Combination of SAXS and Protein Painting Discloses the Three-Dimensional Organization of the Bacterial Cysteine Synthase Complex, a Potential Target for Enhancers of Antibiotic Action

 , , , ,
, , , ,  ,
,  , and
, and 
Abstract
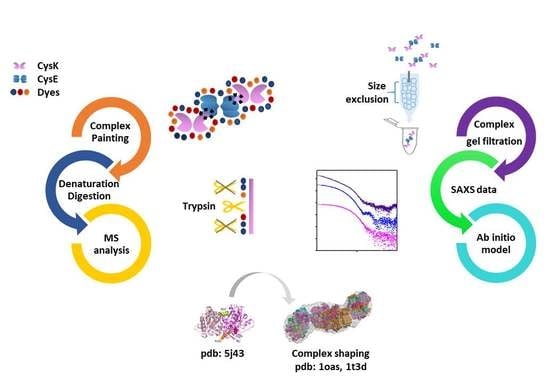
1. Introduction
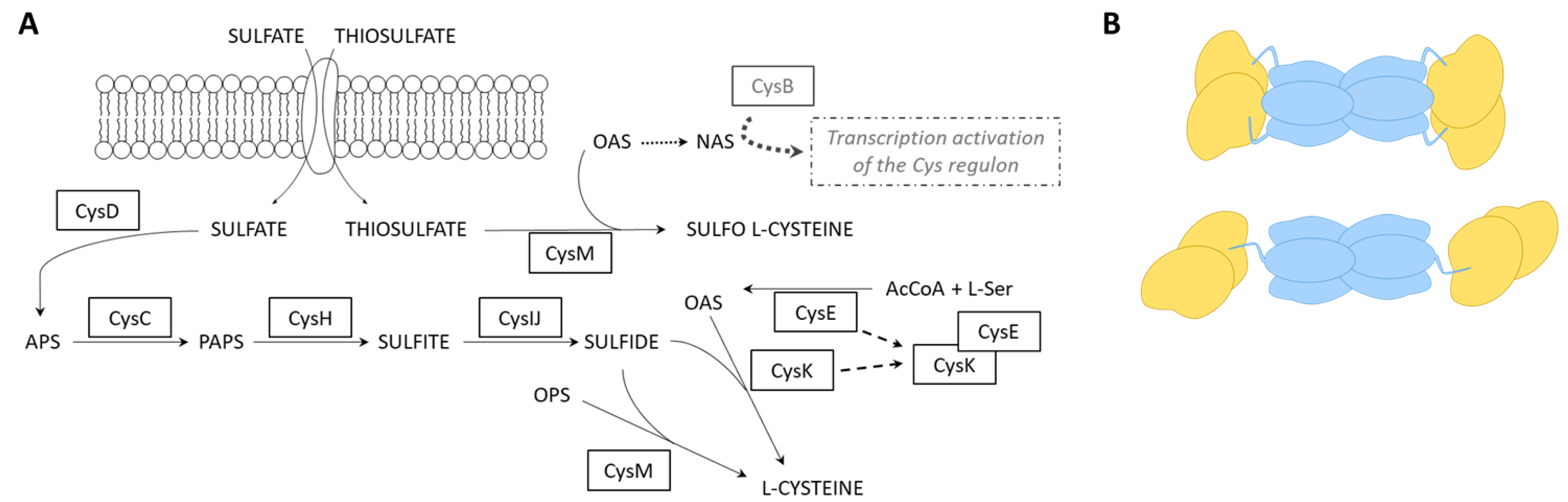
2. Results and Discussion
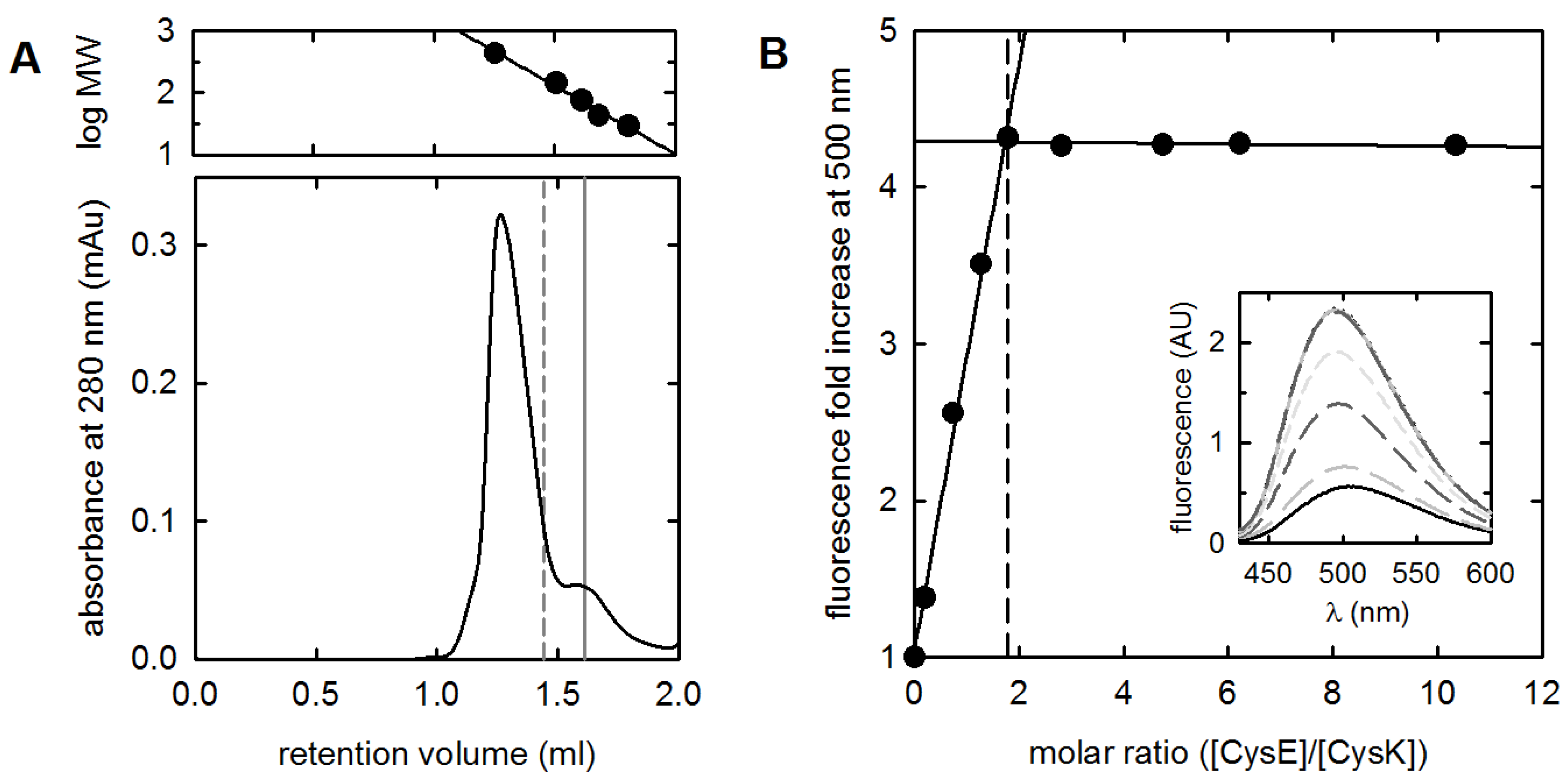
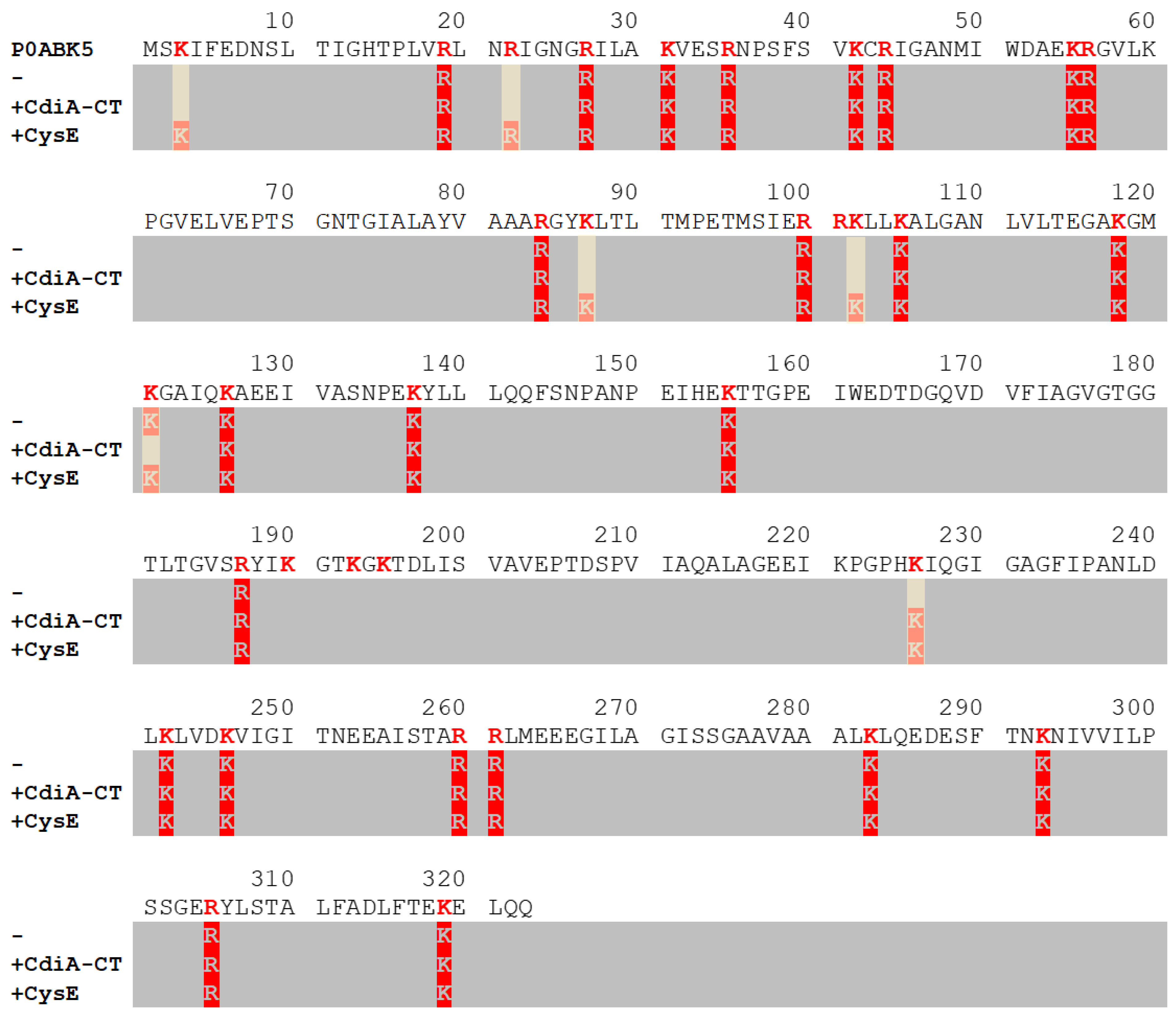
2.1. Validation of the Protein Painting Assay
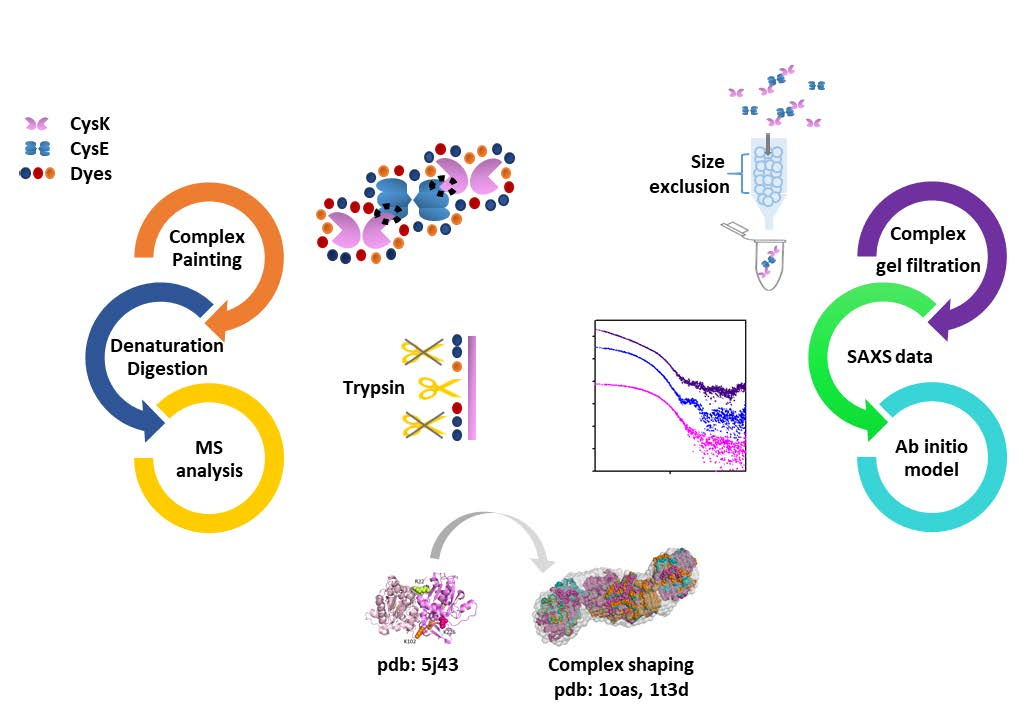
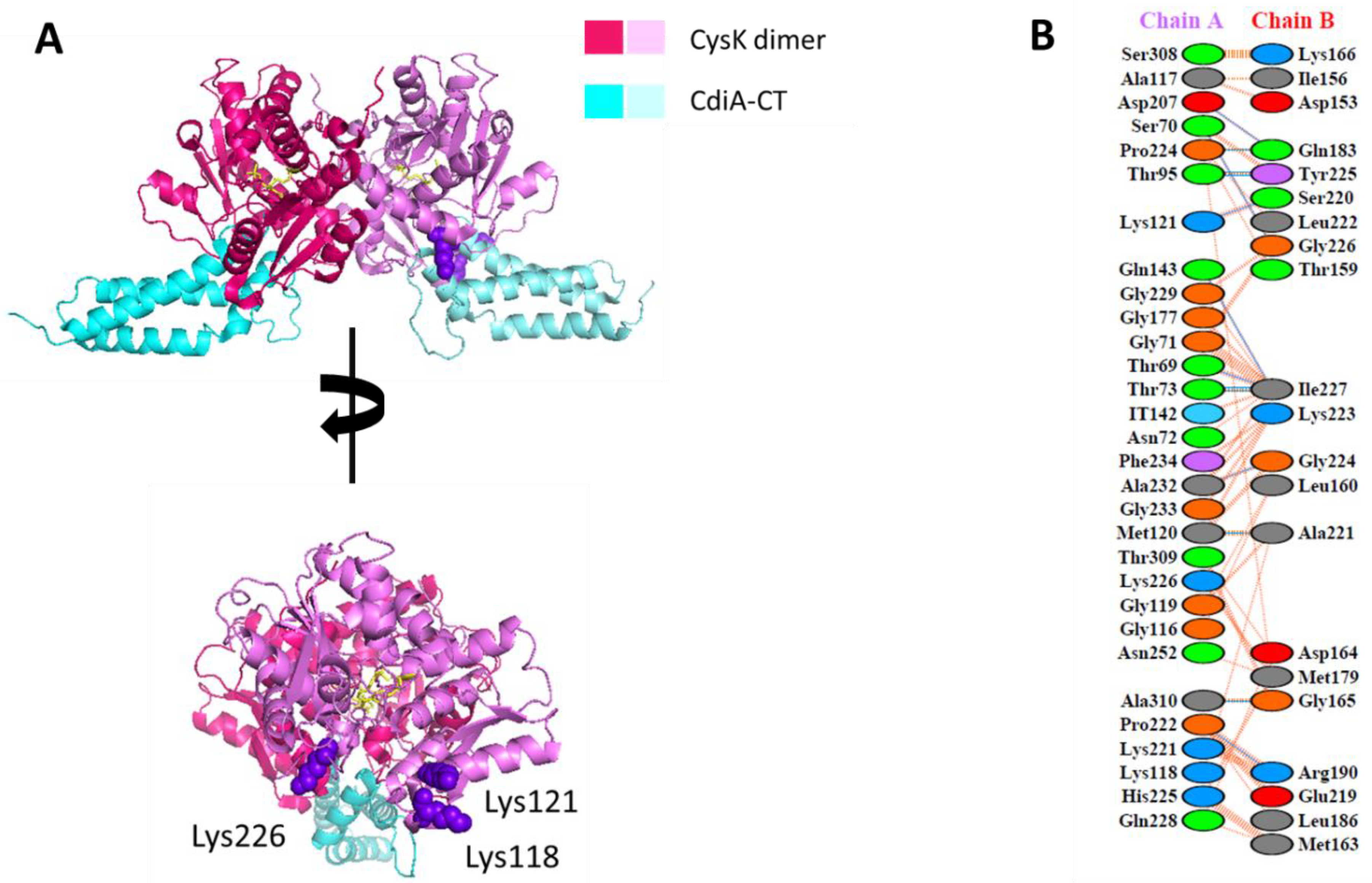
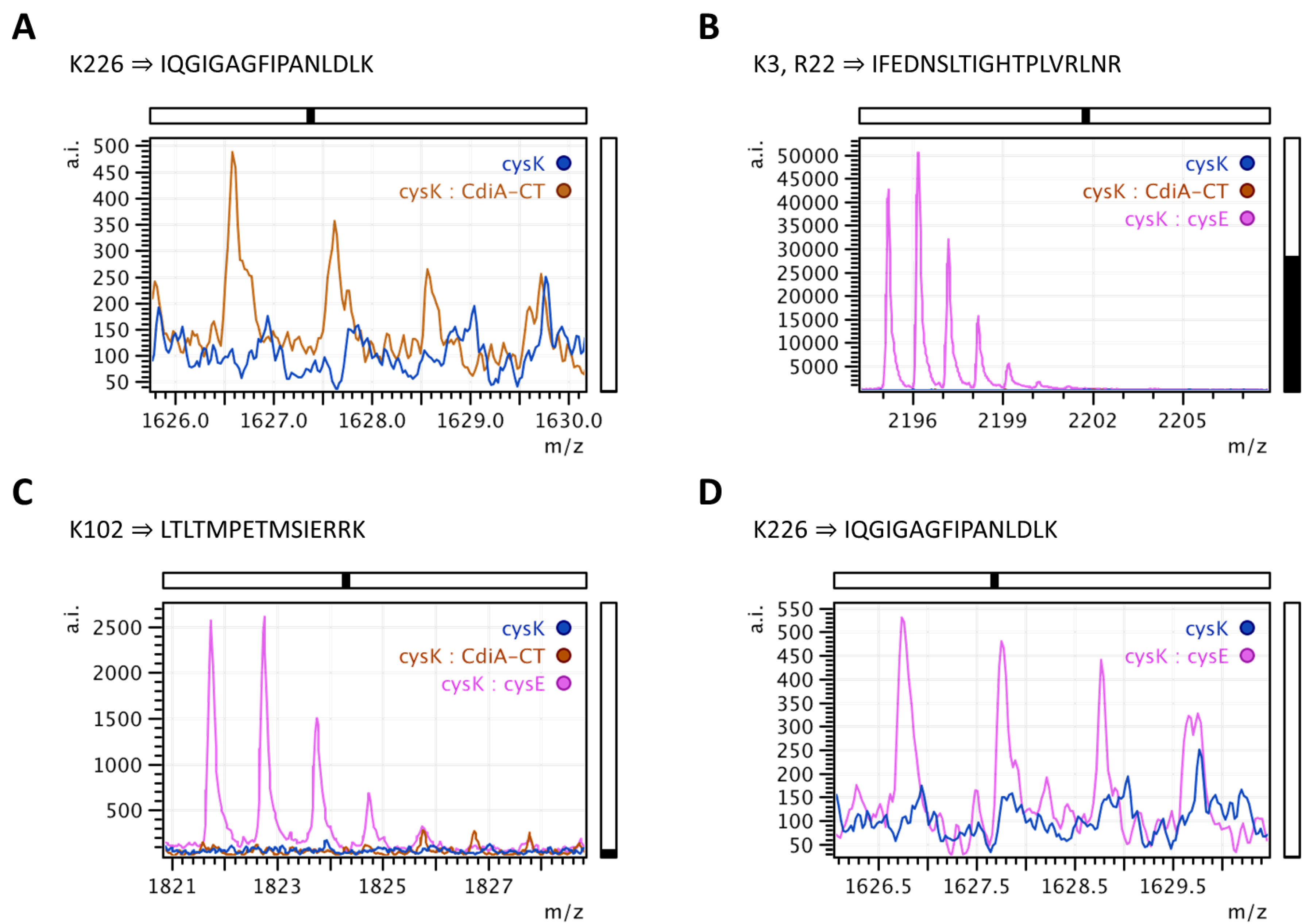
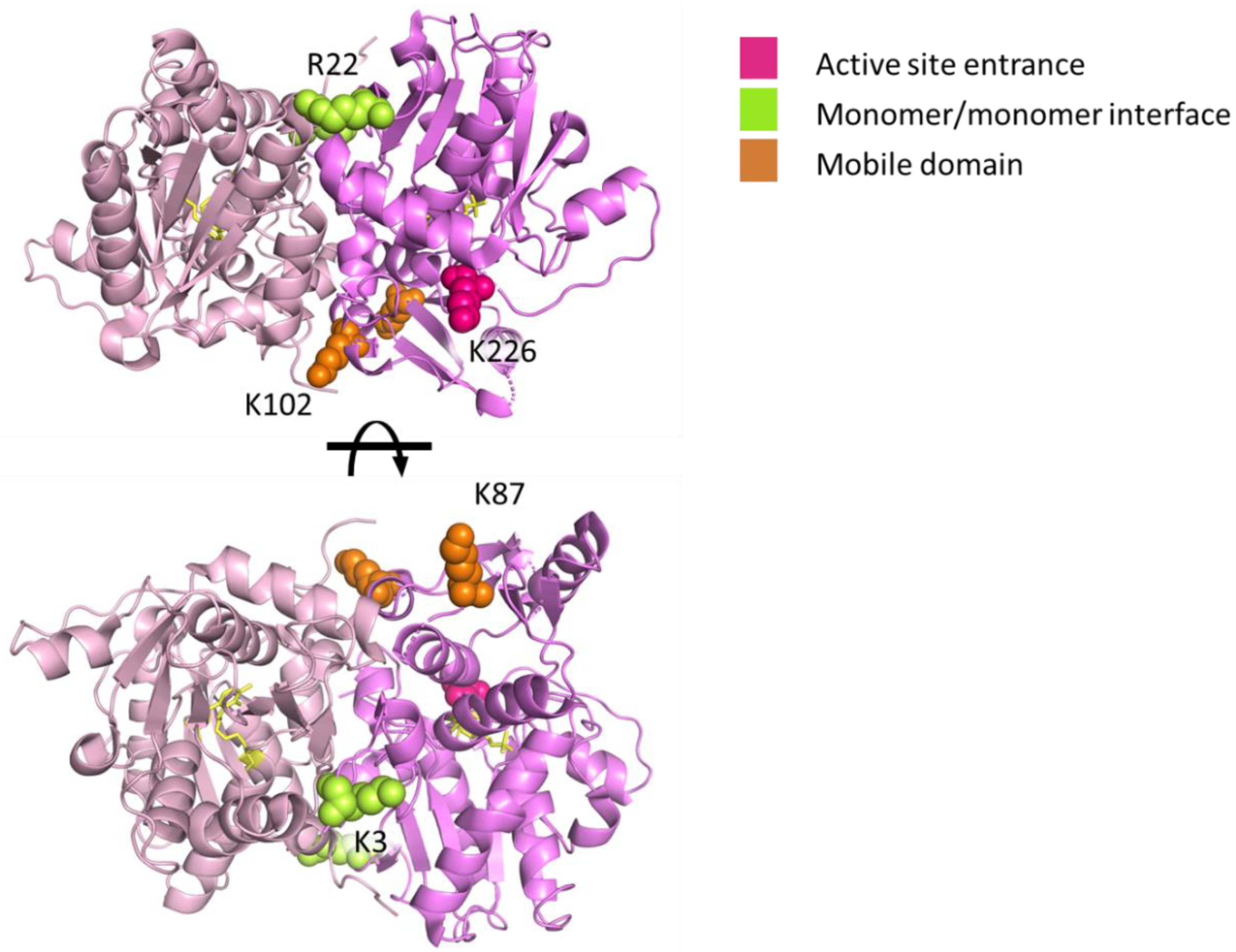
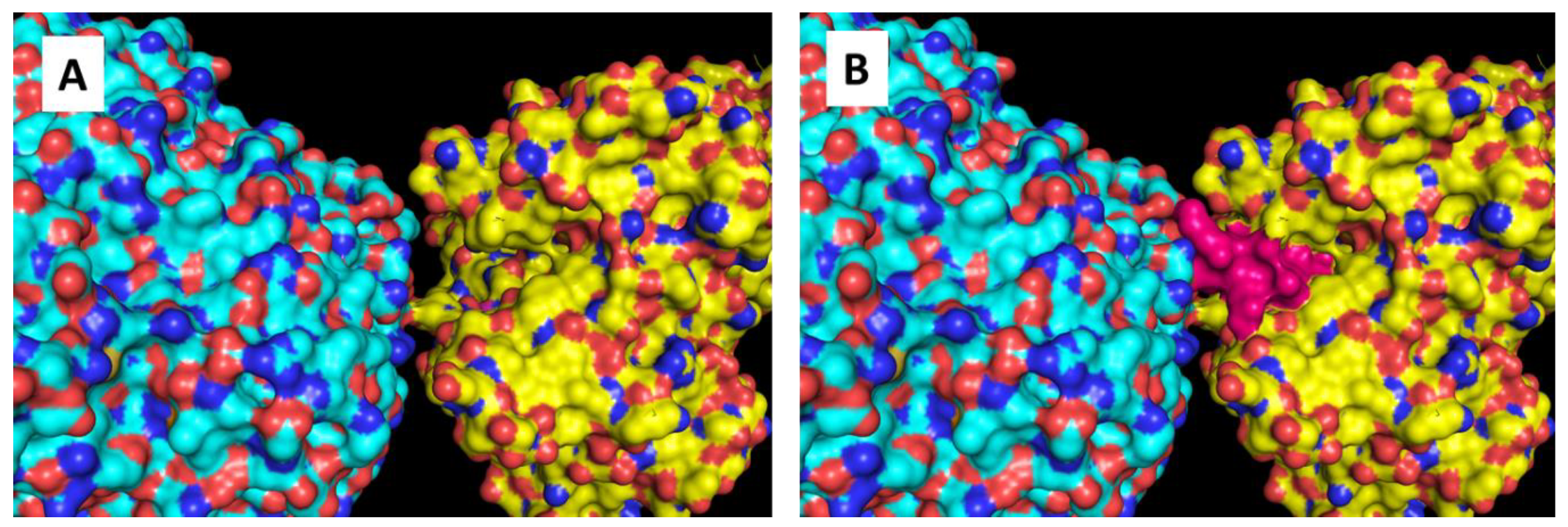
2.2. Mapping Protein–Protein Interaction in Cysteine Synthase
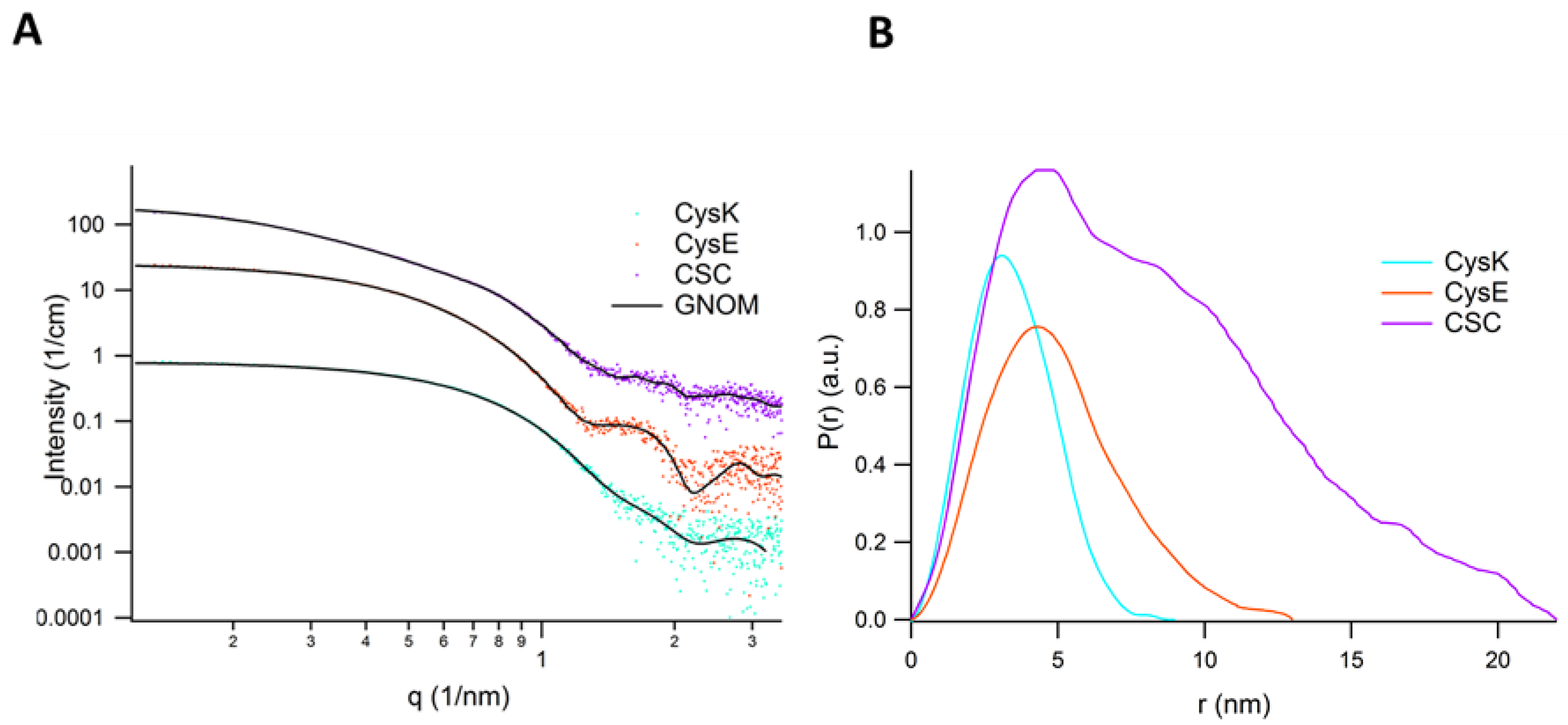
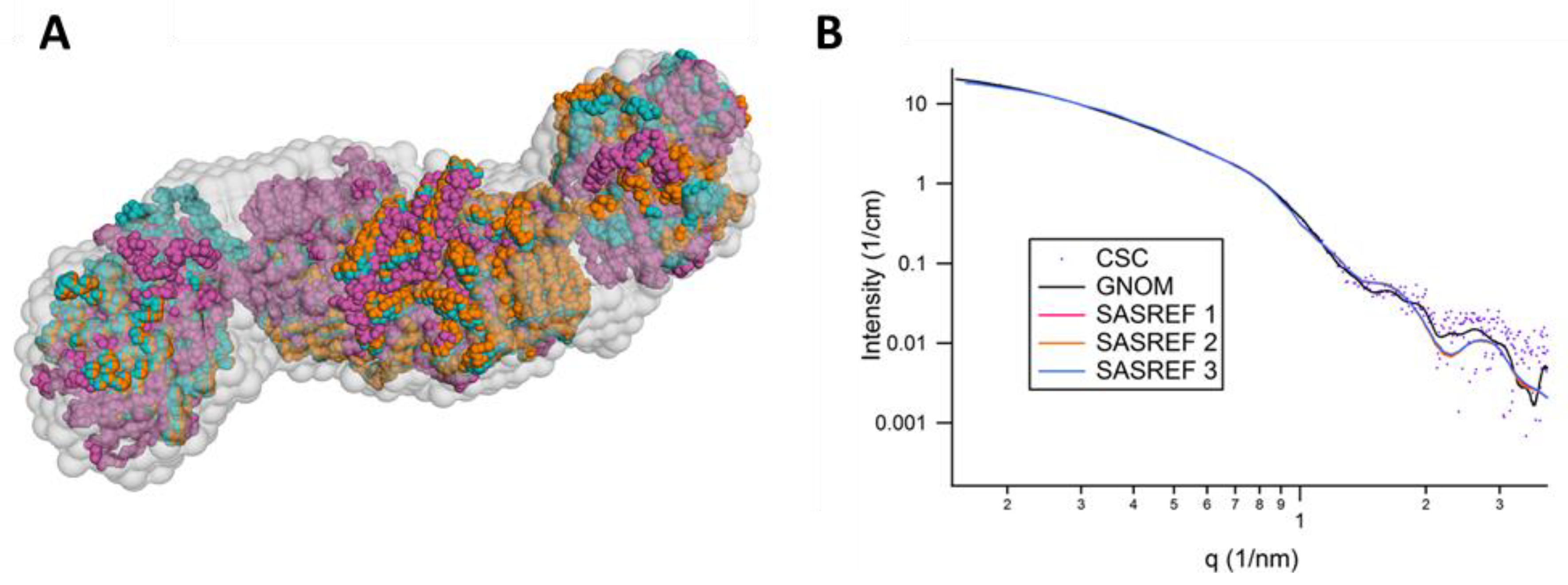
2.3. SAXS Analysis of CysK, CysE, and CS
3. Materials and Methods
3.1. Proteins Expression and Purification
3.2. Spectroscopy
3.3. Size Exclusion Chromatography
3.4. Protein Painting with Small Molecule Dyes
3.5. SAXS Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AcCoA | acetyl coenzyme A |
| Acid Fuchsin | 2-amino-5-[(4-amino-3-sulfophenyl)(4-imino-3-sulfo-2,5-cyclohexadien-1-ylidene)methyl]- 3-methyl-benzenesulfonic acid, sodium salt |
| ACTH | adrenocorticotropic hormone |
| ANSA | 8-Anilino-1-naphthalenesulfonic acid ammonium salt |
| AO50 | sodium 4-(4-(benzyl-et-amino)-ph-azo)-2,5-di-cl-benzenesulfonate |
| BSA | bovine serum albumin |
| DTT | dithiothreitol |
| EDTA | ethylenediaminetetraacetic acid |
| Eosin B | 4’,5’-dibromo-3’,6’-dihydroxy-2’,7’-dinitro-spiro[isobenzofuran-1(3H),9’-[9H]xanthen]-3one |
| CS | cysteine synthase |
| CR | disodium 4-amino-3-[[4-[4-[(1-amino-4-sulfonatonaphthalen-2yl) diazenyl] phenyl] phenyl] diazenyl] naphthalene-1-sulfonate |
| CysD | ATP sulfurylase |
| CysE | serine acetyltransferase |
| CysH | phosphoadenosine phosphosulphate reductase |
| CysK | O-acetylserine sulfhydrylase isozyme A |
| CysM | O-acetylserine sulfhydrylase isozyme B (O-phosphoserine sulfhydrylase) |
| CysN | CysD-associated GTPase |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| HCCA | α-cyano-4-hydroxycinnamic acid |
| HPLC-SEC | high-pressure liquid chromatography-size exclusion chromatography |
| IPGT | isopropyl β-D-1-thiogalactopyranoside |
| MALDI TOF/TOF | matrix-assisted laser desorption ionization-time of flight/time of flight |
| OAS | O-acetylserine |
| NAS | N-acetylserine |
| PDDF | pair distance distribution function |
| PLP | pyridoxal 5′-phosphate |
| PPIs | protein–protein interactions |
| RBB | disodium 1-amino-9,10-dioxo-4-[3-(2-sulfonatooxyethylsulfonyl)anilino]anthracene-2-sulfonate |
| SAXS | small angle X-ray scattering |
| TCEP | tris(2-carboxyethyl)phosphine) |
| TFA | trifluoroacetic acid |
References
- Hara, M.R.; Agrawal, N.; Kim, S.F.; Cascio, M.B.; Fujimuro, M.; Ozeki, Y.; Takahashi, M.; Cheah, J.H.; Tankou, S.K.; Hester, L.D.; et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 2005, 7, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Romberg, L.; Levin, P.A. Assembly dynamics of the bacterial cell division protein FTSZ: Poised at the edge of stability. Annu. Rev. Microbiol. 2003, 57, 125–154. [Google Scholar] [CrossRef] [PubMed]
- Sweetlove, L.J.; Fernie, A.R. The role of dynamic enzyme assemblies and substrate channelling in metabolic regulation. Nat. Commun. 2018, 9, 2136. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Rolland, T.; Tasan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A proteome-scale map of the human interactome network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef]
