajina, K.; Marsland, A.L. Peripheral Inflammation and Cognitive Aging. In Modern Trends in Psychiatry; Karger Publishers: Basel, Switzerland, 2013; Volume 28, pp. 175–187. ISBN 2504-0464. [Google Scholar]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef]
- Gupta, A.; Iadecola, C. Impaired Aβ clearance: A potential link between atherosclerosis and Alzheimer’s disease. Front. Aging Neurosci. 2015, 7, 115. [Google Scholar] [CrossRef]
- Yarchoan, M.; Xie, S.X.; Kling, M.A.; Toledo, J.B.; Wolk, D.A.; Lee, E.B.; Van Deerlin, V.; Lee, V.M.-Y.; Trojanowski, J.Q.; Arnold, S.E. Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain 2012, 135, 3749–3756. [Google Scholar] [CrossRef]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef]
- Ide, M.; Harris, M.; Stevens, A.; Sussams, R.; Hopkins, V.; Culliford, D.; Fuller, J.; Ibbett, P.; Raybould, R.; Thomas, R. Periodontitis and Cognitive Decline in Alzheimer’s Disease. PLoS ONE 2016, 11, e0151081. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.-S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Semmler, A.; Okulla, T.; Sastre, M.; Dumitrescu-Ozimek, L.; Heneka, M.T. Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J. Chem. Neuroanat. 2005, 30, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Nishioku, T.; Dohgu, S.; Takata, F.; Eto, T.; Ishikawa, N.; Kodama, K.B.; Nakagawa, S.; Yamauchi, A.; Kataoka, Y. Detachment of brain pericytes from the basal lamina is involved in disruption of the blood-brain barrier caused by lipopolysaccharide-induced sepsis in mice. Cell. Mol. Neurobiol. 2009, 29, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Irwin, M.G.; Wong, G.T.C.; Chang, R.C.C. Evidence of the impact of systemic inflammation on neuroinflammation from a non-bacterial endotoxin animal model. J. Neuroinflammation 2018, 15, 147. [Google Scholar] [CrossRef] [PubMed]
- Hoogland, I.C.M.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflammation 2015, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef]
- Shad, K.F.; Aghazadeh, Y.; Ahmad, S.; Kress, B. Peripheral markers of Alzheimer’s disease: Surveillance of white blood cells. Synapse 2013, 67, 541–543. [Google Scholar] [CrossRef]
- Dong, Y.; Lagarde, J.; Xicota, L.; Corne, H.; Chantran, Y.; Chaigneau, T.; Crestani, B.; Bottlaender, M.; Potier, M.-C.; Aucouturier, P.; et al. Neutrophil hyperactivation correlates with Alzheimer’s disease progression. Ann. Neurol. 2018, 83, 387–405. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Clancy, J.W.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123, 1603–1611. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Nieuwland, R.; Berckmans, R.J.; McGregor, S.; Boing, A.N.; Romijn, F.P.; Westendorp, R.G.; Hack, C.E.; Sturk, A. Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood 2000, 95, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Prakash, P.S.; Caldwell, C.C.; Lentsch, A.B.; Pritts, T.A.; Robinson, B.R.H. Human microparticles generated during sepsis in patients with critical illness are neutrophil-derived and modulate the immune response. J. Trauma Acute Care Surg. 2012, 73, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Chironi, G.; Simon, A.; Hugel, B.; Del Pino, M.; Gariepy, J.; Freyssinet, J.-M.; Tedgui, A. Circulating leukocyte-derived microparticles predict subclinical atherosclerosis burden in asymptomatic subjects. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2775–2780. [Google Scholar] [CrossRef]
