Protein and Proteome Atlas for Plants under Stresses: New Highlights and Ways for Integrated Omics in Post-Genomics Era


Abstract
:
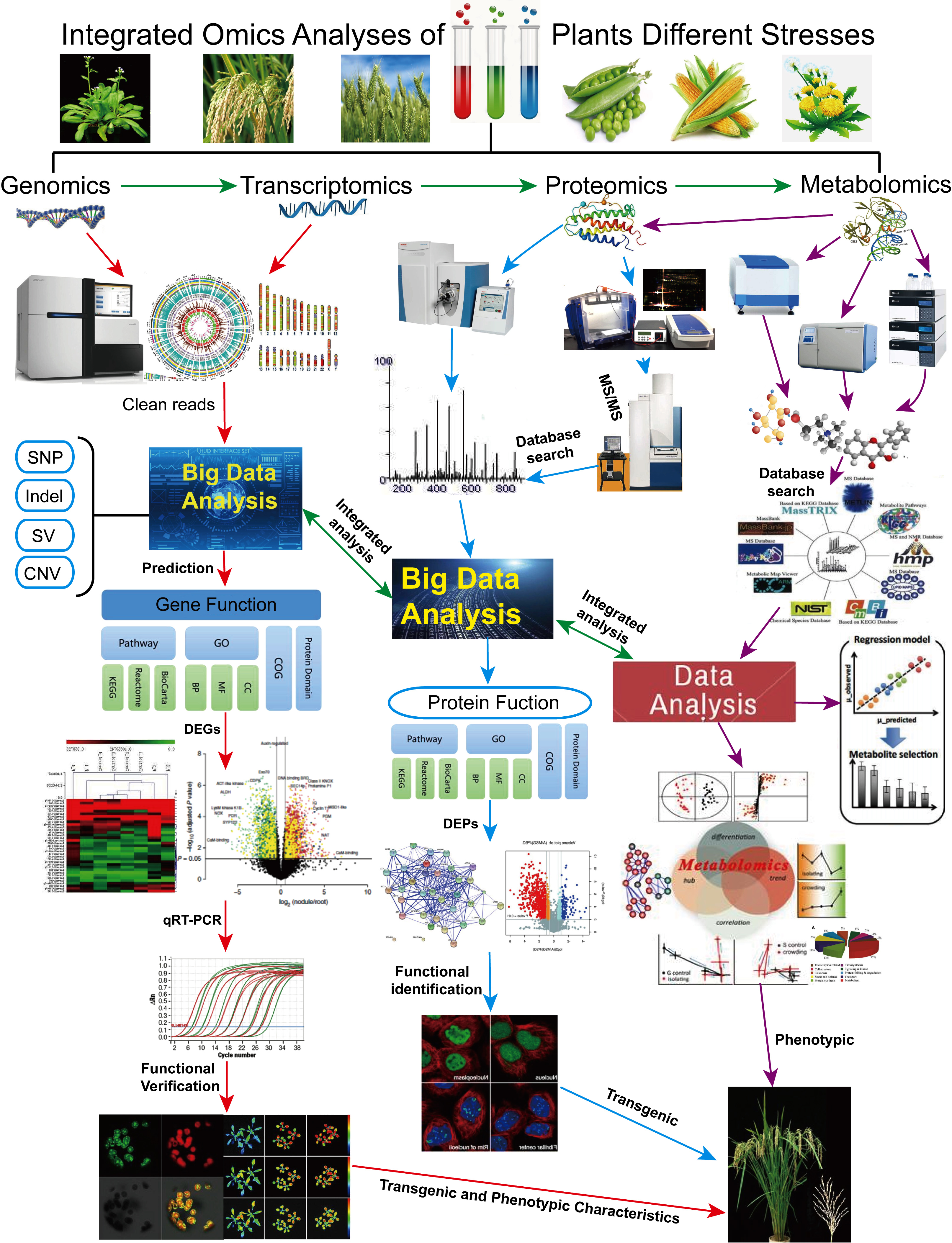{kind=link}
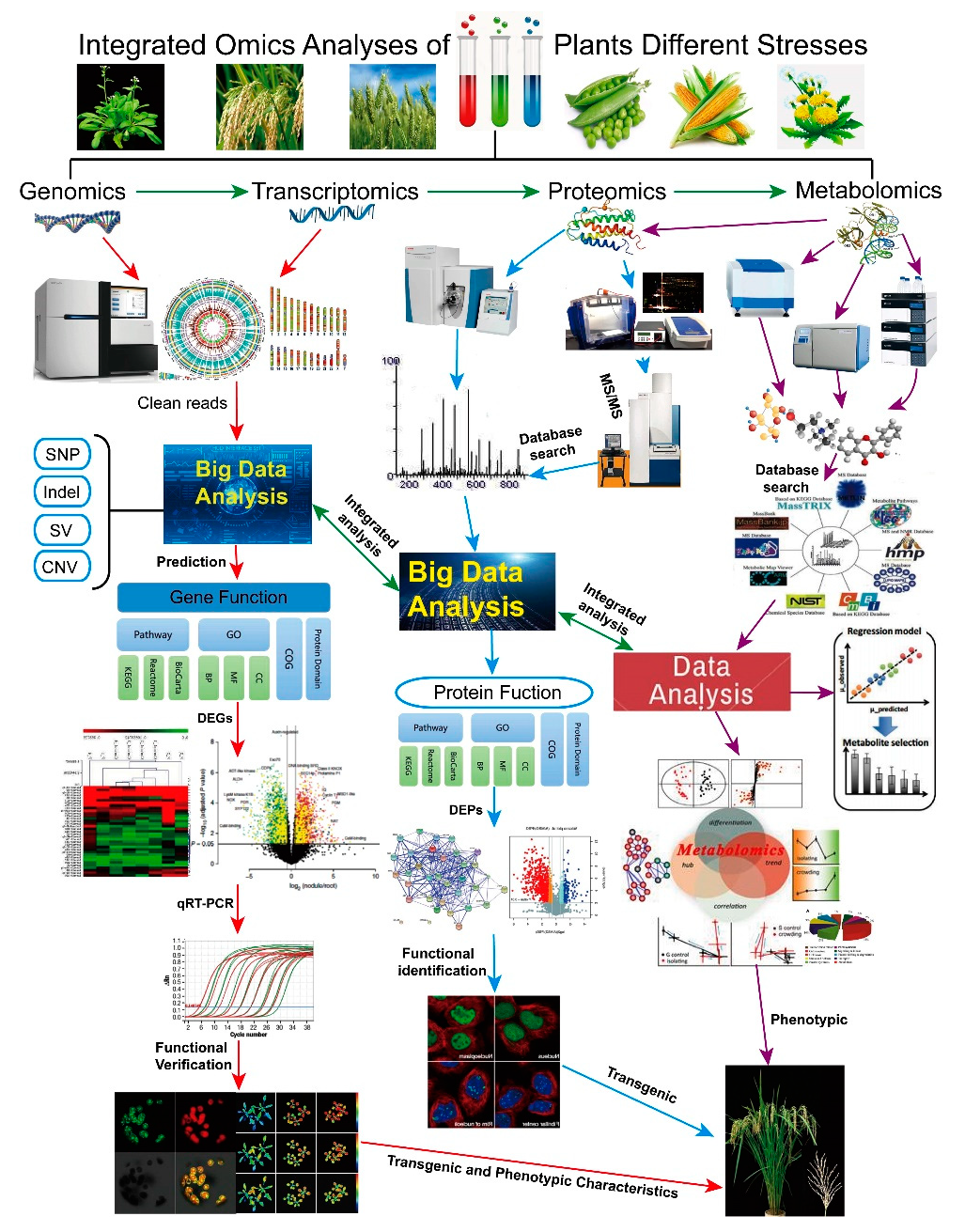{kind=link}
Funding
Acknowledgments
Conflicts of Interest
References
- Aldhous, P. Genomics: Beyond the book of life. Nature 2000, 405, 894–896. [Google Scholar] [CrossRef]
- Wang, W.S.; Mauleon, R.; Hu, Z.Q.; Chebotarov, D.; Tai, S.; Wu, Z.C.; Li, M.; Zheng, T.Q.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Walley, J.W.; Sartor, R.C.; Shen, Z.X.; Schmitz, R.J.; Wu, K.J.; Urich, M.A.; Nery, J.R.; Smith, L.G.; Schnable, J.C.; Ecker, J.R.; et al. Integration of omic networks in a developmental atlas of maize. Science 2016, 353, 814–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Wang, S.; Huang, A.; Zhang, S.; Liao, Q.; Zhang, C.; Lin, T.; Peng, M.; Yang, C.; Cao, X.; et al. Rewiring of the fruit metabolome in tomato breeding. Cell 2018, 172, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.; Yu, J.J.; Li, Y.; Zhang, H.; Bao, X.S.; Bian, J.Y.; Xu, C.X.; Wang, X.L.; Cai, X.F.; Wang, Q.H.; et al. Heat-responsive proteomics of a heat-sensitive spinach variety. Int. J. Mol. Sci. 2019, 20, 3872. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.Y.; Wang, J.; Xie, S.L.; Zhao, M.R.; Nie, L.B.; Zheng, L.B.; Zhu, S.D.; Hou, J.F.; Chen, G.H.; Wang, C.G. Comparative proteomics indicates that redox homeostasis is involved in high- and low-temperature stress tolerance in a novel Wucai (Brassica campestris L.) genotype. Int. J. Mol. Sci. 2019, 20, 3760. [Google Scholar] [CrossRef]
- Wang, S.P.; Zhang, Y.X.; Fang, Z.W.; Zhang, Y.M.; Song, Q.L.; Hou, Z.H.; Sun, K.K.; Song, Y.L.; Li, Y.; Ma, D.F.; et al. Cytological and proteomic analysis of wheat pollen abortion induced by chemical hybridization agent. Int. J. Mol. Sci. 2019, 20, 1615. [Google Scholar] [CrossRef]