- Caufield, J.H.; Wimble, C.; Shary, S.; Wuchty, S.; Uetz, P. Bacterial protein meta-interactomes predict cross-species interactions and protein function. BMC Bioinform. 2017, 18, 171. [Google Scholar] [CrossRef]
- Crua Asensio, N.; Munoz Giner, E.; de Groot, N.S.; Torrent Burgas, M. Centrality in the host-pathogen interactome is associated with pathogen fitness during infection. Nat. Commun. 2017, 8, 14092. [Google Scholar] [CrossRef]
- Rajagopala, S.V.; Sikorski, P.; Kumar, A.; Mosca, R.; Vlasblom, J.; Arnold, R.; Franca-Koh, J.; Pakala, S.B.; Phanse, S.; Ceol, A.; et al. The binary protein-protein interaction landscape of Escherichia coli. Nat. Biotechnol. 2014, 32, 285–290. [Google Scholar] [CrossRef]
- Hauser, R.; Ceol, A.; Rajagopala, S.V.; Mosca, R.; Siszler, G.; Wermke, N.; Sikorski, P.; Schwarz, F.; Schick, M.; Wuchty, S.; et al. A second-generation protein-protein interaction network of Helicobacter pylori. Mol. Cell. Proteomics 2014, 13, 1318–1329. [Google Scholar] [CrossRef]
- Cong, Q.; Anishchenko, I.; Ovchinnikov, S.; Baker, D. Protein interaction networks revealed by proteome coevolution. Science 2019, 365, 185–189. [Google Scholar]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Carro, L. Protein-protein interactions in bacteria: A promising and challenging avenue towards the discovery of new antibiotics. Beilstein J. Org. Chem. 2018, 14, 2881–2896. [Google Scholar] [CrossRef] [PubMed]
- Cossar, P.J.; Lewis, P.J.; McCluskey, A. Protein-protein interactions as antibiotic targets: A medicinal chemistry perspective. Med. Res. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gagarinova, A.; Phanse, S.; Cygler, M.; Babu, M. Insights from protein-protein interaction studies on bacterial pathogenesis. Expert Rev. Proteomic. 2017, 14, 779–797. [Google Scholar] [CrossRef] [PubMed]
- Voter, A.F.; Keck, J.L. Development of protein–protein interaction inhibitors for the treatment of infectious diseases. Adv. Protein Chem. Struct. Biol. 2018, 111, 197–222. [Google Scholar] [PubMed]
- Zoraghi, R.; Reiner, N.E. Protein interaction networks as starting points to identify novel antimicrobial drug targets. Curr. Opin. Microbiol. 2013, 16, 566–572. [Google Scholar] [CrossRef]
- Campanini, B.; Pieroni, M.; Raboni, S.; Bettati, S.; Benoni, R.; Pecchini, C.; Costantino, G.; Mozzarelli, A. Inhibitors of the sulfur assimilation pathway in bacterial pathogens as enhancers of antibiotic therapy. Curr. Med. Chem. 2015, 22, 187–213. [Google Scholar] [CrossRef]
- Singh, K.; Ali, V.; Pratap Singh, K.; Gupta, P.; Suman, S.S.; Ghosh, A.K.; Bimal, S.; Pandey, K.; Das, P. Deciphering the interplay between cysteine synthase and thiol cascade proteins in modulating Amphotericin B resistance and survival of Leishmania donovani under oxidative stress. Redox Biol. 2017, 12, 350–366. [Google Scholar] [CrossRef]
- Turnbull, A.L.; Surette, M.G. L-Cysteine is required for induced antibiotic resistance in actively swarming Salmonella enterica serovar Typhimurium. Microbiology 2008, 154, 3410–3419. [Google Scholar] [CrossRef]
- Oppezzo, O.J.; Anton, D.N. Involvement of cysB and cysE genes in the sensitivity of Salmonella typhimurium to mecillinam. J. Bacteriol. 1995, 177, 4524–4527. [Google Scholar] [CrossRef]
- Fravega, J.; Alvarez, R.; Diaz, F.; Inostroza, O.; Tejias, C.; Rodas, P.I.; Paredes-Sabja, D.; Fuentes, J.A.; Calderon, I.L.; Gil, F. Salmonella Typhimurium exhibits fluoroquinolone resistance mediated by the accumulation of the antioxidant molecule H2S in a CysK-dependent manner. J. Antimicrob. Chemother. 2016, 71, 3409–3415. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, R.; Frávega, J.; Rodas, P.I.; Fuentes, J.A.; Paredes-Sabja, D.; Calderón, I.L.; Gil, F. Participation of S. Typhimurium cysJIH operon in the H2S-mediated ciprofloxacin resistance in presence of sulfate as sulfur source. Antibiotics 2015, 4, 321–328. [Google Scholar] [CrossRef]