- Gasser, O.; Schifferli, J.A. Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood 2004, 104, 2543–2548. [Google Scholar] [CrossRef]
- Hess, C.; Sadallah, S.; Hefti, A.; Landmann, R.; Schifferdi, J.A. Ectosomes released by human neutrophils are specialized functional units. Mol. Immunol. 1998, 6, 354. [Google Scholar] [CrossRef]
- Hong, Y.; Eleftheriou, D.; Hussain, A.A.K.; Price-Kuehne, F.E.; Savage, C.O.; Jayne, D.; Little, M.A.; Salama, A.D.; Klein, N.J.; Brogan, P.A. Anti-neutrophil cytoplasmic antibodies stimulate release of neutrophil microparticles. J. Am. Soc. Nephrol. 2012, 23, 49–62. [Google Scholar] [CrossRef]
- Jaffe, E.A.; Nachman, R.L.; Becker, C.G.; Minick, C.R. Culture of Human Endothelial Cells Derived from Umbilical Veins. Identification by Morphologic and Immunologic Criteria. J. Clin. Invest. 1973, 52, 2745–2756. [Google Scholar] [CrossRef]
- Gomez, I.; Ward, B.; Souilhol, C.; Recarti, C.; Ariaans, M.; Johnston, J.; Burnett, A.; Mahmoud, M.; Luong, L.A.; West, L.; et al. Neutrophil microvesicles drive atherosclerosis by delivering miR-155 to atheroprone endothelium. bioRxiv 2018, 319392. [Google Scholar] [CrossRef]
- Dalli, J.; Norling, L.V.; Renshaw, D.; Cooper, D.; Leung, K.-Y.; Perretti, M. Annexin 1 mediates the rapid anti-inflammatory effects of neutrophil-derived microparticles. Blood 2008, 112, 2512–2519. [Google Scholar] [CrossRef]
- Rhys, H.I.; Dell’Accio, F.; Pitzalis, C.; Moore, A.; Norling, L.V.; Perretti, M. Neutrophil Microvesicles from Healthy Control and Rheumatoid Arthritis Patients Prevent the Inflammatory Activation of Macrophages. EBioMedicine 2018, 29, 60–69. [Google Scholar] [CrossRef]
- Pitanga, T.N.; de Aragão França, L.; Rocha, V.C.J.; Meirelles, T.; Borges, V.M.; Gonçalves, M.S.; Pontes-de-Carvalho, L.C.; Noronha-Dutra, A.A.; dos-Santos, W.L.C. Neutrophil-derived microparticles induce myeloperoxidase-mediated damage of vascular endothelial cells. BMC Cell Biol. 2014, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Gyorgy, B.; Szabo, T.G.; Pasztoi, M.; Pal, Z.; Misjak, P.; Aradi, B.; Laszlo, V.; Pallinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.; Dorovini-Zis, K.; Vincent, S.R. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood–brain barrier. Exp. Neurol. 2004, 190, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, P.J.; de Boer, A.; Bert, G.; Breimer, D.D. Pharmacological investigations on lipopolysaccharide-induced permeability changes in the blood–brain barrier in vitro. Microvasc. Res. 2003, 65, 24–31. [Google Scholar] [CrossRef]
- Marsland, A.L.; Gianaros, P.J.; Kuan, D.C.-H.; Sheu, L.K.; Krajina, K.; Manuck, S.B. Brain morphology links systemic inflammation to cognitive function in midlife adults. Brain. Behav. Immun. 2015, 48, 195–204. [Google Scholar] [CrossRef]
- King, E.; O’Brien, J.T.; Donaghy, P.; Morris, C.; Barnett, N.; Olsen, K.; Martin-Ruiz, C.; Taylor, J.-P.; Thomas, A.J. Peripheral inflammation in prodromal Alzheimer’s and Lewy body dementias. J. Neurol. Neurosurg. Psychiatry 2018, 89, 339–345. [Google Scholar] [CrossRef]
- Teixeira, F.B.; Saito, M.T.; Matheus, F.C.; Prediger, R.D.; Yamada, E.S.; Maia, C.S.F.; Lima, R.R. Periodontitis and Alzheimer’s Disease: A Possible Comorbidity between Oral Chronic Inflammatory Condition and Neuroinflammation. Front. Aging Neurosci. 2017, 9, 327. [Google Scholar] [CrossRef]
