Depicting Conformational Ensembles of α-Synuclein by Single Molecule Force Spectroscopy and Native Mass Spectroscopy

 , ,
, ,  ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
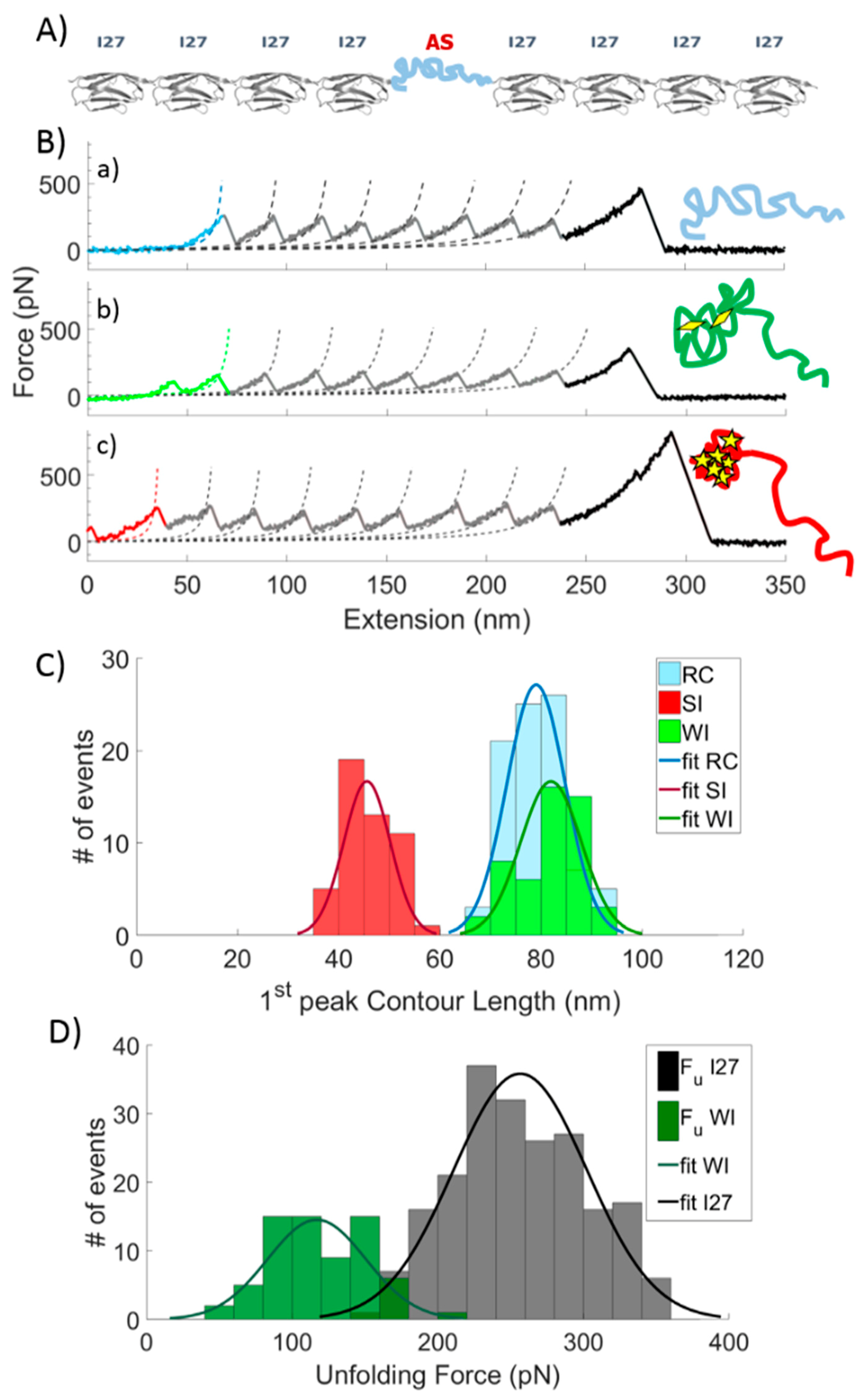
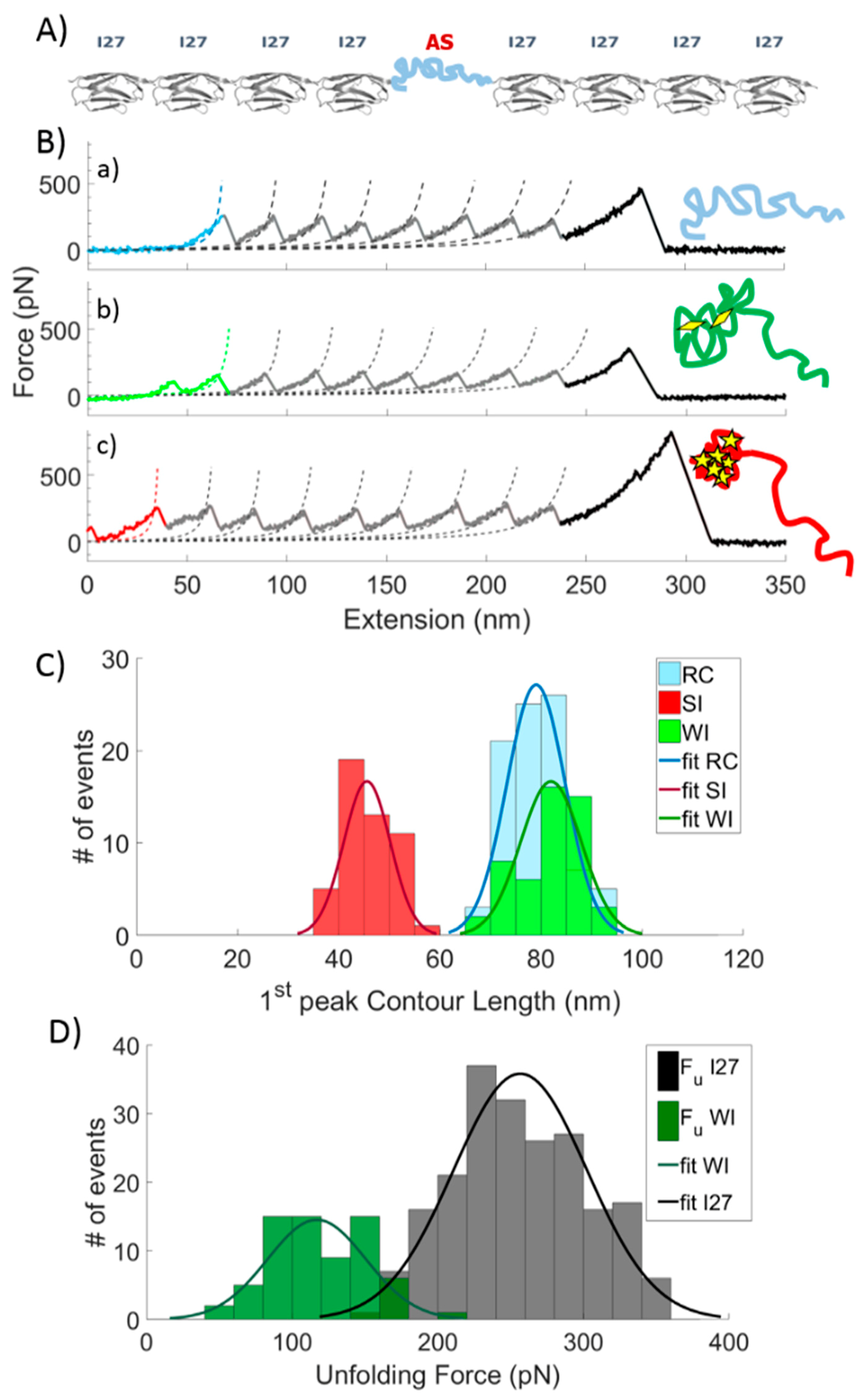
2.1. Single Molecule Force Spectroscopy (SMSF)
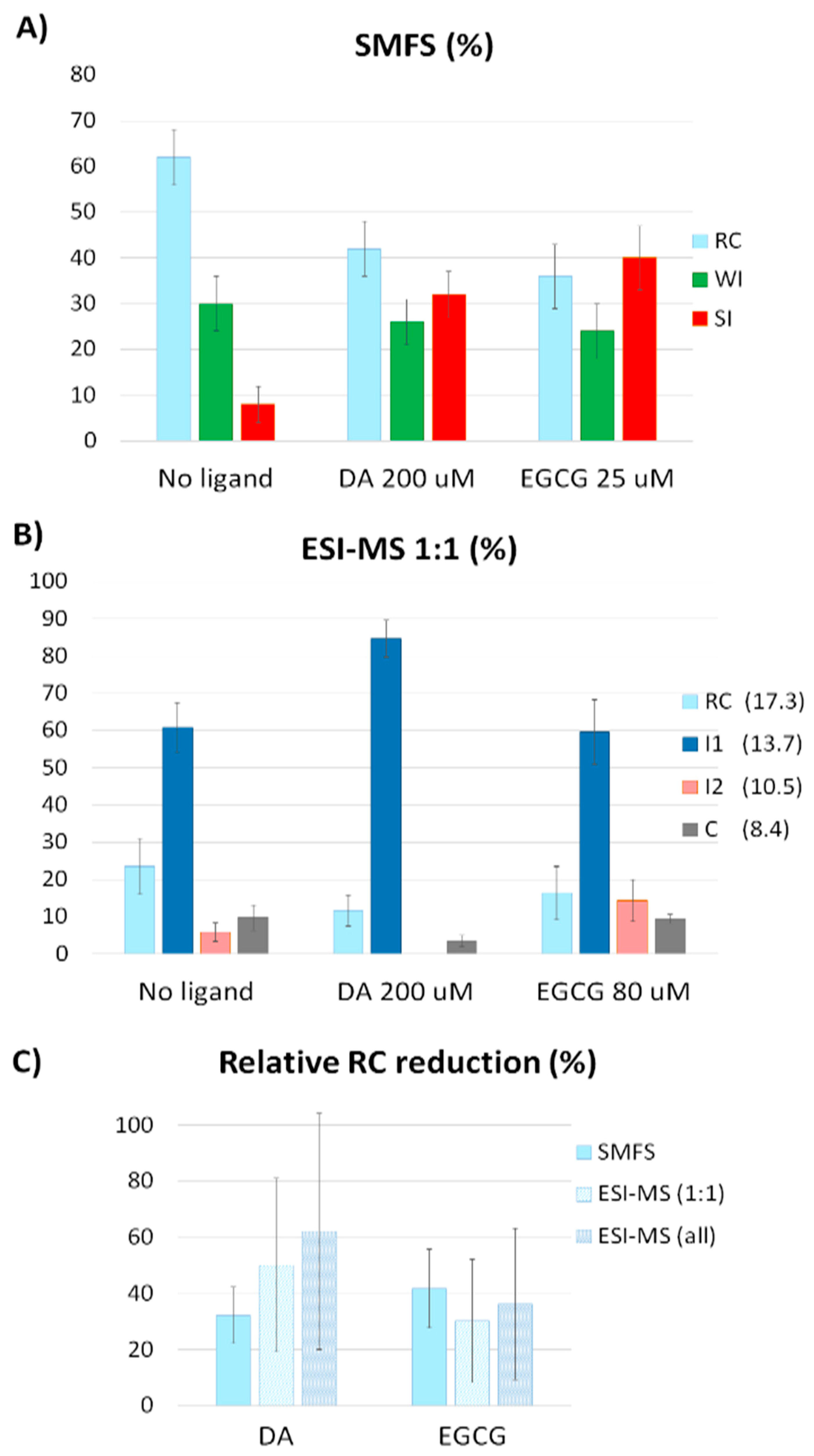
2.2. Effect of DA and EGCG on the Conformational Ensemble
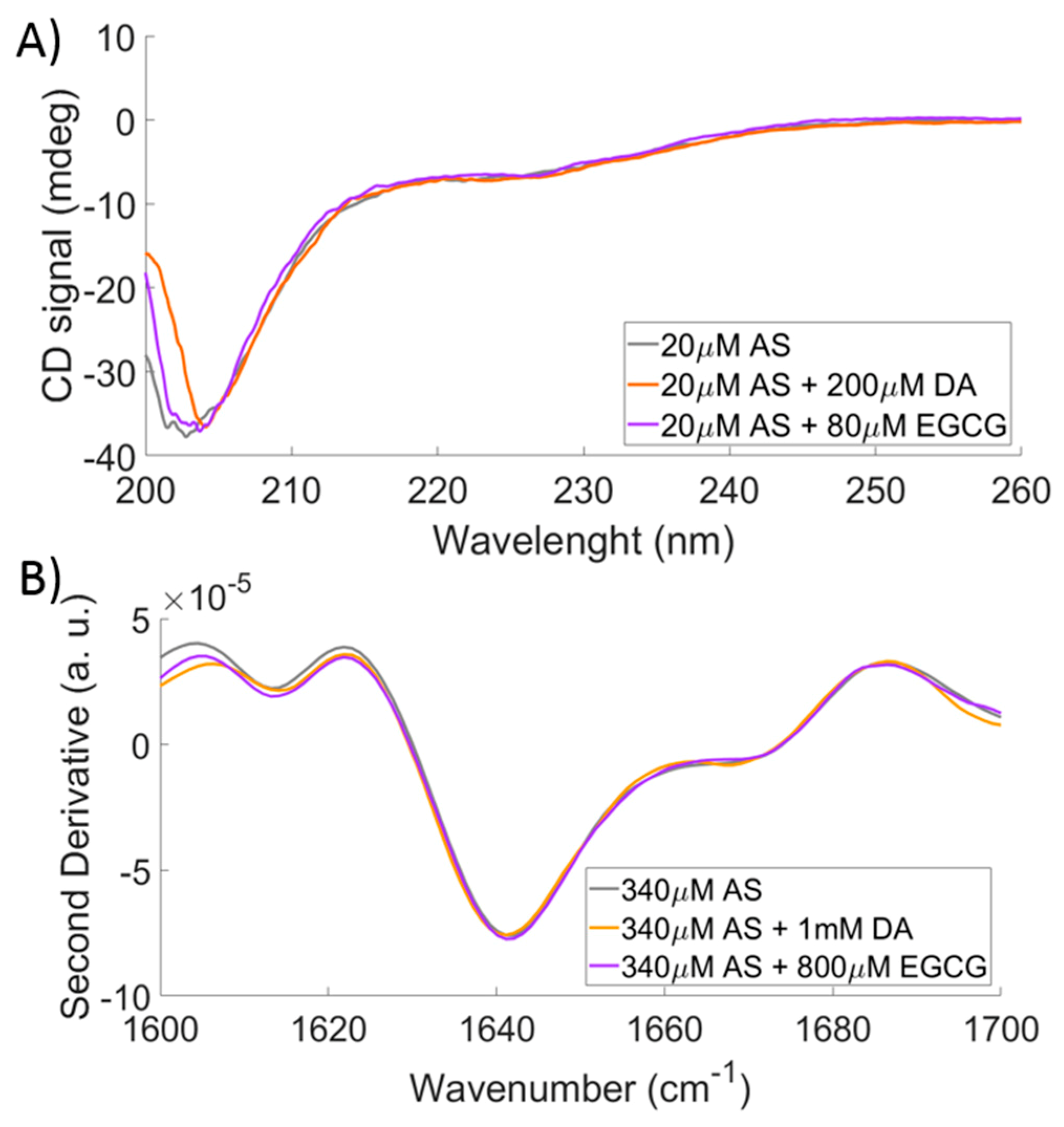
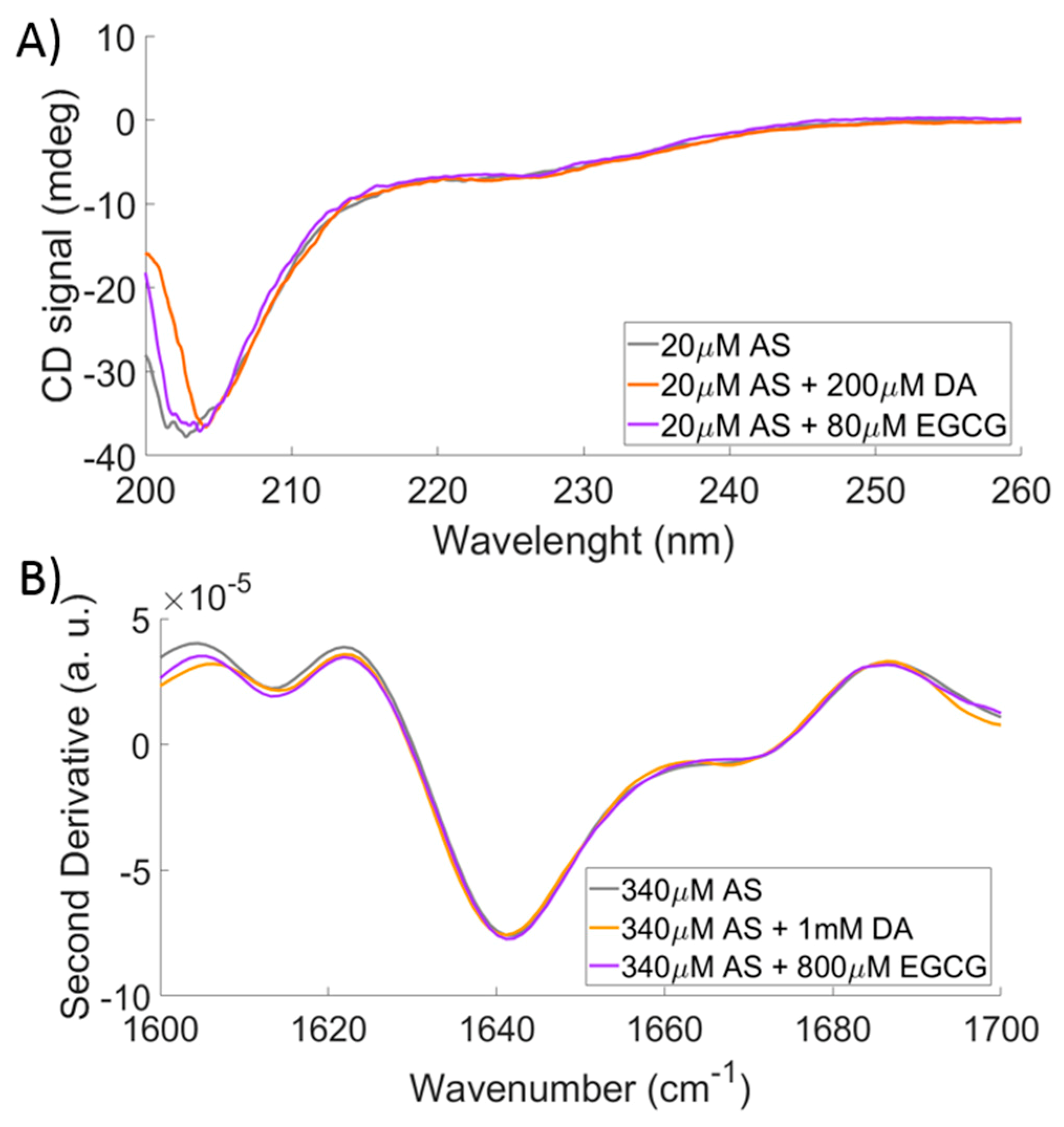
2.3. Comparison to CD and FTIR
3. Discussion
4. Materials and Methods
4.1. Cloning, Expression, and Purification of the (I27)4_AS_(I27)4 Polyprotein
4.2. AFM—Single Molecule Force Spectroscopy
4.3. AFM Data Analysis
4.4. Native-MS Experiments
4.5. CD and FTIR Experiments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AS | α-synuclein |
| C | compact structure detected in native MS |
| CD | circular dichroism |
| CSDs | charge state distributions |
| DA | dopamine |
| EGCG | epigallocatechin-3-gallate |
| ESI-MS | electrospray ionization mass spectrometry |
| F | unfolding force |
| FTIR | Fourier-transform infrared spectroscopy |
| I1, I2 | Intermediate 1 and 2 detected in native MS |
| I27 | 27th titin immunoglobulin-like domain |
| IDP | intrinsically disordered protein |
| IDPs | intrinsically disordered proteins |
| LC | contour length |
| LP | persistence length |
| native MS | native mass spectrometry |
| NMR | nuclear magnetic resonance |
| RC | random coil |
| SAXS-EOM | small-angle X-ray scattering and ensemble-optimization method |
| SI | strong interactions |
| SMFS | single molecule force spectroscopy |
| WI | weak interactions |
| WLC | worm-like-chain |
References
- Rezaei-Ghaleh, N.; Parigi, G.; Soranno, A.; Holla, A.; Becker, S.; Schuler, B.; Luchinat, C.; Zweckstetter, M. Local and Global Dynamics in Intrinsically Disordered Synuclein. Angew. Chem. Int. Ed. Engl. 2018, 57, 15262–15266. [Google Scholar] [CrossRef] [PubMed]