- Sturgill, G.; Toutain, C.M.; Komperda, J.; O’Toole, G.A.; Rather, P.N. Role of CysE in production of an extracellular signaling molecule in Providencia stuartii and Escherichia coli: Loss of CysE enhances biofilm formation in Escherichia coli. J. Bacteriol. 2004, 186, 7610–7617. [Google Scholar] [CrossRef] [PubMed]
- Benoni, R.; Beck, C.M.; Garza-Sànchez, F.; Bettati, S.; Mozzarelli, A.; Hayes, C.S.; Campanini, B. Activation of an anti-bacterial toxin by the biosynthetic enzyme CysK: Mechanism of binding, interaction specificity and competition with cysteine synthase. Sci. Rep. 2017, 7, 8817. [Google Scholar] [CrossRef]
- Diner, E.J.; Beck, C.M.; Webb, J.S.; Low, D.A.; Hayes, C.S. Identification of a target cell permissive factor required for contact-dependent growth inhibition (CDI). Genes Dev. 2012, 26, 515–525. [Google Scholar] [CrossRef]
- Magalhaes, J.; Franko, N.; Annunziato, G.; Pieroni, M.; Benoni, R.; Nikitjuka, A.; Mozzarelli, A.; Bettati, S.; Karawajczyk, A.; Jirgensons, A.; et al. Refining the structure-activity relationships of 2-phenylcyclopropane carboxylic acids as inhibitors of O-acetylserine sulfhydrylase isoforms. J. Enzyme Inhib. Med. Chem. 2019, 34, 31–43. [Google Scholar] [CrossRef]
- Magalhaes, J.; Franko, N.; Annunziato, G.; Welch, M.; Dolan, S.K.; Bruno, A.; Mozzarelli, A.; Armao, S.; Jirgensons, A.; Pieroni, M.; et al. Discovery of novel fragments inhibiting O-acetylserine sulphhydrylase by combining scaffold hopping and ligand-based drug design. J. Enzyme Inhib. Med. Chem. 2018, 33, 1444–1452. [Google Scholar] [CrossRef]
- Franko, N.; Grammatoglou, K.; Campanini, B.; Costantino, G.; Jirgensons, A.; Mozzarelli, A. Inhibition of O-acetylserine sulfhydrylase by fluoroalanine derivatives. J. Enzyme Inhib. Med. Chem. 2018, 33, 1343–1351. [Google Scholar] [CrossRef]
- Pieroni, M.; Annunziato, G.; Beato, C.; Wouters, R.; Benoni, R.; Campanini, B.; Pertinhez, T.A.; Bettati, S.; Mozzarelli, A.; Costantino, G. Rational Design, synthesis, and preliminary structure-activity relationships of alpha-substituted-2-phenylcyclopropane carboxylic acids as inhibitors of Salmonella typhimurium O-acetylserine sulfhydrylase. J. Med. Chem. 2016, 59, 2567–2578. [Google Scholar] [CrossRef]
- Annunziato, G.; Pieroni, M.; Benoni, R.; Campanini, B.; Pertinhez, T.A.; Pecchini, C.; Bruno, A.; Magalhaes, J.; Bettati, S.; Franko, N.; et al. Cyclopropane-1,2-dicarboxylic acids as new tools for the biophysical investigation of O-acetylserine sulfhydrylases by fluorimetric methods and saturation transfer difference (STD) NMR. J. Enzyme Inhib. Med. Chem. 2016, 31, 78–87. [Google Scholar] [CrossRef]
- Spyrakis, F.; Singh, R.; Cozzini, P.; Campanini, B.; Salsi, E.; Felici, P.; Raboni, S.; Benedetti, P.; Cruciani, G.; Kellogg, G.E.; et al. Isozyme-specific ligands for O-acetylserine sulfhydrylase, a novel antibiotic target. PloS ONE 2013, 8, e77558. [Google Scholar] [CrossRef] [PubMed]
- Spyrakis, F.; Felici, P.; Bayden, A.S.; Salsi, E.; Miggiano, R.; Kellogg, G.E.; Cozzini, P.; Cook, P.F.; Mozzarelli, A.; Campanini, B. Fine tuning of the active site modulates specificity in the interaction of O-acetylserine sulfhydrylase isozymes with serine acetyltransferase. Biochim. Biophys. Acta 2013, 1834, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Amori, L.; Katkevica, S.; Bruno, A.; Campanini, B.; Felici, P.; Mozzarelli, A.; Costantino, G. Design and synthesis of trans-2-substituted-cyclopropane-1-carboxylic acids as the first non-natural small molecule inhibitors of O-acetylserine sulfhydrylase. Med. Chem. Comm. 2012, 3, 1111–1116. [Google Scholar] [CrossRef]