- Le Page, A.; Dupuis, G.; Frost, E.H.; Pawelec, G.P.; Witkowski, J.M.; Larbi, A.; Fülöp, T. Role of the Innate Immune Response in the Progression of Alzheimer’s Disease. Innov. Aging 2017, 1, 1151. [Google Scholar] [CrossRef][Green Version]
- Pliyev, B.K.; Kalintseva, M.V.; Abdulaeva, S.V.; Yarygin, K.N.; Savchenko, V.G. Neutrophil microparticles modulate cytokine production by natural killer cells. Cytokine 2014, 65, 126–129. [Google Scholar] [CrossRef]
- Nolan, S.; Dixon, R.; Norman, K.; Hellewell, P.; Ridger, V. Nitric Oxide Regulates Neutrophil Migration through Microparticle Formation. Am. J. Pathol. 2008, 172, 265–273. [Google Scholar] [CrossRef]
- Porro, C.; Lepore, S.; Trotta, T.; Castellani, S.; Ratclif, L.; Battaglino, A.; Di Gioia, S.; Martínez, M.C.; Conese, M.; Maffione, A.B. Isolation and characterization of microparticles in sputum from cystic fibrosis patients. Respir. Res. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Montero-Melendez, T.; Norling, L.V.; Yin, X.; Hinds, C.; Haskard, D.; Mayr, M.; Perretti, M. Heterogeneity in neutrophil microparticles reveals distinct proteome and functional properties. Mol. Cell. Proteomics 2013, 12, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Haynes, C.L. On-chip evaluation of neutrophil activation and neutrophil-endothelial cell interaction during neutrophil chemotaxis. Anal. Chem. 2013, 85, 10787–10796. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, Y.; Liu, Y.; Dai, X.; Li, W.; Cai, X.; Yin, Y.; Wang, Q.; Xue, Y.; Wang, C. Microvesicle-mediated transfer of microRNA-150 from monocytes to endothelial cells promotes angiogenesis. J. Biol. Chem. 2013, 288, 23586–23596. [Google Scholar] [CrossRef]
- Faille, D.; El-Assaad, F.; Mitchell, A.J.; Alessi, M.-C.; Chimini, G.; Fusai, T.; Grau, G.E.; Combes, V. Endocytosis and intracellular processing of platelet microparticles by brain endothelial cells. J. Cell. Mol. Med. 2012, 16, 1731–1738. [Google Scholar] [CrossRef]
- Kawamoto, T.; Ohga, N.; Akiyama, K.; Hirata, N.; Kitahara, S.; Maishi, N.; Osawa, T.; Yamamoto, K.; Kondoh, M.; Shindoh, M.; et al. Tumor-Derived Microvesicles Induce Proangiogenic Phenotype in Endothelial Cells via Endocytosis. PLoS ONE 2012, 7, e34045. [Google Scholar] [CrossRef]
- Fitzner, D.; Schnaars, M.; van Rossum, D.; Krishnamoorthy, G.; Dibaj, P.; Bakhti, M.; Regen, T.; Hanisch, U.-K.; Simons, M. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 2011, 124, 447–458. [Google Scholar] [CrossRef]
- Wilson, J.M.; de Hoop, M.; Zorzi, N.; Toh, B.H.; Dotti, C.G.; Parton, R.G. EEA1, a tethering protein of the early sorting endosome, shows a polarized distribution in hippocampal neurons, epithelial cells, and fibroblasts. Mol. Biol. Cell 2000, 11, 2657–2671. [Google Scholar] [CrossRef]
- Schneider, D.J.; Speth, J.M.; Penke, L.R.; Wettlaufer, S.H.; Swanson, J.A.; Peters-Golden, M. Mechanisms and modulation of microvesicle uptake in a model of alveolar cell communication. J. Biol. Chem. 2017, 292, 20897–20910. [Google Scholar] [CrossRef]
- Faille, D.; Combes, V.; Mitchell, A.J.; Fontaine, A.; Juhan-Vague, I.; Alessi, M.-C.; Chimini, G.; Fusai, T.; Grau, G.E. Platelet microparticles: A new player in malaria parasite cytoadherence to human brain endothelium. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 3449–3458. [Google Scholar] [CrossRef]
- Cvjetkovic, A.; Jang, S.C.; Konečná, B.; Höög, J.L.; Sihlbom, C.; Lässer, C.; Lötvall, J. Detailed Analysis of Protein Topology of Extracellular Vesicles–Evidence of Unconventional Membrane Protein Orientation. Sci. Rep. 2016, 6, 36338. [Google Scholar] [CrossRef]