- Borgia, A.; Kemplen, K.R.; Borgia, M.B.; Soranno, A.; Shammas, S.; Wunderlich, B.; Nettels, D.; Best, R.B.; Clarke, J.; Schuler, B. Transient misfolding dominates multidomain protein folding. Nat. Commun. 2015, 6, 8861. [Google Scholar] [CrossRef] [PubMed]
- Gruebele, M.; Dave, K.; Sukenik, S. Globular Protein Folding In Vitro and In Vivo. Annu. Rev. Biophys. 2016, 45, 233–251. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell. Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Oberhauser, A.F.; Marszalek, P.E.; Carrion-Vazquez, M.; Fernandez, J.M. Single protein misfolding events captured by atomic force microscopy. Nat. Struct. Biol. 1999, 6, 102510–102528. [Google Scholar] [CrossRef]
- Rounsevell, R.; Forman, J.R.; Clarke, J. Atomic force microscopy: Mechanical unfolding of proteins. J. Methods 2004, 34, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, A.C.M.; Deniz, A.A. Protein folding at single-molecule resolution. Biochim. Biophys. Acta 2011, 1814, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Junker, J.P.; Rief, M. Single-molecule force spectroscopy distinguishes target binding modes of calmodulin. Proc. Natl. Acad. Sci. USA 2009, 106, 14361–14366. [Google Scholar] [CrossRef] [Green Version]
- Cassina, V.; Manghi, M.; Salerno, D.; Tempestini, A.; Iadarola, V.; Nardo, L.; Brioschi, S.; Mantegazza, F. Effects of cytosine methylation on DNA morphology: An atomic force microscopy study. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 1–7. [Google Scholar] [CrossRef]
- Cassina, V.; Seruggia, D.; Beretta, G.L.; Salerno, D.; Brogioli, D.; Manzini, S.; Zunino, F.; Mantegazza, F. Atomic force microscopy study of DNA conformation in the presence of drugs. Eur. Biophys. J. 2011, 40, 59–68. [Google Scholar] [CrossRef]
- Beedle, A.E.M.; Lezamiz, A.; Stirnemann, G.; Garcia-Manyes, S. The mechanochemistry of copper reports on the directionality of unfolding in model cupredoxin proteins. Nat. Commun. 2015, 6, 7894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walder, R.; LeBlanc, M.A.; Van Patten, W.J.; Edwards, D.T.; Greenberg, J.A.; Adhikari, A.; Okoniewski, S.R.; Sullan, R.M.A.; Rabuka, D.; Sousa, M.C.; et al. Rapid Characterization of a Mechanically Labile α-Helical Protein Enabled by Efficient Site-Specific Bioconjugation. J. Am. Chem. Soc. 2017, 39, 9867–9875. