Microcirculatory Changes in Experimental Models of Stroke and CNS-Injury Induced Immunodepression



Abstract
1. Introduction
2. Animal Models of Ischemic Stroke
2.1. Middle Cerebral Artery Occlusion Model
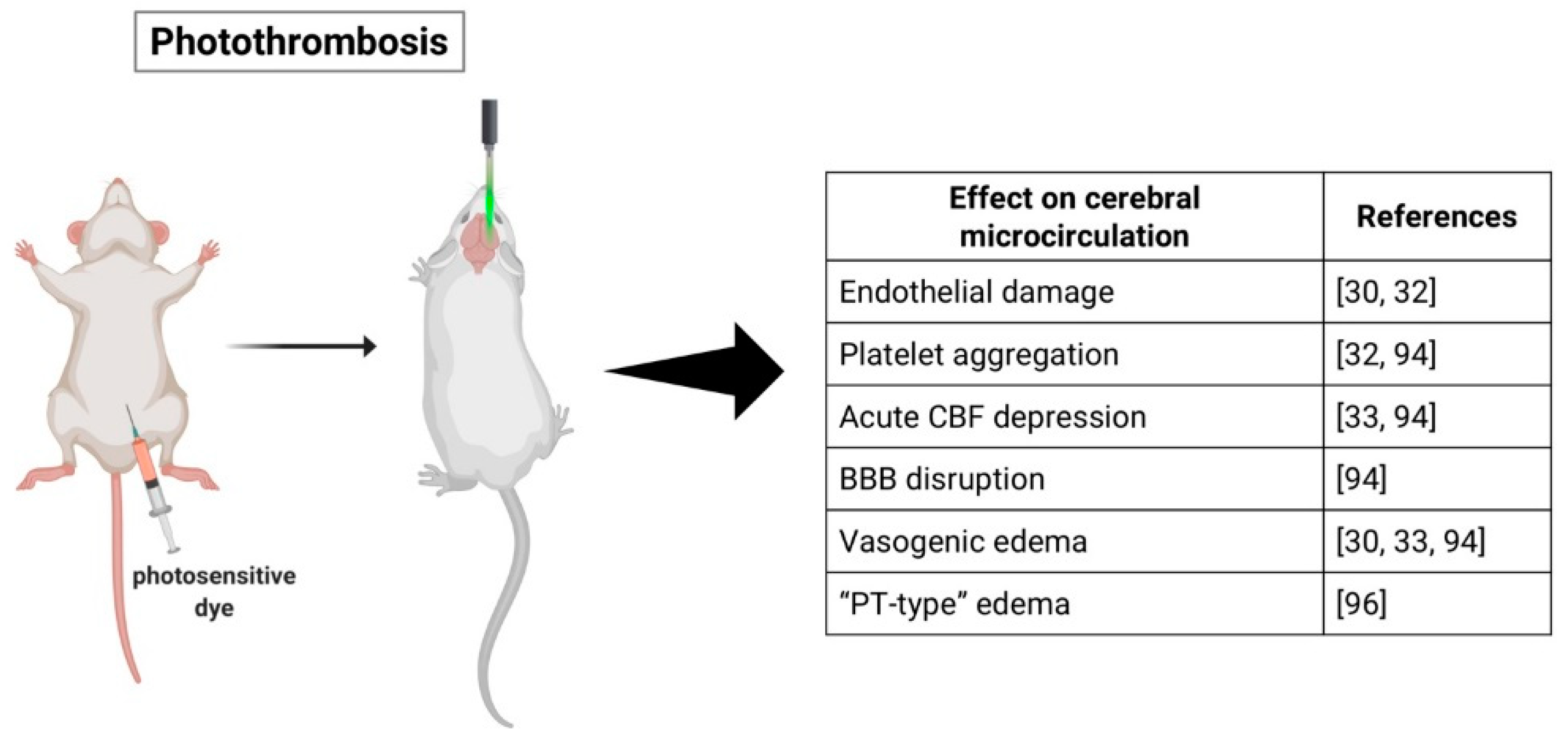
2.2. Photothrombotic Stroke Model
2.3. Endothelin-1 Model
2.4. Hypoxia-Ischemia Model
3. Cerebral Microcirculation in Experimental Stroke
3.1. Middle Cerebral Artery Occlusion Model
3.2. Photothrombotic Stroke Model
3.3. Endothelin-1 Model
3.4. Hypoxia-Ischemia Model
4. Peripheral Immune Response
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CNS | Central nervous system |
| DALYs | Disability-adjusted life-years |
| HPA | Hypothalamic-pituitary-adrenal |
| SNS | Sympathetic nervous system |
| CIDS | CNS injury-induced immunodepression syndrome |
| MCAO | Middle cerebral artery occlusion |
| ICA | Internal carotid artery |
| MCA | Middle cerebral artery |
| SAH | Subarachnoid hemorrhage |
| PTS | Photothrombotic stroke |
| CBF | Cerebral blood flow |
| BBB | Blood-brain barrier |
| ET-1 | Endothelin-1 |
| HI | Hypoxia-ischemia |
| CCA | Common carotid artery |
| ECA | External carotid artery |
| CBV | Cerebral blood volume |
| ACA | Anterior cerebral artery |
| CCs | Collateral channels |
| ROS | Reactive oxygen species |
| IL-6 | Interleukin-6 |
| IL-1β | Interleukin-1β |
| NK | Natural killer |
| IFN-γ | Interferon-γ |
| TNF-α | Tumor necrosis factor alpha |
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; De Ferranti, S.; Després, J.P.; Fullerton, H.J.; et al. Heart disease and stroke statistics–2016 Update: A Report From the American Heart Association. Circulation 2016, 133, e38–e360. [Google Scholar] [CrossRef] [PubMed]
- Naghavi, M.; Alemu Abajobir, A.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Ferede Abera, S.; Aboyans, V.; Adetokunboh, O.; Afshin, A.; Agrawal, A.; et al. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1151–1210. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; De Ferranti, S.; Després, J.P.; Fullerton, H.J.; et al. Executive summary: Heart disease and stroke statistics-2016 update: A Report from the American Heart Association. Circulation 2016, 133, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Guzik, A. & Bushnell, C. Stroke Epidemiology and risk factors. Contin. Lifelong Learn. Neurol. 2017, 23, 15–39. [Google Scholar]
- Johnson, C.O.; Nguyen, M.; Roth, G.A.; Nichols, E.; Alam, T.; Abate, D.; Abd-Allah, F.; Abdelalim, A.; Abraha, H.N.; Abu-Rmeileh, N.M.; et al. Global, regional, and national burden of stroke, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 439–458. [Google Scholar] [CrossRef]
- GBD 2016 Lifetime Risk of Stroke Collaborators Global, Regional, and Country-Specific Lifetime Risks of Stroke, 1990 and 2016. N. Engl. J. Med. 2018, 379, 2429–2437.