- Yuan, D.; Zhao, Y.; Banks, W.A.; Bullock, K.M.; Haney, M.; Batrakova, E.; Kabanov, A. V Macrophage exosomes as natural nanocarriers for protein delivery to inflamed brain. Biomaterials 2017, 142, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lamaze, C.; Fujimoto, L.M.; Yin, H.L.; Schmid, S.L. The actin cytoskeleton is required for receptor-mediated endocytosis in mammalian cells. J. Biol. Chem. 1997, 272, 20332–20335. [Google Scholar] [CrossRef] [PubMed]
- Macia, E.; Ehrlich, M.; Massol, R.; Boucrot, E.; Brunner, C.; Kirchhausen, T. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 2006, 10, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.A.; Vanhecke, D.; Michen, B.; Blank, F.; Gehr, P.; Petri-Fink, A.; Rothen-Rutishauser, B. Different endocytotic uptake mechanisms for nanoparticles in epithelial cells and macrophages. Beilstein J. Nanotechnol. 2014, 5, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Koivusalo, M.; Welch, C.; Hayashi, H.; Scott, C.C.; Kim, M.; Alexander, T.; Touret, N.; Hahn, K.M.; Grinstein, S. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol. 2010, 188, 547–563. [Google Scholar] [CrossRef]
- Sun, X.-Y.; Gan, Q.-Z.; Ouyang, J.-M. Size-dependent cellular uptake mechanism and cytotoxicity toward calcium oxalate on Vero cells. Sci. Rep. 2017, 7, 41949. [Google Scholar] [CrossRef]
- Rejman, J.; Oberle, V.; Zuhorn, I.S.; Hoekstra, D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem. J. 2004, 377, 159–169. [Google Scholar] [CrossRef]
- Morelli, A.E.; Larregina, A.T.; Shufesky, W.J.; Sullivan, M.L.G.; Stolz, D.B.; Papworth, G.D.; Zahorchak, A.F.; Logar, A.J.; Wang, Z.; Watkins, S.C. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood 2004, 104, 3257–3266. [Google Scholar] [CrossRef]
- Mesri, M.; Altieri, D.C. Endothelial cell activation by leukocyte microparticles. J. Immunol. 1998, 161, 4382–4387. [Google Scholar]
- Mesri, M.; Altieri, D.C. Leukocyte microparticles stimulate endothelial cell cytokine release and tissue factor induction in a JNK1 signaling pathway. J. Biol. Chem. 1999, 274, 23111–23118. [Google Scholar] [CrossRef] [PubMed]
- Eken, C.; Martin, P.J.; Sadallah, S.; Treves, S.; Schaller, M.; Schifferli, J.A. Ectosomes released by polymorphonuclear neutrophils induce a MerTK-dependent anti-inflammatory pathway in macrophages. J. Biol. Chem. 2010, 285, 39914–39921. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Goudeva, L.; Immenschuh, S.; Schambach, A.; Skokowa, J.; Eiz-Vesper, B.; Blasczyk, R.; Figueiredo, C. miR-155 is associated with the leukemogenic potential of the class IV granulocyte colony-stimulating factor receptor in CD34+ progenitor cells. Mol. Med. 2015, 20, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Walshe, T.E.; Saint-Geniez, M.; Maharaj, A.S.R.; Sekiyama, E.; Maldonado, A.E.; D’Amore, P.A. TGF-beta is required for vascular barrier function, endothelial survival and homeostasis of the adult microvasculature. PLoS ONE 2009, 4, e5149. [Google Scholar] [CrossRef] [PubMed]
- Yeretssian, G.; Correa, R.G.; Doiron, K.; Fitzgerald, P.; Dillon, C.P.; Green, D.R.; Reed, J.C.; Saleh, M. Non-apoptotic role of BID in inflammation and innate immunity. Nature 2011, 474, 96. [Google Scholar] [CrossRef]
- Korsmeyer, S.J.; Wei, M.C.; Saito, M.; Weiler, S.; Oh, K.J.; Schlesinger, P.H. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000, 7, 1166–1173. [Google Scholar] [CrossRef]