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manyes, S.; Kuo, T.L.; Fernández, J.M. Contrasting the individual reactive pathways in protein unfolding and disulfide bond reduction observed within a single protein. J. Am. Chem. Soc. 2011, 133, 3104–3113. [Google Scholar] [CrossRef] [PubMed]
- Sandal, M.; Valle, F.; Tessari, I.; Mammi, S.; Bergantino, E.; Musiani, F.; Brucale, M.; Bubacco, L.; Samorì, B. Conformational equilibria in monomeric alpha-synuclein at the single-molecule level. PLoS Biol. 2008, 6, 99–108. [Google Scholar] [CrossRef]
- Brucale, M.; Sandal, M.; Di Maio, S.; Rampion, A.; Tessari, I.; Tosatto, L.; Bisaglia, M.; Bubacco, L.; Samorì, B. Pathogenic mutations shift the equilibria of alpha-synuclein single molecules towards structured conformers. ChemBioChem 2009, 10, 176–183. [Google Scholar] [CrossRef]
- Hervàs, R.; Oroz, J.; Galera-Prat, A.; Goñi, O.; Valbuena, A.; Vera, A.M.; Gòmez-Sicilia, A.; Losada-Urzáiz, F.; Uversky, V.N.; Menéndez, M.; et al. Common features at the start of the neurodegeneration cascade. PLoS Biol. 2012, 10, 1001335. [Google Scholar] [CrossRef]
- Zhang, Y.; Hashemi, M.; Lv, Z.; Williams, B.; Popov, K.I.; Dokholyan, N.V.; Lyubchenko, Y.L. High-speed atomic force microscopy reveals structural dynamics of α-synuclein monomers and dimers. J. Chem. Phys. 2018, 148, 123322. [Google Scholar] [CrossRef]
- Stephens, A.D.; Zacharopoulou, M.; Kaminski Schierle, G.S. The Cellular Environment Affects Monomeric α-Synuclein Structure. Trends Biochem. Sci. 2019, 44, 453–466. [Google Scholar] [CrossRef]
- Curtain, C.C.; Kirby, N.M.; Mertens, H.D.; Barnham, K.J.; Knott, R.B.; Masters, C.L.; Cappai, R.; Rekas, A.; Kenche, V.B.; Ryan, T. α-synuclein oligomers and fibrils originate in two distinct conformer pools: A small angle X-ray scattering and ensemble optimisation modelling study. Mol. Biosyst. 2015, 11, 190–196. [Google Scholar] [CrossRef]
- Theillet, F.X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef]