- Feigin, V.L.; Krishnamurthi, R.V.; Theadom, A.M.; Abajobir, A.A.; Mishra, S.R.; Ahmed, M.B.; Abate, K.H.; Mengistie, M.A.; Wakayo, T.; Abd-Allah, F.; et al. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef]
- Disease Control Priorities, Third Edition (Volume 5): Cardiovascular, Respiratory, and Related Disorders; Prabhakaran, D., Anand, S., Watkins, D.A., Gaziano, T., Wu, Y., Mbanya, J.-C., Nugent, R., Eds.; The International Bank for Reconstruction and Development/The World Bank: Washington, DC, USA, 2017. [Google Scholar]
- Kauhanen, M.L.; Korpelainen, J.T.; Hiltunen, P.; Brusin, E.; Mononen, H.; Määttä, R.; Nieminen, P.; Sotaniemi, K.A.; Myllylä, V.V. Poststroke depression correlates with cognitive impairment and neurological deficits. Stroke 1999, 30, 1875–1880. [Google Scholar] [CrossRef]
- Langhorne, P.; Stott, D.J.; Robertson, L.; MacDonald, J.; Jones, L.; McAlpine, C.; Dick, F.; Taylor, G.S.; Murray, G. Medical complications after stroke: A multicenter study. Stroke 2000, 31, 1223–1229. [Google Scholar] [CrossRef]
- Johnston, K.C.; Li, J.Y.; Lyden, P.D.; Hanson, S.K.; Feasby, T.E.; Adams, R.J.; Faught, R.E.; Haley, E.C. Medical and Neurological Complications of Ischemic Stroke. Stroke 2011, 29, 447–453. [Google Scholar] [CrossRef]
- Heuschmann, P.U.; Kolominsky-Rabas, P.L.; Misselwitz, B.; Hermanek, P.; Leffmann, C.; Janzen, R.W.; Rother, J.; Buecker-Nott, H.J.; Berger, K. Predictors of in-hospital mortality and attributable risks of death after ischemic stroke: The German Stroke Registers Study Group. Arch. Intern. Med. 2004, 164, 1761–1768. [Google Scholar] [CrossRef] [PubMed]
- Prass, K.; Meisel, C.; Höflich, C.; Braun, J.; Halle, E.; Wolf, T.; Ruscher, K.; Victorov, I.V.; Priller, J.; Dirnagl, U.; et al. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J. Exp. Med. 2003, 198, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, K.G.; Schmidt, W.U.H.; Föhring, F.; Meisel, C.; Helms, T.; Jungehulsing, G.J.; Nolte, C.H.; Schmolke, K.; Wegner, B.; Meisel, A.; et al. Cellular immunodepression preceding infectious complications after acute ischemic stroke in humans. Cerebrovasc. Dis. 2008, 25, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, B.A.; Neuhaus, A.A.; Couch, Y.; Balami, J.S.; Deluca, G.C.; Hadley, G.; Harris, S.L.; Grey, A.N.; Buchan, A.M. The transient intraluminal filament middle cerebral artery occlusion model as a model of endovascular thrombectomy in stroke. J. Cereb. Blood Flow Metab. 2016, 36, 363–369. [Google Scholar] [CrossRef]
- Durukan, A.; Tatlisumak, T. Acute ischemic stroke: Overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol. Biochem. Behav. 2007, 87, 179–197. [Google Scholar] [CrossRef]
- Koizumi, J.; Yoshida, Y.; Nakazawa, T.; Ooneda, G. Experimental studies of ischemic brain edema: 1. A new experimental model of cerebral embolism in rats in which recirculation can be introduced in the ischemic area. Jpn. J. Stroke 1986, 8, 1–8. [Google Scholar] [CrossRef]
- Fluri, F.; Schuhmann, M.; Kleinschnitz, C. Animal models of ischemic stroke and their application in clinical research. Drug Des. Devel. Ther. 2015, 9, 3445–3454. [Google Scholar]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible Middle Cerebral Artery Occlusion Without Craniectomy in Rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef]
- Hata, R.; Mies, G.; Wiessner, C.; Fritze, K.; Hesselbarth, D.; Brinker, G.; Hossmann, K.-A. A Reproducible Model of Middle Cerebral Artery Occlusion in Mice: Hemodynamic, Biochemical, and Magnetic Resonance Imaging. J. Cereb. Blood Flow Metab. 2003, 18, 367–375. [Google Scholar] [CrossRef]
- Sommer, C.J. Ischemic stroke: Experimental models and reality. Acta Neuropathol. 2017, 133, 245–261. [Google Scholar] [CrossRef]
- McCabe, C.; Arroja, M.M.; Reid, E.; Macrae, I.M. Animal models of ischaemic stroke and characterisation of the ischaemic penumbra. Neuropharmacology 2018, 134, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Olsen, T.S.; Skriver, E.B.; Herning, M. Cause of cerebral infarction in the carotid territory. Its relation to the size and the location of the infarct and to the underlying vascular lesion. Stroke 1985, 16, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Schmid-elsaesser, R.; Zausinger, S.; Hungerhuber, E.; Baethmann, A.; Reulen, H.-J. A Critical Reevaluation of the Intraluminal Thread Model of Focal Cerebral Ischemia: Evidence of Inadvertent Premature Reperfusion and Subarachnoid Hemorrhage in Rats by Laser-Doppler Flowmetry. Stroke 1998, 29, 2162–2170. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Omae, T.; Fisher, M. Spontaneous Hyperthermia and Its Mechanism in the Intraluminal Suture Middle Cerebral Artery Occlusion Model of Rats. Stroke 1999, 30, 2464–2471. [Google Scholar] [CrossRef]