- Satheeswaran, T.; Shang, X.M.; Sun, J.; Liu, H.J. Quantitative proteomic analysis reveals novel insights into intracellular silicate stress-responsive mechanisms in the diatom Skeletonema dohrnii. Int. J. Mol. Sci. 2019, 20, 2540. [Google Scholar]
- Huo, J.Q.; Huang, D.J.; Zhang, J.; Fang, H.; Wang, B.; Wang, C.L.; Ma, Z.J.; Liao, W.B. Comparative proteomic analysis during the involvement of nitric oxide in hydrogen gas-improved postharvest freshness in cut lilies. Int. J. Mol. Sci. 2018, 19, 3955. [Google Scholar] [CrossRef]
- Xie, Q.L.; Ding, G.H.; Zhu, L.P.; Yu, L.; Yuan, B.X.; Gao, X.Q.; Wang, D.; Sun, Y.; Liu, Y.; Li, H.B.; et al. Proteomic landscape of the mature roots in a rubber-producing grass Taraxacum Kok-saghyz. Int. J. Mol. Sci. 2019, 20, 2596. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Zhang, X.Y.; Huang, G.R.; Feng, F.; Liu, X.Y.; Guo, R.; Gu, F.X.; Zhong, X.L.; Mei, X.R. iTRAQ-Based quantitative analysis of responsive proteins under PEG-induced drought stress in wheat leaves. Int. J. Mol. Sci. 2019, 20, 2621. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.B.; Xu, Y.H.; Li, J.J.; Ren, Y.Z.; Wang, Z.Q.; Xin, Z.Y.; Lin, T.B. Identification of two novel wheat drought tolerance-related proteins by comparative proteomic analysis combined with virus-induced gene silencing. Int. J. Mol. Sci. 2018, 19, 4020. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Qiu, L.; Song, Q.L.; Wang, S.P.; Wang, Y.J.; Ge, Y.H. Root proteomics reveals the effects of wood vinegar on wheat growth and subsequent tolerance to drought stress. Int. J. Mol. Sci. 2019, 20, 943. [Google Scholar] [CrossRef]
- Wang, X.; Tinashe, Z.; Liu, S.T.; Liu, G.; Jin, H.Y.; Dai, L.; Dong, A.Y.; Yang, Y.T.; Duan, H.J. Comparative proteomics and physiological analyses reveal important maize filling-kernel drought-responsive genes and metabolic pathways. Int. J. Mol. Sci. 2019, 20, 3743. [Google Scholar] [CrossRef]