- Zhao, J.; Liang, Q.; Sun, Q.; Chen, C.; Xu, L.; Ding, Y.; Zhou, P. (-)-Epigallocatechin-3-gallate (EGCG) inhibits fibrillation, disaggregates amyloid fibrils of α-synuclein, and protects PC12 cells against alpha-synuclein-induced toxicity. RSC Adv. 2017, 7, 32508–32517. [Google Scholar] [CrossRef]
- Lee, H.J.; Baek, S.M.; Ho, D.H.; Suk, J.E.; Cho, E.D.; Lee, S.J. Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Exp. Mol. Med. 2011, 43, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Santambrogio, C.; Natalello, A.; Brocca, S.; Ponzini, E.; Grandori, R. Conformational Characterization and Classification of Intrinsically Disordered Proteins by Native Mass Spectrometry and Charge-State Distribution. Proteomics 2019, 19, 1800060. [Google Scholar] [CrossRef] [PubMed]
- Lössl, P.; Van de Waterbeemd, M.; Heck, A.J. The diverse and expanding role of mass spectrometry in structural and molecular biology. EMBO J. 2016, 35, 2634–2657. [Google Scholar] [CrossRef]
- Konijnenberg, A.; Butterer, A.; Sobott, F. Native ion mobility-mass spectrometry and related methods in structural biology. Biochim. Biophys. Acta 2013, 1834, 1239–1256. [Google Scholar] [CrossRef]
- Loo, R.R.; Loo, J.A. Salt Bridge Rearrangement (SaBRe) Explains the Dissociation Behavior of Noncovalent Complexes. J. Am. Soc. Mass Spectrom. 2016, 27, 975–990. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, S.K.; Katta, V.; Chait, B.T. Probing conformational changes in proteins by mass spectrometry. J. Am. Chem. Soc. 1990, 112, 9012–9013. [Google Scholar] [CrossRef]
- Grandori, R.; Matecko, I.; Mayr, P.; Müller, N. Probing protein stabilization by glycerol using electrospray mass spectrometry. J. Mass Spectrom. 2001, 36, 918–922. [Google Scholar] [CrossRef]
- Natalello, A.; Benetti, F.; Doglia, S.M.; Legname, G.; Grandori, R. Compact conformations of α-synuclein induced by alcohols and copper. Proteins 2011, 79, 611–621. [Google Scholar] [CrossRef]