- Gerriets, T.; Stolz, E.; Walberer, M.; Müller, C.; Rottger, C.; Kluge, A.; Kaps, M.; Fisher, M.; Bachmann, G. Complications and Pitfalls in Rat Stroke Models for Middle Cerebral Artery Occlusion: A Comparison Between the Suture and the Macrosphere Model Using Magnetic Resonance Angiography. Stroke 2004, 35, 2372–2377. [Google Scholar] [CrossRef]
- Hossmann, K.A. The two pathophysiologies of focal brain ischemia: Implications for translational stroke research. J. Cereb. Blood Flow Metab. 2012, 32, 1310–1316. [Google Scholar] [CrossRef]
- Berkhemer, O.A.; Fransen, P.S.S.; Beumer, D.; van den Berg, L.A.; Lingsma, H.F. A randomized Trial of Intraarterial Treatment for Acute Ischemic Stroke. N. Engl. J. Med. 2015, 372, 11–20. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; Mitchell, P.J.; Kleinig, T.J.; Dewey, H.M.; Churilov, L.; Yassi, N.; Yan, B.; Dowling, R.J.; Parsons, M.W.; Oxley, T.J.; et al. Endovascular Therapy for Ischemic Stroke with Perfusion-Imaging Selection. N. Engl. J. Med. 2015, 372, 1009–1018. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, R.L.; Jiang, Q.; Ding, G.; Chopp, M.; Zhang, Z.G. Focal embolic cerebral ischemia in the rat. Nat. Protoc. 2015, 10, 539–547. [Google Scholar] [CrossRef]
- Macrae, I.M. Preclinical stroke research—Advantages and disadvantages of the most common rodent models of focal ischaemia. Br. J. Pharmacol. 2011, 164, 1062–1078. [Google Scholar] [CrossRef]
- Howells, D.W.; Porritt, M.J.; Rewell, S.S.; O’Collins, V.; Sena, E.S.; van Der Worp, H.B.; Traystman, R.J.; MacLeod, M.R. Different strokes for different folks: The rich diversity of animal models of focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 1412–1431. [Google Scholar] [CrossRef] [PubMed]
- Watson, B.D.; Dietrich, W.D.; Busto, R.; Wachtel, M.S.; Ginsberg, M.D. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann. Neurol. 1985, 17, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Sugawara, T.; Chan, P.H. Involvement of oxidative stress and caspase-3 in cortical infarction after photothrombotic ischemia in mice. J. Cereb. Blood Flow Metab. 2000, 20, 1690–1701. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, W.D.; Watson, B.D.; Busto, R.; Ginsberg, M.D.; Bethea, J.R. Photochemically induced cerebral infarction. I. Early microvascular alterations. Acta Neuropathol. 1987, 72, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, W.D.; Ginsberg, M.D.; Busto, R.; Watson, B.D. Photochemically induced cortical infarction in the rat. 1. Time course of hemodynamic consequences. J. Cereb. Blood Flow Metab. 1986, 6, 184–194. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Braeuninger, S.; Pham, M.; Austinat, M.; Nölte, I.; Renné, T.; Nieswandt, B.; Bendszus, M.; Stoll, G. Blocking of platelets or intrinsic coagulation pathway-driven thrombosis does not prevent cerebral infarctions induced by photothrombosis. Stroke 2008, 39, 1262–1268. [Google Scholar] [CrossRef]
- Labat-gest, V.; Tomasi, S. Photothrombotic ischemia: A minimally invasive and reproducible photochemical cortical lesion model for mouse stroke studies. J. Vis. Exp. 2013. [Google Scholar] [CrossRef]
- Uzdensky, A.B. Photothrombotic Stroke as a Model of Ischemic Stroke. Transl. Stroke Res. 2018, 9, 437–451. [Google Scholar] [CrossRef]
- Klatzo, I. Brain oedema following brain ischaemia and the influence of therapy. Br. J. Anaesth. 1985, 57, 18–22. [Google Scholar] [CrossRef][Green Version]
- Lee, V.M.; Burdett, N.; Carpenter, T.A.; Hall, L.D.; Pambakian, P.S.; Patel, S.; Wood, N.I.; James, M.F. Evolution of Photochemically Induced Focal Cerebral Ischemia in the Rat: Magnetic Resonance Imaging and Histology. Stroke 1996, 27, 2110–2118. [Google Scholar] [CrossRef]