- Jia, T.T.; Wang, J.; Chang, W.; Fan, X.X.; Sui, X.; Song, F.Q. Proteomics analysis of E. angustifolia seedlings inoculated with Arbuscular Mycorrhizal fungi under salt stress. Int. J. Mol. Sci. 2019, 20, 788. [Google Scholar] [CrossRef]
- Wang, Y.; Cong, Y.T.; Wang, Y.H.; Guo, Z.H.; Yue, J.R.; Xing, Z.Y.; Gao, X.N.; Chai, X.J. Identification of early salinity stress-responsive proteins in Dunaliella salina by isobaric tags for relative and absolute quantitation (iTRAQ)-based quantitative proteomic analysis. Int. J. Mol. Sci. 2019, 20, 599. [Google Scholar] [CrossRef]
- Liu, H.; Wang, F.F.; Peng, X.J.; Huang, J.H.; Shen, S.H. Global phosphoproteomic analysis reveals the defense and response mechanisms of Jatropha Curcas seedling under chilling stress. Int. J. Mol. Sci. 2019, 20, 208. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Bai, X.; Jiang, C.F.; Li, Z. Phosphoproteomic analysis of two contrasting maize inbred lines provides insights into the mechanism of salt-stress tolerance. Int. J. Mol. Sci. 2019, 20, 1886. [Google Scholar] [CrossRef]
- Yu, G.W.; Lv, Y.N.; Shen, L.Y.; Wang, Y.B.; Qing, Y.; Wu, N.; Li, Y.P.; Huang, H.H.; Zhang, N.; Liu, Y.H.; et al. The proteomic analysis of maize endosperm protein enriched by phos-tagtm reveals the phosphorylation of brittle-2 subunit of ADP-Glc pyrophosphorylase in starch biosynthesis process. Int. J. Mol. Sci. 2019, 20, 986. [Google Scholar] [CrossRef]
- Guo, H.X.; Guo, H.H.; Zhang, L.; Fan, Y.P.; Fan, Y.P.; Tang, Z.M.; Zeng, F.C. Dynamic TMT-based quantitative proteomics analysis of critical initiation process of totipotency during cotton somatic embryogenesis transdifferentiation. Int. J. Mol. Sci. 2019, 20, 1691. [Google Scholar]