- Salsi, E.; Bayden, A.S.; Spyrakis, F.; Amadasi, A.; Campanini, B.; Bettati, S.; Dodatko, T.; Cozzini, P.; Kellogg, G.E.; Cook, P.F.; et al. Design of O-acetylserine sulfhydrylase inhibitors by mimicking nature. J. Med. Chem. 2010, 53, 345–356. [Google Scholar] [CrossRef]
- Poyraz, O.; Jeankumar, V.U.; Saxena, S.; Schnell, R.; Haraldsson, M.; Yogeeswari, P.; Sriram, D.; Schneider, G. Structure-guided design of novel thiazolidine inhibitors of O-acetyl serine sulfhydrylase from Mycobacterium tuberculosis. J. Med. Chem. 2013, 56, 6457–6466. [Google Scholar] [CrossRef]
- Kumar, J.V.U.; Poyraz, O.; Saxena, S.; Schnell, R.; Yogeeswari, P.; Schneider, G.; Sriram, D. Discovery of novel inhibitors targeting the Mycobacterium tuberculosis O-acetylserine sulfhydrylase (CysK1) using virtual high-throughput screening. Bioorg. Med. Chem. Lett. 2013, 23, 1182–1186. [Google Scholar] [CrossRef]
- Brunner, K.; Maric, S.; Reshma, R.S.; Almqvist, H.; Seashore-Ludlow, B.; Gustavsson, A.L.; Poyraz, O.; Yogeeswari, P.; Lundback, T.; Vallin, M.; et al. Inhibitors of the cysteine synthase CysM with antibacterial potency against dormant Mycobacterium tuberculosis. J. Med. Chem. 2016, 59, 6848–6859. [Google Scholar] [CrossRef]
- Palde, P.B.; Bhaskar, A.; Pedro Rosa, L.E.; Madoux, F.; Chase, P.; Gupta, V.; Spicer, T.; Scampavia, L.; Singh, A.; Carroll, K.S. First-in-class inhibitors of sulfur metabolism with bactericidal activity against non-replicating M. tuberculosis. ACS Chem. Biol. 2016, 11, 172–184. [Google Scholar] [CrossRef]
- Campanini, B.; Speroni, F.; Salsi, E.; Cook, P.F.; Roderick, S.L.; Huang, B.; Bettati, S.; Mozzarelli, A. Interaction of serine acetyltransferase with O-acetylserine sulfhydrylase active site: Evidence from fluorescence spectroscopy. Protein Sci. 2005, 14, 2115–2124. [Google Scholar] [CrossRef]
- Mori, M.; Tsuge, S.; Fukasawa, W.; Jeelani, G.; Nakada-Tsukui, K.; Nonaka, K.; Matsumoto, A.; Omura, S.; Nozaki, T.; Shiomi, K. Discovery of antiamebic compounds that inhibit cysteine synthase from the enteric parasitic protist Entamoeba histolytica by screening of microbial secondary metabolites. Front. Cell. Infect. Mi. 2018, 8, 409. [Google Scholar] [CrossRef]
- Brunner, K.; Steiner, E.M.; Reshma, R.S.; Sriram, D.; Schnell, R.; Schneider, G. Profiling of in vitro activities of urea-based inhibitors against cysteine synthases from Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2017, 27, 4582–4587. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, I.; Raj, I.; Subbarao, N.; Gourinath, S. Virtual screening, identification and in vitro testing of novel inhibitors of O-acetyl-L-serine sulfhydrylase of Entamoeba histolytica. PloS ONE 2012, 7, e30305. [Google Scholar] [CrossRef] [PubMed]
- Kredich, N.M.; Becker, M.A.; Tomkins, G.M. Purification and characterization of cysteine synthetase, a bifunctional protein complex, from Salmonella typhimurium. J. Biol. Chem. 1969, 244, 2428–2439. [Google Scholar] [PubMed]
- Campanini, B.; Benoni, R.; Bettati, S.; Beck, C.M.; Hayes, C.S.; Mozzarelli, A. Moonlighting O-acetylserine sulfhydrylase: New functions for an old protein. Biochim. Biophys. Acta 2015, 1854, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Baecker, P.A.; Wedding, R.T. Purification of serine acetyltransferase, a component of a multienzyme complex, by immunoadsorption and selective dissociation of the complex. Anal. Biochem. 1980, 102, 16–21. [Google Scholar] [CrossRef]