- D’Urzo, A.; Konijnenberg, A.; Rossetti, G.; Habchi, J.; Li, J.; Carloni, P.; Sobott, F.; Longhi, S.; Grandori, R. Molecular basis for structural heterogeneity of an intrinsically disordered protein bound to a partner by combined ESI-IM-MS and modeling. J. Am. Soc. Mass Spectrom. 2015, 26, 472–481. [Google Scholar] [CrossRef]
- Wongkongkathep, P.; Han, J.Y.; Choi, T.S.; Yin, S.; Kim, H.I.; Loo, J.A. Native Top-Down Mass Spectrometry and Ion Mobility MS for Characterizing the Cobalt and Manganese Metal Binding of α-Synuclein Protein. J. Am. Soc. Mass Spectrom. 2018, 29, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Testa, L.; Brocca, S.; Santambrogio, C.; D’Urzo, A.; Habchi, J.; Longhi, S.; Uversky, V.N.; Grandori, R. Extracting structural information from charge-state distributions of intrinsically disordered proteins by non-denaturing electrospray-ionization mass spectrometry. Intrinsic. Disord. Proteins 2013, 1, 25068. [Google Scholar] [CrossRef] [PubMed]
- Frimpong, A.K.; Abzalimov, R.R.; Uversky, V.N.; Kaltashov, I.A. Characterization of intrinsically disordered proteins with electrospray ionization mass spectrometry: Conformational heterogeneity of α-synuclein. Proteins 2010, 78, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, U.H.; Kebarle, P. Ion-ion and ion-molecule reactions at the surface of proteins produced by nanospray. Information on the number of acidic residues and control of the number of ionized acidic and basic residues. J. Am. Soc. Mass Spectrom. 2005, 16, 1325–1341. [Google Scholar] [CrossRef]
- Testa, L.; Brocca, S.; Grandori, R. Charge-surface correlation in electrospray ionization of folded and unfolded proteins. Anal. Chem. 2011, 83, 6459–6463. [Google Scholar] [CrossRef]
- Kaltashov, I.A.; Mohimen, A. Estimates of protein surface areas in solution by electrospray ionization mass spectrometry. Anal. Chem. 2005, 77, 5370–5379. [Google Scholar] [CrossRef]
- Hall, Z.; Robinson, C.V. Do charge state signatures guarantee protein conformations? J. Am. Soc. Mass Spectrom. 2012, 23, 1161–1168. [Google Scholar] [CrossRef]
- Li, J.; Santambrogio, C.; Brocca, S.; Rossetti, G.; Carloni, P.; Grandori, R. Conformational effects in protein electrospray-ionization mass spectrometry. Mass Spectrom. Rev. 2016, 35, 111–122. [Google Scholar] [CrossRef]