- Lin, Z.Y.; Zhang, C.; Cao, D.D.; Damaris, R.N.; Yang, P.F. The latest studies on lotus (Nelumbo nucifera), an emerging horticultural model plant. Int. J. Mol. Sci. 2019, 20, 3680. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Q.; Lyu, C.C.; Li, J.H.; Tong, Z.; Lu, Y.F.; Wang, X.Y.; Ni, S.; Yang, S.M.; Zeng, F.H.; Lu, L.M. Physiological analysis and proteome quantification of alligator weed stems in response to potassium deficiency stress. Int. J. Mol. Sci. 2019, 20, 221. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.H.; Guo, H.X.; Zhang, L.; Tang, Z.M.; Yu, X.M.; Wu, J.F.; Zeng, F.C. Metabolome and transcriptome association analysis reveals dynamic regulation of purine metabolism and flavonoid synthesis in transdifferentiation during somatic embryogenesis in cotton. Int. J. Mol. Sci. 2019, 20, 2070. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Qin, L.; Dai, X.R.; Ding, Z.D.; Bi, R.; Liu, P.; Chen, Y.H.; Thomas, P.B.; Wang, X.L.; Li, P.H. Transcriptomic analysis of leaf sheath maturation in maize. Int. J. Mol. Sci. 2019, 20, 2472. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, W.; Su, X.; Ge, P.F.; Zhou, Y.; Hao, Y.Y.; Shu, H.Y.; Gao, C.L.; Cheng, S.H.; Zhu, G.P.; et al. Early response of radish to heat stress by strand-specific transcriptome and miRNA analysis. Int. J. Mol. Sci. 2019, 20, 3321. [Google Scholar] [CrossRef]
- Jin, Y.; Liu, L.; Hao, X.H.; David, E.H.; Zheng, Y.Z.; Huang, T.B.; Huang, J.Z. Unravelling the microRNA-mediated gene regulation in developing Pongamia seeds by high-throughput small RNA profiling. Int. J. Mol. Sci. 2019, 20, 3509. [Google Scholar] [CrossRef]
- Tang, J.C.; Sun, Z.G.; Chen, Q.H.; Rebecca, N.D.; Lu, B.L.; Hu, Z.R. Nitrogen fertilizer induced alterations in the root proteome of two rice cultivars. Int. J. Mol. Sci. 2019, 20, 3674. [Google Scholar] [CrossRef]