- Hell, R.; Jost, R.; Berkowitz, O.; Wirtz, M. Molecular and biochemical analysis of the enzymes of cysteine biosynthesis in the plant Arabidopsis thaliana. Amino Acids 2002, 22, 245–257. [Google Scholar] [CrossRef]
- Feldman-Salit, A.; Wirtz, M.; Hell, R.; Wade, R.C. A mechanistic model of the cysteine synthase complex. J. Mol. Biol. 2009, 386, 37–59. [Google Scholar] [CrossRef]
- Feldman-Salit, A.; Wirtz, M.; Lenherr, E.D.; Throm, C.; Hothorn, M.; Scheffzek, K.; Hell, R.; Wade, R.C. Allosterically gated enzyme dynamics in the cysteine synthase complex regulate cysteine biosynthesis in Arabidopsis thaliana. Structure 2012, 20, 292–302. [Google Scholar] [CrossRef]
- Mino, K.; Yamanoue, T.; Sakiyama, T.; Eisaki, N.; Matsuyama, A.; Nakanishi, K. Effects of bienzyme complex formation of cysteine synthetase from Escherichia coli on some properties and kinetics. Biosci. Biotechnol. Biochem. 2000, 64, 1628–1640. [Google Scholar] [CrossRef]
- Benoni, R.; De Bei, O.; Paredi, G.; Hayes, C.S.; Franko, N.; Mozzarelli, A.; Bettati, S.; Campanini, B. Modulation of Escherichia coli serine acetyltransferase catalytic activity in the cysteine synthase complex. FEBS Lett. 2017, 591, 1212–1224. [Google Scholar] [CrossRef]
- Salsi, E.; Campanini, B.; Bettati, S.; Raboni, S.; Roderick, S.L.; Cook, P.F.; Mozzarelli, A. A two-step process controls the formation of the bienzyme cysteine synthase complex. J. Biol. Chem. 2010, 285, 12813–12822. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Leyh, T.S. Three-stage assembly of the cysteine synthase complex from Escherichia coli. J. Biol. Chem. 2012, 287, 4360–4367. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Vetting, M.W.; Roderick, S.L. The active site of O-acetylserine sulfhydrylase is the anchor point for bienzyme complex formation with serine acetyltransferase. J. Bacteriol. 2005, 187, 3201–3205. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, S.; Yi, H.; Krishnan, H.B.; Jez, J.M. Assembly of the cysteine synthase complex and the regulatory role of protein-protein interactions. J. Biol. Chem. 2009, 284, 10268–10275. [Google Scholar] [CrossRef]
- Mino, K.; Hiraoka, K.; Imamura, K.; Sakiyama, T.; Eisaki, N.; Matsuyama, A.; Nakanishi, K. Characteristics of serine acetyltransferase from Escherichia coli deleting different lengths of amino acid residues from the C-terminus. Biosci. Biotechnol. Biochem. 2000, 64, 1874–1880. [Google Scholar] [CrossRef]
- Mino, K.; Imamura, K.; Sakiyama, T.; Eisaki, N.; Matsuyama, A.; Nakanishi, K. Increase in the stability of serine acetyltransferase from Escherichia coli against cold inactivation and proteolysis by forming a bienzyme complex. Biosci. Biotechnol. Biochem. 2001, 65, 865–874. [Google Scholar] [CrossRef]
- Becker, M.A.; Kredich, N.M.; Tomkins, G.M. The purification and characterization of O-acetylserine sulfhydrylase-A from Salmonella typhimurium. J. Biol. Chem. 1969, 244, 2418–2427. [Google Scholar]
- Droux, M.; Ruffet, M.L.; Douce, R.; Job, D. Interactions between serine acetyltransferase and O-acetylserine (thiol) lyase in higher plants - Structural and kinetic properties of the free and bound enzymes. Eur. J. Biochem. 1998, 255, 235–245. [Google Scholar] [CrossRef]
- Hell, R.; Hillebrand, H. Plant concepts for mineral acquisition and allocation. Curr. Opin. Biotechnol. 2001, 12, 161–168. [Google Scholar] [CrossRef]
- Kumada, Y.; Zhao, C.; Ishimura, R.; Imanaka, H.; Imamura, K.; Nakanishi, K. Protein-protein interaction analysis using an affinity peptide tag and hydrophilic polystyrene plate. J. Biotechnol. 2006, 128, 354–361. [Google Scholar] [CrossRef]