- Winter-Vann, A.M.; Johnson, G.L. Integrated activation of MAP3Ks balances cell fate in response to stress. J. Cell. Biochem. 2007, 102, 848–858. [Google Scholar] [CrossRef]
- Edrissi, H.; Schock, S.C.; Hakim, A.M.; Thompson, C.S. Microparticles generated during chronic cerebral ischemia increase the permeability of microvascular endothelial barriers in vitro. Brain Res. 2016, 1634, 83–93. [Google Scholar] [CrossRef]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome Delivered Anticancer Drugs Across the Blood-Brain Barrier for Brain Cancer Therapy in Danio Rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef]
- Eigenmann, D.E.; Xue, G.; Kim, K.S.; Moses, A.V.; Hamburger, M.; Oufir, M. Comparative study of four immortalized human brain capillary endothelial cell lines, hCMEC/D3, hBMEC, TY10, and BB19, and optimization of culture conditions, for an in vitro blood-brain barrier model for drug permeability studies. Fluids Barriers CNS 2013, 10, 33. [Google Scholar] [CrossRef]
- Weksler, B.; Romero, I.A.; Couraud, P.-O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 2013, 10, 16. [Google Scholar] [CrossRef]
- Butt, A.M.; Jones, H.C.; Abbott, N.J. Electrical resistance across the blood-brain barrier in anaesthetized rats: A developmental study. J. Physiol. 1990, 429, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Hatherell, K.; Couraud, P.-O.; Romero, I.A.; Weksler, B.; Pilkington, G.J. Development of a three-dimensional, all-human in vitro model of the blood–brain barrier using mono-, co-, and tri-cultivation Transwell models. J. Neurosci. Methods 2011, 199, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Cucullo, L.; Couraud, P.-O.; Weksler, B.; Romero, I.-A.; Hossain, M.; Rapp, E.; Janigro, D. Immortalized human brain endothelial cells and flow-based vascular modeling: A marriage of convenience for rational neurovascular studies. J. Cereb. Blood Flow Metab. 2008, 28, 312–328. [Google Scholar] [CrossRef]
- Papadia, K.; Markoutsa, E.; Antimisiaris, S.G. How do the physicochemical properties of nanoliposomes affect their interactions with the hCMEC/D3 cellular model of the BBB? Int. J. Pharm. 2016, 509, 431–438. [Google Scholar] [CrossRef]
- Ni, Y.; Teng, T.; Li, R.; Simonyi, A.; Sun, G.Y.; Lee, J.C. TNFα alters occludin and cerebral endothelial permeability: Role of p38MAPK. PLoS ONE 2017, 12, e0170346. [Google Scholar] [CrossRef] [PubMed]
- Markoutsa, E.; Pampalakis, G.; Niarakis, A.; Romero, I.A.; Weksler, B.; Couraud, P.-O.; Antimisiaris, S.G. Uptake and permeability studies of BBB-targeting immunoliposomes using the hCMEC/D3 cell line. Eur. J. Pharm. Biopharm. 2011, 77, 265–274. [Google Scholar] [CrossRef]
- Densmore, J.C.; Signorino, P.R.; Ou, J.; Hatoum, O.A.; Rowe, J.J.; Shi, Y.; Kaul, S.; Jones, D.W.; Sabina, R.E.; Pritchard, K.A., Jr. Endothelium-derived microparticles induce endothelial dysfunction and acute lung injury. Shock 2006, 26, 464–471. [Google Scholar] [CrossRef]
- Andrews, A.M.; Lutton, E.M.; Merkel, S.F.; Razmpour, R.; Ramirez, S.H. Mechanical Injury Induces Brain Endothelial-Derived Microvesicle Release: Implications for Cerebral Vascular Injury during Traumatic Brain Injury. Front. Cell. Neurosci. 2016, 10, 43. [Google Scholar] [CrossRef]
- Tai, L.M.; Holloway, K.A.; Male, D.K.; Loughlin, A.J.; Romero, I.A. Amyloid-β-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J. Cell. Mol. Med. 2010, 14, 1101–1112. [Google Scholar] [CrossRef]