- Song, Q.; Lv, F.; Muhammad, T.Q.; Xing, F.; Zhou, R.; Li, H.; Chen, L.L. Identification and analysis of micro-exon genes in the rice genome. Int. J. Mol. Sci. 2019, 20, 2685. [Google Scholar] [CrossRef]
- Ji, M.C.; Wang, K.; Wang, L.; Chen, S.X.; Li, H.Y.; Ma, C.Q.; Wang, Y.G. Overexpression of a S-adenosylmethionine decarboxylase from sugar beet M14 increased Araidopsis salt tolerance. Int. J. Mol. Sci. 2019, 20, 1990. [Google Scholar] [CrossRef]
- Ma, R.D.; Song, W.Y.; Wang, F.; Cao, A.; Xie, S.Q.; Chen, X.F.; Jin, X.; Li, H.B. A cotton (Gossypium hirsutum) Myo-inositol-1-phosphate synthase (GhMIPS1D) gene promotes root cell elongation in Arabidopsis. Int. J. Mol. Sci. 2019, 20, 1224. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Kong, D.F.; Li, Q.H.; Sun, S.; Song, J.L.; Zhu, Y.B.; Liang, K.J.; Ke, Q.M.; Lin, W.X.; Huang, J.W. The function of inositol phosphatases in plant tolerance to abiotic stress. Int. J. Mol. Sci. 2019, 20, 3999. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X. Protein and Proteome Atlas for Plants under Stresses: New Highlights and Ways for Integrated Omics in Post-Genomics Era. Int. J. Mol. Sci. 2019, 20, 5222. https://doi.org/10.3390/ijms20205222
Wang X. Protein and Proteome Atlas for Plants under Stresses: New Highlights and Ways for Integrated Omics in Post-Genomics Era. International Journal of Molecular Sciences. 2019; 20(20):5222. https://doi.org/10.3390/ijms20205222
Chicago/Turabian StyleWang, Xuchu. 2019. "Protein and Proteome Atlas for Plants under Stresses: New Highlights and Ways for Integrated Omics in Post-Genomics Era" International Journal of Molecular Sciences 20, no. 20: 5222. https://doi.org/10.3390/ijms20205222
APA StyleWang, X. (2019). Protein and Proteome Atlas for Plants under Stresses: New Highlights and Ways for Integrated Omics in Post-Genomics Era. International Journal of Molecular Sciences, 20(20), 5222. https://doi.org/10.3390/ijms20205222