2.1. Sequence-based Analysis
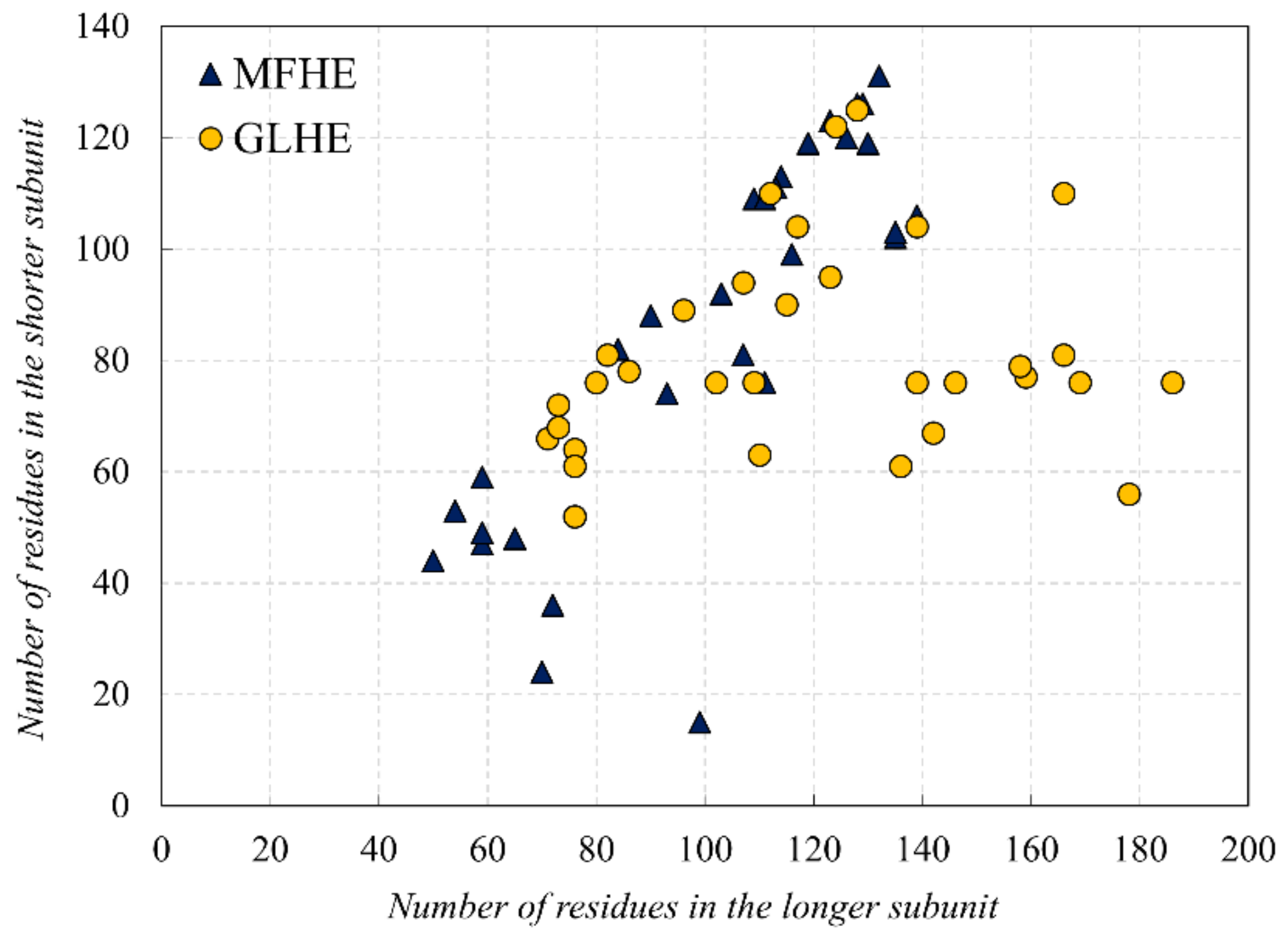
In this study, first, we examined the amino acid composition of the MFIB heterodimeric (MFHE) complexes, which were compared with a globular heterodimeric reference dataset (GLHE), which has similar size distribution for the heterodimeric state (see
Figure 1). Note that all GLHE subunits are more than 40 residues away from both axes, while the closest distance of an MFHE chain from the x-axis is less than 20 residues. Also, we will show later (see Figure 5) that the smallest identified globular monomer has 35 residues. In some cases, heterodimeric MSF complexes do not have enough amino acids for creating a hydrophobic core, but in most cases, they have as many residues as globular proteins have, thus other factors might also be responsible for the disordered nature of MFHE proteins.