- Natalello, A.; Santambrogio, C.; Grandori, R. Are Charge-State Distributions a Reliable Tool Describing Molecular Ensembles of Intrinsically Disordered Proteins by Native MS? J. Am. Soc. Mass Spectrom. 2017, 28, 21–28. [Google Scholar] [CrossRef]
- Beveridge, R.; Migas, L.G.; Das, R.K.; Pappu, R.V.; Kriwacki, R.W.; Barran, P.E. Ion Mobility Mass Spectrometry Uncovers the Impact of the Patterning of Oppositely Charged Residues on the Conformational Distributions of Intrinsically Disordered Proteins. J. Am. Chem. Soc. 2019, 141, 4908–4918. [Google Scholar] [CrossRef] [Green Version]
- Stuchfield, D.; Barran, P. Unique insights to intrinsically disordered proteins provided by ion mobility mass spectrometry. Curr. Opin. Chem. Biol. 2018, 42, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Konijnenberg, A.; Ranica, S.; Narkiewicz, J.; Legname, G.; Grandori, R.; Sobott, F.; Natalello, A. Opposite Structural Effects of Epigallocatechin-3-gallate and Dopamine Binding to α-Synuclein. Anal. Chem. 2016, 88, 8468–8475. [Google Scholar] [CrossRef] [PubMed]
- Ponzini, E.; De Palma, A.; Cerboni, L.; Natalello, A.; Rossi, R.; Moons, R.; Konijnenberg, A.; Narkiewicz, J.; Legname, G.; Sobott, F.; et al. Methionine oxidation in α-synuclein inhibits its propensity for ordered secondary structure. J. Biol. Chem. 2019, 294, 5657–5665. [Google Scholar] [CrossRef] [PubMed]
- Borysik, A.J.; Kovacs, D.; Guharoy, M.; Tompa, P. Ensemble Methods Enable a New Definition for the Solution to Gas-Phase Transfer of Intrinsically Disordered Proteins. J. Am. Chem. Soc. 2015, 137, 13807–13817. [Google Scholar] [CrossRef]
- Steward, A.; Toca-Herrera, J.L.; Clarke, J. Versatile cloning system for construction of multimeric proteins for use in atomic force microscopy. J. Protein Sci. 2002, 11, 2179–2183. [Google Scholar] [CrossRef]
- Hoffman, T.; Dougan, L. Single molecule force spectroscopy using polyproteins. Chem. Soc. Rev. 2012, 41, 4781–4796. [Google Scholar] [CrossRef]