- Zhao, C.; Moriga, Y.; Feng, B.; Kumada, Y.; Imanaka, H.; Imamura, K.; Nakanishi, K. On the interaction site of serine acetyltransferase in the cysteine synthase complex from Escherichia coli. Biochem. Biophys. Res. Commun. 2006, 341, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Campanini, B.; Raboni, S.; Vaccari, S.; Zhang, L.; Cook, P.F.; Hazlett, T.L.; Mozzarelli, A.; Bettati, S. Surface-exposed tryptophan residues are essential for O-acetylserine sulfhydrylase structure, function, and stability. J. Biol. Chem. 2003, 278, 37511–37519. [Google Scholar] [CrossRef] [PubMed]
- Francois, J.A.; Kumaran, S.; Jez, J.M. Structural basis for interaction of o-acetylserine sulfhydrylase and serine acetyltransferase in the Arabidopsis cysteine synthase complex. Plant Cell 2006, 18, 3647–3655. [Google Scholar] [CrossRef] [PubMed]
- Jez, J.M.; Dey, S. The cysteine regulatory complex from plants and microbes: What was old is new again. Curr. Opin. Struct. Biol. 2013, 23, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, M.; Berkowitz, O.; Droux, M.; Hell, R. The cysteine synthase complex from plants. Mitochondrial serine acetyltransferase from Arabidopsis thaliana carries a bifunctional domain for catalysis and protein-protein interaction. Eur. J. Biochem. 2001, 268, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, M.; Hell, R. Functional analysis of the cysteine synthase protein complex from plants: Structural, biochemical and regulatory properties. J. Plant Physiol. 2006, 163, 273–286. [Google Scholar] [CrossRef]
- Wirtz, M.; Birke, H.; Heeg, C.; Muller, C.; Hosp, F.; Throm, C.; Konig, S.; Feldman-Salit, A.; Rippe, K.; Petersen, G.; et al. Structure and function of the hetero-oligomeric cysteine synthase complex in plants. J. Biol. Chem. 2010, 285, 32810–32817. [Google Scholar] [CrossRef]
- Mino, K.; Yamanoue, T.; Sakiyama, T.; Eisaki, N.; Matsuyama, A.; Nakanishi, K. Purification and characterization of serine acetyltransferase from Escherichia coli partially truncated at the C-terminal region. Biosci. Biotechnol. Biochem. 1999, 63, 168–179. [Google Scholar] [CrossRef]
- Hindson, V.J.; Moody, P.C.; Rowe, A.J.; Shaw, W.V. Serine acetyltransferase from Escherichia coli is a dimer of trimers. J. Biol. Chem. 2000, 275, 461–466. [Google Scholar] [CrossRef]
- Johnson, P.M.; Gucinski, G.C.; Garza-Sanchez, F.; Wong, T.; Hung, L.W.; Hayes, C.S.; Goulding, C.W. Functional diversity of cytotoxic tRNase/immunity protein complexes from Burkholderia pseudomallei. J. Biol. Chem. 2016, 291, 19387–19400. [Google Scholar] [CrossRef]
- Becker, M.A.; Tomkins, G.M. Pleiotrophy in a cysteine-requiring mutant of Samonella typhimurium resulting from altered protein-protein interaction. J. Biol. Chem. 1969, 244, 6023–6030. [Google Scholar] [PubMed]
- Hulanicka, M.D.; Hallquist, S.G.; Kredich, N.M.; Mojica, A.T. Regulation of O-acetylserine sulfhydrylase B by L-cysteine in Salmonella typhimurium. J. Bacteriol. 1979, 140, 141–146. [Google Scholar] [PubMed]
- Berkowitz, O.; Wirtz, M.; Wolf, A.; Kuhlmann, J.; Hell, R. Use of biomolecular interaction analysis to elucidate the regulatory mechanism of the cysteine synthase complex from Arabidopsis thaliana. J. Biol. Chem. 2002, 277, 30629–30634. [Google Scholar] [CrossRef] [PubMed]
- Luchini, A.; Espina, V.; Liotta, L.A. Protein painting reveals solvent-excluded drug targets hidden within native protein-protein interfaces. Nat. Commun. 2014, 5, 4413. [Google Scholar] [CrossRef] [PubMed]