- Wester, P.; Watson, B.D.; Prado, R.; Dietrich, W.D. A Photothrombotic ‘Ring’ Model of Rat Stroke-in-Evolution Displaying Putative Penumbral Inversion. Stroke 1995, 26, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Hilger, T.; Blunk, J.A.; Hoehn, M.; Mies, G.; Wester, P. Characterization of a novel chronic photothrombotic ring stroke model in rats by magnetic resonance imaging, biochemical imaging, and histology. J. Cereb. Blood Flow Metab. 2004, 24, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Pevsner, P.H.; Eichenbaum, J.W.; Miller, D.C.; Pivawer, G.; Eichenbaum, K.D.; Stern, A.; Zakian, K.L.; Koutcher, J.A. A photothrombotic model of small early ischemic infarcts in the rat brain with histologic and MRI correlation. J. Pharmacol. Toxicol. Methods 2001, 45, 227–233. [Google Scholar] [CrossRef]
- Qian, C.; Li, P.C.; Jiao, Y.; Yao, H.H.; Chen, Y.C.; Yang, J.; Ding, J.; Yang, X.Y.; Teng, G.J. Precise characterization of the penumbra revealed by MRI: A modified photothrombotic stroke model study. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Lu, H.; Li, Y.; Yuan, L.; Li, H.; Lu, X.; Tong, S. Induction and imaging of photothrombotic stroke in conscious and freely moving rats. J. Biomed. Opt. 2014, 19, 096013. [Google Scholar] [CrossRef]
- Yu, C.L.; Zhou, H.; Chai, A.P.; Yang, Y.X.; Mao, R.R.; Xu, L. Whole-scale neurobehavioral assessments of photothrombotic ischemia in freely moving mice. J. Neurosci. Methods 2015, 239, 100–107. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Goto, K.; Masaki, T. A novel peptide vasoconstrictor, endothelin, is produced by vascular endothelium and modulates smooth muscle Ca2+ channels. J. Hypertens. Suppl. Off. J. Int. Soc. Hipertens. 1988, 6, S188–S191. [Google Scholar] [CrossRef]
- Robinson, M.J.; Macrae, I.M.; Todd, M.; Reid, J.L.; McCulloch, J. Reduction of local cerebral blood flow to pathological levels by endothelin-1 applied to the middle cerebral artery in the rat. Neurosci. Lett. 1990, 118, 269–272. [Google Scholar] [CrossRef]
- Sharkey, J.; Ritchie, I.M.; Kelly, P.A.T. Perivascular microapplication of endothelin-1: A new model of focal cerebral ischaemia in the rat. J. Cereb. Blood Flow Metab. 1993, 13, 865–871. [Google Scholar] [CrossRef]
- Hughes, P.M.; Anthony, D.C.; Ruddin, M.; Botham, M.S.; Rankine, E.L.; Sablone, M.; Baumann, D.; Mir, A.K.; Perry, V.H. Focal lesions in the rat central nervous system induced by endothelin-1. J. Neuropathol. Exp. Neurol. 2003, 62, 1276–1286. [Google Scholar] [CrossRef]
- Macrae, I.M.; Robinson, M.J.; Graham, D.I.; Reid, J.L.; McCulloch, J. Endothelin-1-induced reductions in cerebral blood flow: Dose dependency, time course, and neuropathological consequences. J. Cereb. Blood Flow Metab. 1993, 13, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Biernaskie, J.; Corbett, D.; Peeling, J.; Wells, J.; Lei, H. A serial MR study of cerebral blood flow changes and lesion development following endothelin-1-induced ischemia in rats. Magn. Reson. Med. 2001, 46, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, J.; Butcher, S.P. Characterisation of an experimental model of stroke produced by intracerebral microinjection of endothelin-1 adjacent to the rat middle cerebral artery. J. Neurosci. Methods 1995, 60, 125–131. [Google Scholar] [CrossRef]
- Horie, N.; Maag, A.L.; Hamilton, S.A.; Shichinohe, H.; Bliss, T.M.; Steinberg, G.K. Mouse model of focal cerebral ischemia using endothelin-1. J. Neurosci. Methods 2008, 173, 286–290. [Google Scholar] [CrossRef]
- Fuxe, K.; Bjelke, B.; Andbjer, B.; Grahn, H.; Rimondini, R.; Agnati, L.F. Endothelin-1 induced lesions of the frontoparietal cortex of the rat. A possible model of focal cortical ischemia. Neuroreport 1997, 8, 2623–2629. [Google Scholar] [CrossRef]
- Moyanova, S.; Kortenska, L.; Kirov, R.; Iliev, I. Quantitative Electroencephalographic Changes Due To Middle Cerebral Artery Occlusion By Endothelin 1 in Conscious Rats. Arch. Physiol. Biochem. 1998, 106, 384–391. [Google Scholar] [CrossRef]
- Nakagomi, S.; Kiryu-Seo, S.; Kiyama, H. Endothelin-converting enzymes and endothelin receptor B messenger RNAs are expressed in different neural cell species and these messenger RNAs are coordinately induced in neurons and astrocytes respectively following nerve injury. Neuroscience 2000, 101, 441–449. [Google Scholar] [CrossRef]
- Uesugi, M.; Kasuya, Y.; Hayashi, K.; Goto, K. SB209670, a potent endothelin receptor antagonist, prevents or delays axonal degeneration after spinal cord injury. Brain Res. 1998, 786, 235–239. [Google Scholar] [CrossRef]