- Best, R.B.; Brockwell, D.J.; Toca-Herrera, J.L.; Blake, A.W.; Smith, A.; Radford, S.E.; Clarke, J. Force mode atomic force microscopy as a tool for protein folding studies. J. Anal. Chim. Acta 2003, 479, 87–105. [Google Scholar] [CrossRef]
- Bustamante, C.; Marko, J.F.; Siggia, E.D.; Smith, S. Entropic elasticity of lambda-phage DNA. Science 1994, 265, 1599–1600. [Google Scholar] [CrossRef]
- Borgia, M.B.; Borgia, A.; Best, R.B.; Steward, A.; Nettels, D.; Wunderlich, B.; Schuler, B.; Clarke, J. Single-molecule fluorescence reveals sequence-specific misfolding in multidomain proteins. Nature 2011, 474, 662–665. [Google Scholar] [CrossRef]
- Heller, G.T.; Bonomi, M.; Vendruscolo, M. Structural Ensemble Modulation upon Small-Molecule Binding to Disordered Proteins. J. Mol. Biol. 2018, 430, 2288–2292. [Google Scholar] [CrossRef]
- Rossetti, G.; Musiani, F.; Abad, E.; Dibenedetto, D.; Mouhib, H.; Fernandez, C.O.; Carloni, P. Conformational ensemble of human α-synuclein physiological form predicted by molecular simulations. Phys. Chem. Chem. Phys. 2016, 18, 5702–5706. [Google Scholar] [CrossRef] [PubMed]
- Balupuri, A.; Choi, K.E.; Kang, N.S. Computational insights into the role of α-strand/sheet in aggregation of α-synuclein. Sci. Rep. 2019, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Brodie, N.I.; Popov, K.I.; Petrotchenko, E.V.; Dokholyan, N.V.; Borchers, C.H. Conformational ensemble of native α-synuclein in solution as determined by short-distance crosslinking constraint-guided discrete molecular dynamics simulations. PLoS Comput. Biol. 2019, 15, e1006859. [Google Scholar] [CrossRef] [PubMed]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef]
- Kuprowski, M.C.; Konermann, L. Signal response of coexisting protein conformers in electrospray mass spectrometry. Anal. Chem. 2007, 79, 2499–2506. [Google Scholar] [CrossRef]
- Samalikova, M.; Matecko, I.; Müller, N.; Grandori, R. Interpreting conformational effects in protein nano-ESI-MS spectra. Anal. Bioanal. Chem. 2004, 378, 1112–1123. [Google Scholar] [CrossRef]