- Haymond, A.; Dey, D.; Carter, R.; Dailing, A.; Nara, V.; Nara, P.; Venkatayogi, S.; Paige, M.; Liotta, L.; Luchini, A. Protein painting, an optimized MS-based technique, reveals functionally relevant interfaces of the PD-1/PD-L1 complex and the YAP2/ZO-1 complex. J. Biol. Chem. 2019, 294, 11180–11198. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Hutchinson, E.G.; Michie, A.D.; Wallace, A.C.; Jones, M.L.; Thornton, J.M. PDBsum: A Web-based database of summaries and analyses of all PDB structures. Trends Biochem. Sci 1997, 22, 488–490. [Google Scholar] [CrossRef]
- Johnson, P.M.; Beck, C.M.; Morse, R.P.; Garza-Sanchez, F.; Low, D.A.; Hayes, C.S.; Goulding, C.W. Unraveling the essential role of CysK in CDI toxin activation. Proc. Natl. Acad. Sci. USA 2016, 113, 9792–9797. [Google Scholar] [CrossRef]
- Bonner, E.R.; Cahoon, R.E.; Knapke, S.M.; Jez, J.M. Molecular basis of cysteine biosynthesis in plants: Structural and functional analysis of O-acetylserine sulfhydrylase from Arabidopsis thaliana. J. Biol. Chem. 2005, 280, 38803–38813. [Google Scholar] [CrossRef]
- Burkhard, P.; Rao, G.S.; Hohenester, E.; Schnackerz, K.D.; Cook, P.F.; Jansonius, J.N. Three-dimensional structure of O-acetylserine sulfhydrylase from Salmonella typhimurium. J. Mol. Biol. 1998, 283, 121–133. [Google Scholar] [CrossRef]
- Burkhard, P.; Tai, C.H.; Ristroph, C.M.; Cook, P.F.; Jansonius, J.N. Ligand binding induces a large conformational change in O-acetylserine sulfhydrylase from Salmonella typhimurium. J. Mol. Biol. 1999, 291, 941–953. [Google Scholar] [CrossRef]
- Burkhard, P.; Tai, C.H.; Jansonius, J.N.; Cook, P.F. Identification of an allosteric anion-binding site on O-acetylserine sulfhydrylase: Structure of the enzyme with chloride bound. J. Mol. Biol. 2000, 303, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.; Barberato, C.; Koch, M.H.J. CRYSOL—A program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- Valentini, E.; Kikhney, A.G.; Previtali, G.; Jeffries, C.M.; Svergun, D.I. SASBDB, a repository for biological small-angle scattering data. Nucleic Acids Res. 2014, 43, D357–D363. [Google Scholar] [CrossRef] [PubMed]
- Olsen, L.R.; Huang, B.; Vetting, M.W.; Roderick, S.L. Structure of serine acetyltransferase in complexes with CoA and its cysteine feedback inhibitor. Biochemistry 2004, 43, 6013–6019. [Google Scholar] [CrossRef]
- Mylonas, E.; Svergun, D.I. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J. Appl. Crystallogr. 2007, 40, s245–s249. [Google Scholar] [CrossRef]
- Peterson, E.A.; Sober, H.A. Preparation of crystalline phosphorylated derivatives of vitamin B6. J. Am. Chem. Soc. 1954, 76, 169–175. [Google Scholar] [CrossRef]
- Gaitonde, M.K. A spectrophotometric method for the direct determination of cysteine in the presence of other naturally occurring amino acids. Biochem. J. 1967, 104, 627–633. [Google Scholar] [CrossRef]
- Aoki, S.K.; Diner, E.J.; de Roodenbeke, C.t.K.; Burgess, B.R.; Poole, S.J.; Braaten, B.A.; Jones, A.M.; Webb, J.S.; Hayes, C.S.; Cotter, P.A.; et al. A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature 2010, 468, 439–442. [Google Scholar] [CrossRef]
- Luchini, C.; Capelli, P.; Fassan, M.; Simbolo, M.; Mafficini, A.; Pedica, F.; Ruzzenente, A.; Guglielmi, A.; Corbo, V.; Scarpa, A. Next-generation histopathologic diagnosis: A lesson from a hepatic carcinosarcoma. J. Clin. Oncol. 2014, 32, e63–e66. [Google Scholar] [CrossRef]
- Bergmann, A.; Fritz, G.; Glatter, O. Solving the generalized indirect Fourier transformation (GIFT) by Boltzmann simplex simulated annealing (BSSA). J. Appl. Crystallogr. 2000, 33, 1212–1216. [Google Scholar] [CrossRef]
- Svergun, D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Franke, D.; Svergun, D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009, 42, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Volkov, V.V.; Svergun, D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003, 36, 860–864. [Google Scholar] [CrossRef]