- Uesugi, M.; Kasuya, Y.; Hama, H.; Yamamoto, M.; Hayashi, K.; Masaki, T.; Goto, K. Endogenous endothelin-1 initiates astrocytic growth after spinal cord injury. Brain Res. 1996, 728, 255–259. [Google Scholar] [CrossRef]
- Carmichael, S.T. Rodent models of focal stroke: Size, mechanism, and purpose. NeuroRx 2005, 2, 396–409. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, K.; Greenberg, D.A. Neurogenesis associated with endothelin-induced cortical infarction in the mouse. Brain Res. 2007, 1167, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Sozmen, E.G.; Kolekar, A.; Havton, L.A.; Carmichael, S.T. A white matter stroke model in the mouse: Axonal damage, progenitor responses and MRI correlates. J. Neurosci. Methods 2009, 180, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Roome, R.B.; Bartlett, R.F.; Jeffers, M.; Xiong, J.; Corbett, D.; Vanderluit, J.L. A reproducible Endothelin-1 model of forelimb motor cortex stroke in the mouse. J. Neurosci. Methods 2014, 233, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Levine, S. Anoxic-Ischemic encephalopathy in rats. Am. J. Pathol. 1960, 36, 1–17. [Google Scholar] [PubMed]
- Rice, J.E.; Vannucci, R.C.; Brierley, J.B. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 1981, 9, 131–141. [Google Scholar] [CrossRef]
- Adhami, F.; Liao, G.; Morozov, Y.M.; Schloemer, A.; Schmithorst, V.J.; Lorenz, J.N.; Dunn, R.S.; Vorhees, C.V.; Wills-Karp, M.; Degen, J.L.; et al. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am. J. Pathol. 2006, 169, 566–583. [Google Scholar] [CrossRef]
- Edwards, A.B.; Feindel, K.W.; Cross, J.L.; Anderton, R.S.; Clark, V.W.; Knuckey, N.W.; Meloni, B.P. Modification to the Rice-Vannucci perinatal hypoxic-ischaemic encephalopathy model in the P7 rat improves the reliability of cerebral infarct development after 48 hours. J. Neurosci. Methods 2017, 288, 62–71. [Google Scholar] [CrossRef]
- Okusa, C.; Oeschger, F.; Ginet, V.; Wang, W.Z.; Hoerder-Suabedissen, A.; Matsuyama, T.; Truttmann, A.C.; Molnár, Z. Subplate in a rat model of preterm hypoxia–ischemia. Ann. Clin. Transl. Neurol. 2014, 1, 679–691. [Google Scholar] [CrossRef]
- Vannucci, S.J.; Willing, L.B.; Goto, S.; Alkayed, N.J.; Brucklacher, R.M.; Wood, T.L.; Towfighi, J.; Hurn, P.D.; Simpson, I.A. Experimental stroke in the female diabetic, db/db, mouse. J. Cereb. Blood Flow Metab. 2001, 21, 52–60. [Google Scholar] [CrossRef]
- Basu, A.; Lazovic, J.; Krady, J.K.; Mauger, D.T.; Rothstein, R.P.; Smith, M.B.; Levison, S.W. Interleukin-1 and the interleukin-1 type 1 receptor are essential for the progressive neurodegeneration that ensues subsequent to a mild hypoxic/ischemic injury. J. Cereb. Blood Flow Metab. 2005, 25, 17–29. [Google Scholar] [CrossRef]
- O’Donnell, S.L.; Frederick, T.J.; Krady, J.K.; Vannucci, S.J.; Wood, T.L. IGF-I and microglia/macrophage proliferation in the ischemic mouse brain. Glia 2002, 39, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Silverstein, F.S.; Skoff, R.; Barks, J.D.E. Hypoxic-ischemic oligodendroglial injury in neonatal rat brain. Pediatr. Res. 2002, 51, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, X.; Xu, F.; Bahr, B.A.; Shibata, M.; Uchiyama, Y.; Hagberg, H.; Blomgren, K. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ. 2005, 12, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Towfighi, J.; Mauger, D.; Vannucci, R.C.; Vannucci, S.J. Influence of age on the cerebral lesions in an immature rat model of cerebral hypoxia-ischemia: A light microscopic study. Dev. Brain Res. 1997, 100, 149–160. [Google Scholar] [CrossRef]
- Yager, J.Y.; Shuaib, A.; Thornhill, J. The effect of age on susceptibility to brain damage in a model of global hemispheric hypoxia-ischemia. Dev. Brain Res. 1996, 93, 143–154. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Mosinger, J.; Salles, K.; Labruyere, J.; Olney, J. Sensitivity of the developing rat brain to hypobaric/ischemic damage parallels sensitivity to N-methyl-aspartate neurotoxicity. J. Neurosci. 2018, 9, 2809–2818. [Google Scholar] [CrossRef]
- Hagberg, H.; Bona, E.; Gilland, E.; Puka-Sundvall, M. Hypoxia-ischaemia model in the 7-day-old rat: Possibilities and shortcomings. Acta Paediatr. Suppl. 1997, 422, 85–88. [Google Scholar] [CrossRef]
- Theilen, H.; Schröck, H.; Kuschinsky, W. Gross persistence of capillary plasma perfusion after middle cerebral artery occlusion in the rat brain. J. Cereb. Blood Flow Metab. 1994, 14, 1055–1061. [Google Scholar] [CrossRef]