- Marsh, J.A.; Forman-Kay, J.D. Sequence determinants of compaction in intrinsically disordered proteins. Biophys. J. 2010, 98, 2383–2390. [Google Scholar] [CrossRef]
- Cartelli, D.; Aliverti, A.; Barbiroli, C.; Santambrogio, C.; Raggi, E.M.; Casagrande, F.V.M.; Cantele, F.; Beltramone, S.; Marangon, J.; De Gregorio, C.; et al. α-Synuclein is a Novel Microtubule Dynamase. Sci. Rep. 2016, 6, 33289. [Google Scholar] [CrossRef]
- Marszalek, P.E.; Lu, H.; Li, H.; Carrion-Vazquez, M.; Oberhauser, A.F.; Schulten, K.; Fernandez, J.M. Mechanical unfolding intermediates in titin modules. Nature 1999, 402, 100–103. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corti, R.; Marrano, C.A.; Salerno, D.; Brocca, S.; Natalello, A.; Santambrogio, C.; Legname, G.; Mantegazza, F.; Grandori, R.; Cassina, V. Depicting Conformational Ensembles of α-Synuclein by Single Molecule Force Spectroscopy and Native Mass Spectroscopy. Int. J. Mol. Sci. 2019, 20, 5181. https://doi.org/10.3390/ijms20205181
Corti R, Marrano CA, Salerno D, Brocca S, Natalello A, Santambrogio C, Legname G, Mantegazza F, Grandori R, Cassina V. Depicting Conformational Ensembles of α-Synuclein by Single Molecule Force Spectroscopy and Native Mass Spectroscopy. International Journal of Molecular Sciences. 2019; 20(20):5181. https://doi.org/10.3390/ijms20205181
Chicago/Turabian StyleCorti, Roberta, Claudia A. Marrano, Domenico Salerno, Stefania Brocca, Antonino Natalello, Carlo Santambrogio, Giuseppe Legname, Francesco Mantegazza, Rita Grandori, and Valeria Cassina. 2019. "Depicting Conformational Ensembles of α-Synuclein by Single Molecule Force Spectroscopy and Native Mass Spectroscopy" International Journal of Molecular Sciences 20, no. 20: 5181. https://doi.org/10.3390/ijms20205181
APA StyleCorti, R., Marrano, C. A., Salerno, D., Brocca, S., Natalello, A., Santambrogio, C., Legname, G., Mantegazza, F., Grandori, R., & Cassina, V. (2019). Depicting Conformational Ensembles of α-Synuclein by Single Molecule Force Spectroscopy and Native Mass Spectroscopy. International Journal of Molecular Sciences, 20(20), 5181. https://doi.org/10.3390/ijms20205181