- Akdis, M.; Aab, A.; Altunbulakli, C.; Azkur, K.; Costa, R.A.; Crameri, R.; Duan, S.; Eiwegger, T.; Eljaszewicz, A.; Ferstl, R.; et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor beta, and TNF-alpha: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2016, 138, 984–1010. [Google Scholar] [CrossRef] [PubMed]
- Biesmans, S.; Meert, T.F.; Bouwknecht, J.A.; Acton, P.D.; Davoodi, N.; De Haes, P.; Kuijlaars, J.; Langlois, X.; Matthews, L.J.R.; Ver Donck, L. Systemic immune activation leads to neuroinflammation and sickness behavior in mice. Mediators Inflamm. 2013, 2013, 271359. [Google Scholar] [CrossRef]
- Henry, C.J.; Huang, Y.; Wynne, A.M.; Godbout, J.P. Peripheral Lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain. Behav. Immun. 2009, 23, 309–317. [Google Scholar] [CrossRef]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Timár, C.I.; Lőrincz, Á.M.; Csépányi-Kömi, R.; Vályi-Nagy, A.; Nagy, G.; Buzás, E.I.; Iványi, Z.; Kittel, Á.; Powell, D.W.; McLeish, K.R.; et al. Antibacterial effect of microvesicles released from human neutrophilic granulocytes. Blood 2013, 121, 510–518. [Google Scholar] [CrossRef]
- Sahlin, S.; Hed, J.; Runfquist, I. Differentiation between attached and ingested immune complexes by a fluorescence quenching cytofluorometric assay. J. Immunol. Methods 1983, 60, 115–124. [Google Scholar] [CrossRef]
- Zhang, L.; Miles, M.F.; Aldape, K.D. A model of molecular interactions on short oligonucleotide microarrays. Nat. Biotechnol. 2003, 21, 818–821. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional Group | p-Value | Number of Genes |
|---|---|---|
| Tight junction proteins | 0.0062 | 6 |
| Vesicle mediated transport | 0.0074 | 19 |
| Protein transport | 0.0019 | 18 |
| RNA localization | 0.0011 | 9 |
| Metal cluster binding | 0.032 | 4 |
| FY-rich terminals | 0.0043 | 3 |
| Pathway | p-Value |
|---|---|
| Ubiquitin-mediated proteolysis | 0.034 |
| SNARE-mediated vesicular transport | 0.0163 |
| P38 pathway/Regulation of SMAD2/3 signaling | 0.0191 |
| Coenzyme B biosynthesis | 0.0209 |
| Gap junction degradation | 0.0324 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ajikumar, A.; Long, M.B.; Heath, P.R.; Wharton, S.B.; Ince, P.G.; Ridger, V.C.; Simpson, J.E. Neutrophil-Derived Microvesicle Induced Dysfunction of Brain Microvascular Endothelial Cells In Vitro. Int. J. Mol. Sci. 2019, 20, 5227. https://doi.org/10.3390/ijms20205227
Ajikumar A, Long MB, Heath PR, Wharton SB, Ince PG, Ridger VC, Simpson JE. Neutrophil-Derived Microvesicle Induced Dysfunction of Brain Microvascular Endothelial Cells In Vitro. International Journal of Molecular Sciences. 2019; 20(20):5227. https://doi.org/10.3390/ijms20205227
Chicago/Turabian StyleAjikumar, Anjana, Merete B. Long, Paul R. Heath, Stephen B. Wharton, Paul G. Ince, Victoria C. Ridger, and Julie E. Simpson. 2019. "Neutrophil-Derived Microvesicle Induced Dysfunction of Brain Microvascular Endothelial Cells In Vitro" International Journal of Molecular Sciences 20, no. 20: 5227. https://doi.org/10.3390/ijms20205227
APA StyleAjikumar, A., Long, M. B., Heath, P. R., Wharton, S. B., Ince, P. G., Ridger, V. C., & Simpson, J. E. (2019). Neutrophil-Derived Microvesicle Induced Dysfunction of Brain Microvascular Endothelial Cells In Vitro. International Journal of Molecular Sciences, 20(20), 5227. https://doi.org/10.3390/ijms20205227