- Zhang, Z.; Davies, K.; Prostak, J.; Fenstermacher, J.; Chopp, M. Quantitation of microvascular plasma perfusion and neuronal microtubule-associated protein in ischemic mouse brain by laser-scanning confocal microscopy. J. Cereb. Blood Flow Metab. 1999, 19, 68–78. [Google Scholar] [CrossRef]
- Buchweitz-Milton, E.; Weiss, H.R. Perfused microvascular morphometry during middle cerebral artery occlusion. Am. J. Physiol. Circ. Physiol. 1988, 255, H623–H628. [Google Scholar] [CrossRef]
- Vogel, J.; Hermes, A.; Kuschinsky, W. Evolution of microcirculatory disturbances after permanent middle cerebral artery occlusion in rats. J. Cereb. Blood Flow Metab. 1999, 19, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Chang, C.; Cheung, W.M.; Lin, M.H.; Chen, J.J.; Hsu, C.Y.; Chen, J.H.; Lin, T.N. Dynamic changes in vascular permeability, cerebral blood volume, vascular density, and size after transient focal cerebral ischemia in rats: Evaluation with contrast-enhanced magnetic resonance imaging. J. Cereb. Blood Flow Metab. 2008, 28, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Martín, A.; Macé, E.; Boisgard, R.; Montaldo, G.; Thézé, B.; Tanter, M.; Tavitian, B. Imaging of perfusion, angiogenesis, and tissue elasticity after stroke. J. Cereb. Blood Flow Metab. 2012, 32, 1496–1507. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.N.; Sun, S.W.; Cheung, W.M.; Li, F.; Chang, C. Dynamic changes in cerebral blood flow and angiogenesis after transient focal cerebral ischemia in rats: Evaluation with serial magnetic resonance imaging. Stroke 2002, 33, 2985–2991. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.C.; Tsai, C.Y.; Liao, M.C.; Yang, J.L.; Su, C.H.; Chen, J.H. Quantitative susceptibility mapping-based microscopy of magnetic resonance venography (QSM-mMRV) for in vivo morphologically and functionally assessing cerebromicrovasculature in rat stroke model. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Liebeskind, D.S. Collateral circulation. Stroke 2003, 34, 2279–2284. [Google Scholar] [CrossRef] [PubMed]
- Armitage, G.A.; Todd, K.G.; Shuaib, A.; Winship, I.R. Laser speckle contrast imaging of collateral blood flow during acute ischemic stroke. J. Cereb. Blood Flow Metab. 2010, 30, 1432–1436. [Google Scholar] [CrossRef]
- Wang, Z.; Luo, W.; Zhou, F.; Li, P.; Luo, Q. Dynamic change of collateral flow varying with distribution of regional blood flow in acute ischemic rat cortex. J. Biomed. Opt. 2012, 17, 125001. [Google Scholar] [CrossRef]
- Offner, H.; Subramanian, S.; Parker, S.M.; Afentoulis, M.E.; Vandenbark, A.A.; Hurn, P.D. Experimental stroke induces massive, rapid activation of the peripheral immune system. J. Cereb. Blood Flow Metab. 2006, 26, 654–665. [Google Scholar] [CrossRef]
- Ritter, L.S.; Orozco, J.A.; Coull, B.M.; McDonagh, P.F. Leukocyte accumulation and hemodynamic changes in the cerebral microcirculation during early reperfusion after stroke. Stroke 2000, 31, 1153–1161. [Google Scholar] [CrossRef]
- Chopp, M.; Zhang, R.L.; Chen, H.; Li, Y.; Jiang, N.; Rusche, J.R. Postischemic administration of an anti-mac-1 antibody reduces ischemic cell damage after transient middle cerebral artery occlusion in rats. Stroke 1994, 25, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Kihara, T.; Ikeda, M.; Ninomiya, M.; Onodera, H.; Kogure, K. Role of neutrophils in radical production during ischemia and reperfusion of the rat brain: Effect of neutrophil depletion on extracellular ascorbyl radical formation. J. Cereb. Blood Flow Metab. 1995, 15, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Maier, C.M.; Hsieh, L.; Yu, F.; Bracci, P.; Chan, P.H. Matrix Metalloproteinase-9 and Myeloperoxidase Expression. Stroke 2004, 35, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M. Inflammatory responses to ischemia and reperfusion in the cerebral microcirculation. Front. Biosci. 2004, 9, 1339. [Google Scholar] [CrossRef] [PubMed]
- Belayev, L.; Busto, R.; Zhao, W.; Ginsberg, M.D. Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 1996, 739, 88–96. [Google Scholar] [CrossRef]
- Dietrich, W.D.; Busto, R.; Watson, B.D.; Scheinberg, P.; Ginsberg, M.D. Photochemically induced cerebral infarction - II. Edema and blood-brain barrier disruption. Acta Neuropathol. 1987, 72, 326–334. [Google Scholar] [CrossRef]