- Kozin, M.B.; Svergun, D.I. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 2001, 34, 33–41. [Google Scholar] [CrossRef]
- Panjkovich, A.; Svergun, D.I. SASpy: A PyMOL plugin for manipulation and refinement of hybrid models against small angle X-ray scattering data. Bioinformatics 2016, 32, 2062–2064. [Google Scholar] [CrossRef]
- Petoukhov, M.V.; Svergun, D.I. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 2005, 89, 1237–1250. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | CysK | CysE | CS |
|---|---|---|---|
| Guinier Analysis | |||
| Guinier radius Rg (nm) | 2.64 ± 0.05 | 3.90 ± 0.04 | 6.10 ± 0.11 |
| I(0) (cm−1) | 6.71 ± 0.02 | 19.08 ± 0.09 | 27.03 ± 0.23 |
| q-range (nm−1) | 0.22–0.39 | 0.13–0.31 | 0.13–0.2 |
| Quality (%) | 96 | 91 | 99 |
| GNOM | |||
| Max. dimension Dmax (nm) | 8.5 | 13.0 | 22.0 |
| Guinier radius Rg (nm) | 2.623 ± 0.006 | 3.856 ± 0.009 | 6.606 ± 0.003 |
| q-range (nm−1) | 0.1–3.5 | 0.1–3.5 | 0.1–3.5 |
| Porod volume (kDa) | 72 | 189 | 303 |
| CRYSOL | |||
| PDB file | 1oas | Hexamer built from 1t3d | |
| Guinier radius Rg (nm) | 2.572 | 3.361 | |
| Molecular weight (kDa) | 67.57 | 171.6 | |
| Max. dimension Dmax (nm) | 8.675 | 11.0 | |
| DAMMIF (def. parameter, 10 runs) | |||
| q range for fitting (nm−1) | 0.12–3.9 | 0.12–3.9 | 0.12–3.9 |
| Symmetry, anisotropy assumption | P1, unknown | P1, unknown | P1, unknown |
| χ2 range | 1.803–1.833 | 2.044–2.119 | 2.077–3.731 |
| Resolution from SASRES (nm) | 4.6 ± 0.3 | 2.9 ± 0.2 | 9.4 ± 0.2 |
| NSD (standard dev.), no clusters | 0.9 (0.12), 1 | 0.59 (0.01), 1 | 1.43(0.06), 2 |
| MW estimate for proteins (kDa) | 58.6 | 155 | 300 |
| Phase radius of gyration (nm) | 2.62 | 3.86 | 6.61 |
| Maximum phase diameter (nm) | 10.500 | 14.800 | 24.300 |
| SASBDB [83] codes | SASDGW6 | SASDGV6 | SASDGX6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosa, B.; Marchetti, M.; Paredi, G.; Amenitsch, H.; Franko, N.; Benoni, R.; Giabbai, B.; De Marino, M.G.; Mozzarelli, A.; Ronda, L.; et al. Combination of SAXS and Protein Painting Discloses the Three-Dimensional Organization of the Bacterial Cysteine Synthase Complex, a Potential Target for Enhancers of Antibiotic Action. Int. J. Mol. Sci. 2019, 20, 5219. https://doi.org/10.3390/ijms20205219
Rosa B, Marchetti M, Paredi G, Amenitsch H, Franko N, Benoni R, Giabbai B, De Marino MG, Mozzarelli A, Ronda L, et al. Combination of SAXS and Protein Painting Discloses the Three-Dimensional Organization of the Bacterial Cysteine Synthase Complex, a Potential Target for Enhancers of Antibiotic Action. International Journal of Molecular Sciences. 2019; 20(20):5219. https://doi.org/10.3390/ijms20205219
Chicago/Turabian StyleRosa, Brenda, Marialaura Marchetti, Gianluca Paredi, Heinz Amenitsch, Nina Franko, Roberto Benoni, Barbara Giabbai, Maria Giovanna De Marino, Andrea Mozzarelli, Luca Ronda, and et al. 2019. "Combination of SAXS and Protein Painting Discloses the Three-Dimensional Organization of the Bacterial Cysteine Synthase Complex, a Potential Target for Enhancers of Antibiotic Action" International Journal of Molecular Sciences 20, no. 20: 5219. https://doi.org/10.3390/ijms20205219
APA StyleRosa, B., Marchetti, M., Paredi, G., Amenitsch, H., Franko, N., Benoni, R., Giabbai, B., De Marino, M. G., Mozzarelli, A., Ronda, L., Storici, P., Campanini, B., & Bettati, S. (2019). Combination of SAXS and Protein Painting Discloses the Three-Dimensional Organization of the Bacterial Cysteine Synthase Complex, a Potential Target for Enhancers of Antibiotic Action. International Journal of Molecular Sciences, 20(20), 5219. https://doi.org/10.3390/ijms20205219