- Frederix, K.; Chauhan, A.K.; Kisucka, J.; Zhao, B.Q.; Hoff, E.I.; Spronk, H.M.H.; ten Cate, H.; Wagner, D.D. Platelet adhesion receptors do not modulate infarct volume after a photochemically induced stroke in mice. Brain Res. 2007, 1185, 239–245. [Google Scholar] [CrossRef]
- Yushmanov, V.E.; Kharlamov, A.; Simplaceanu, E.; Williams, D.S.; Jones, S.C. Differences between arterial occlusive and cortical photothrombosis stroke models with magnetic resonance imaging and microtubule-associated protein-2 immunoreactivity. Magn. Reson. Imaging 2006, 24, 1087–1093. [Google Scholar] [CrossRef]
- Schrandt, C.J.; Kazmi, S.M.S.; Jones, T.A.; Dunn, A.K. Chronic monitoring of vascular progression after ischemic stroke using multiexposure speckle imaging and two-photon fluorescence microscopy. J. Cereb. Blood Flow Metab. 2015, 35, 933–942. [Google Scholar] [CrossRef]
- Jander, S.; Kraemer, M.; Schroeter, M.; Witte, O.W.; Stoll, G. Lymphocytic Infiltration and Expression of Intercellular Adhesion Molecule-1 in Photochemically Induced Ischemia of the Rat Cortex. J. Cereb. Blood Flow Metab. 1995, 15, 42–51. [Google Scholar] [CrossRef]
- Schroeter, M.; Jander, S.; Huitinga, I.; Witte, O.W.; Stoll, G. Phagocytic Response in Photochemically Induced Infarction of Rat Cerebral Cortex. Stroke 1997, 28, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Cotrina, M.L.; Lou, N.; Tome-Garcia, J.; Goldman, J.; Nedergaard, M. Direct comparison of microglial dynamics and inflammatory profile in photothrombotic and arterial occlusion evoked stroke. Neuroscience 2017, 343, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Liao, S.; Wei, C.; Jia, D.; Wood, K.; Liu, Q.; Wang, X.; Shi, F.D.; Jin, W.N. Infiltration and persistence of lymphocytes during late-stage cerebral ischemia in middle cerebral artery occlusion and photothrombotic stroke models. J. Neuroinflammation 2017, 14, 248. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.A.; Maltby, S.; Plank, M.W.; Kluge, M.; Nilsson, M.; Foster, P.S.; Walker, F.R. Peripheral immune cells infiltrate into sites of secondary neurodegeneration after ischemic stroke. Brain. Behav. Immun. 2018, 67, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Soylu, H.; Zhang, D.; Buist, R.; Martin, M.; Albensi, B.C.; Parkinson, F.E. Intracortical injection of endothelin-1 induces cortical infarcts in mice: Effect of neuronal expression of an adenosine transporter. Exp. Transl. Stroke Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Endepols, H.; Mertgens, H.; Backes, H.; Himmelreich, U.; Neumaier, B.; Graf, R.; Mies, G. Longitudinal assessment of infarct progression, brain metabolism and behavior following anterior cerebral artery occlusion in rats. J. Neurosci. Methods 2015, 253, 279–291. [Google Scholar] [CrossRef][Green Version]
- Weston, R.M.; Jones, N.M.; Jarrott, B.; Callaway, J.K. Inflammatory cell infiltration after endothelin-1-induced cerebral ischemia: Histochemical and myeloperoxidase correlation with temporal changes in brain injury. J. Cereb. Blood Flow Metab. 2007, 27, 100–114. [Google Scholar] [CrossRef]
- Nedelcu, J.; Klein, M.A.; Aguzzi, A.; Boesiger, P.; Martin, E. Biphasic Edema after Hypoxic-Ischemic Brain Injury in Neonatal Rats Reflects Early Neuronal and Late Glial Damage. Pediatr. Res. 1999, 46, 297–304. [Google Scholar] [CrossRef]
- Rumpel, H.; Nedelcu, J.; Aguzzi, A.; Martin, E. Late Glial Swelling after Acute Cerebral Hypoxia-Ischemia in the Neonatal Rat: A Combined Magnetic Resonance and Histochemical Study. Pediatr. Res. 1997, 42, 54–59. [Google Scholar] [CrossRef][Green Version]
- Bona, E.; Andersson, A.-L.; Blomgren, K.; Gilland, E.; Puka-Sundvall, M.; Gustafson, K.; Hagberg, H. Chemokine and Inflammatory Cell Response to Hypoxia-Ischemia in Immature Rats. Pediatr. Res. 1999. [Google Scholar] [CrossRef]
- Meisel, C.; Schwab, J.M.; Prass, K.; Meisel, A.; Dirnagl, U. Central nervous system injury-induced immune deficiency syndrome. Nat. Rev. Neurosci. 2005, 6, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Jin, W.N.; Liu, Y.; Shi, K.; Sun, H.; Zhang, F.; Zhang, C.; Gonzales, R.J.; Sheth, K.N.; la Cava, A.; et al. Brain Ischemia Suppresses Immunity in the Periphery and Brain via Different Neurogenic Innervations. Immunity 2017, 46, 474–487. [Google Scholar] [CrossRef] [PubMed]
- Hug, A.; Dalpke, A.; Wieczorek, N.; Giese, T.; Lorenz, A.; Auffarth, G.; Liesz, A.; Veltkamp, R. Infarct volume is a major determiner of post-stroke immune cell function and susceptibility to infection. Stroke 2009, 40, 3226–3232. [Google Scholar] [CrossRef] [PubMed]
- Offner, H.; Vandenbark, A.A.; Hurn, P.D. Effect of experimental stroke on peripheral immunity: CNS ischemia induces profound immunosuppression. Neuroscience 2009, 158, 1098–1111. [Google Scholar] [CrossRef]
- Burkovskiy, I.; Zhou, J.; Lehmann, C. Experimental Cannabinoid 2 Receptor Inhibition in CNS Injury-Induced Immunodeficiency Syndrome. Microcirculation 2016, 23, 283–292. [Google Scholar] [CrossRef]
- Burkovskiy, Y. Cannabinoid 2 receptor-based modulation of the immune response in experimental models of CNS injury; Dalhousie University: Halifax, Nova Scotia, Canada, 2019. [Google Scholar]
- O’Collins, V.E.; Macleod, M.R.; Donnan, G.A.; Horky, L.L.; Van Der Worp, B.H.; Howells, D.W. 1,026 Experimental treatments in acute stroke. Ann. Neurol. 2006, 59, 467–477. [Google Scholar] [CrossRef]
- Cheng, Y.D.; Al-Khoury, L.; Zivin, J.A. Neuroprotection for Ischemic Stroke: Two Decades of Success and Failure. NeuroRx 2004, 1, 36–45. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Alfieri, A.; Allan, S.M.; Carswell, H.V.O.; Deuchar, G.A.; Farr, T.D.; Flecknell, P.; Gallagher, L.; Gibson, C.L.; Haley, M.J.; et al. The IMPROVE Guidelines (Ischaemia Models: Procedural Refinements Of in Vivo Experiments). J. Cereb. Blood Flow Metab. 2017, 37, 3488–3517. [Google Scholar] [CrossRef]
- Goodall, S.; Reggia, J.A.; Chen, Y.; Ruppin, E.; Whitney, C. A computational model of acute focal cortical lesions. Stroke 1997, 28, 101–109. [Google Scholar] [CrossRef]
- Dronne, M.A.; Boissel, J.P.; Grenier, E. A mathematical model of ion movements in grey matter during a stroke. J. Theor. Biol. 2006, 240, 599–615. [Google Scholar] [CrossRef]
- Traystman, R.J. Animal models of focal and global cerebral ischemia. ILAR J. 2003, 44, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Liesz, A.; Hagmann, S.; Zschoche, C.; Adamek, J.; Zhou, W.; Sun, L.; Hug, A.; Zorn, M.; Dalpke, A.; Nawroth, P.; et al. The Spectrum of Systemic Immune Alterations After Murine Focal Ischemia. Stroke 2009, 40, 2849–2858. [Google Scholar] [CrossRef] [PubMed]
- Willing, A.E. Experimental models: Help or hindrance. Stroke 2009, 40, 152–154. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Findings | References |
|---|---|---|
| MCAO | Long-lasting depression of cell-mediated immunity resulting in spontaneous bacterial infection | [13] |
| Reduced production of IFN-γ by impaired NK and T cells | [13] | |
| Reduction in activation of T cells and loss of T and B cells in the spleen and thymus | [115] | |
| Rapid decline in NK cells in the spleen and spleen atrophy in acute phase of stroke | [113] | |
| Blockage of adrenergic and HPA axis innervation of NK cells in the periphery enhanced immune defense mediated by NK cells | [113] | |
| Pharmacological inhibition of the sympathetic nervous system (SNS) enhanced cellular immune responses following MCAO | [13] | |
| HI | Reduction in the number of adhering leukocytes in intestinal microcirculation | [116] |
| Decrease in levels of pro-inflammatory cytokines | [116] | |
| HI and ET-1 | Impairment of leukocyte–endothelial interactions in intestinal microcirculation | [117] |
| Increase in infarct size correlated with a weaker immune response | [117] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lunardi Baccetto, S.; Lehmann, C. Microcirculatory Changes in Experimental Models of Stroke and CNS-Injury Induced Immunodepression. Int. J. Mol. Sci. 2019, 20, 5184. https://doi.org/10.3390/ijms20205184
Lunardi Baccetto S, Lehmann C. Microcirculatory Changes in Experimental Models of Stroke and CNS-Injury Induced Immunodepression. International Journal of Molecular Sciences. 2019; 20(20):5184. https://doi.org/10.3390/ijms20205184
Chicago/Turabian StyleLunardi Baccetto, Sarah, and Christian Lehmann. 2019. "Microcirculatory Changes in Experimental Models of Stroke and CNS-Injury Induced Immunodepression" International Journal of Molecular Sciences 20, no. 20: 5184. https://doi.org/10.3390/ijms20205184
APA StyleLunardi Baccetto, S., & Lehmann, C. (2019). Microcirculatory Changes in Experimental Models of Stroke and CNS-Injury Induced Immunodepression. International Journal of Molecular Sciences, 20(20), 5184. https://doi.org/10.3390/ijms20205184