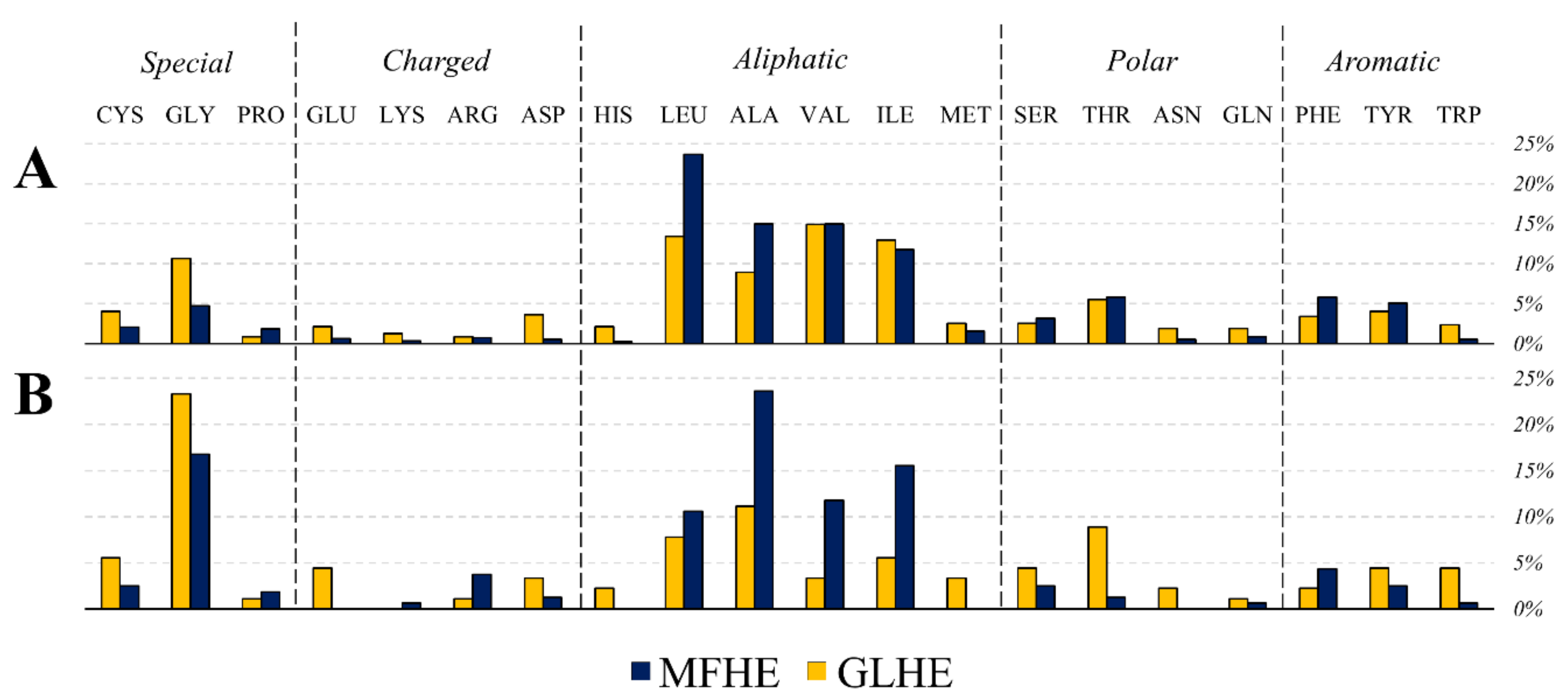
Since the beginning of the studies on IDPs, it is known they generally lack hydrophobic residues although alanine has a notably higher content in MFHE complexes compared to GLHE complexes, while the content of other aliphatic residues was similar among the two datasets (see
Figure 2A). MFHE complexes have a high net charge, like non-MSF IDPs [
11,
12].
The amino acid composition of the MFHE and GLHE heterodimers was depicted by a rank-based, indirect gradient analysis method, called Nonmetric MultiDimensional Scaling (NMDS), which creates an ordination based on a distance or dissimilarity matrix, thus it allows decreasing a multidimensional and quantitative, semi-quantitative, qualitative, or mixed variables data set to two dimensions [
13]. NMDS demonstrated a separation of MFHE and GLHE complexes and subunits (see
Figure 2B,C). The amino acid composition of the subunits, whether globular or MSF complexes are formed, have equal distances from each other as the amino acid compositions of the complexes. Some differences are revealed between the two data sets—the NMDS of the amino acid composition of the MSF heterodimeric complexes showed smaller variation from the globular heterodimeric complexes (see
Figure 2C), than the amino acid composition of the MSF subunits from the globular subunits (see
Figure 2B). These differences can be explained by the fact that although the amino acid composition of the MSF subunits differs slightly from globular proteins, they are unable to fold into an ordered structure independently. The folding of an MSF subunit requires another partner, which in this case has a different amino acid composition, that could form MSF complexes which have similar amino acid composition than the globular subunits. NMDS also pointed out that MFHE is a diverse group based on their amino acid composition, and these complexes are also clustered according to their structural classes in MFIB [
9].
The determination of the amino acids that contribute mainly to the observed difference was revealed by using SIMPER (similarity percentage) analyses. These amino acids were lysine (7.40%; 8.04%), alanine (7.30%; 7.90), leucine (7.14%; 6.64%), glycine (6.86%; 5.83%), arginine (6.39%; 6.70%), and glutamine (6.29%; 6.42%), which values support the similarity of the objects. Mostly aromatic and hydrophobic amino acids cause the amino acid compositions to separate (in slightly different proportions, See
Table S1), which case is more common in heterodimeric MSF subunits and complexes if the MSF data were grouped via MFIB for comparison was considered, for the MFIB structural classes (see
Figure 2,
Table S1), with the exception of glutamine.
Most of the heterodimers from MFIB are histone-type proteins with their high content of lysine and arginine. Acetylated lysine and methylated arginine may interact with proteins containing bromodomains and Tudor domains within the disordered proteins that affect nucleic acid binding and RNA pathways [
14].
The amino acid composition of the homodimeric complexes from MFIB (MFHO), heterodimeric MSF complexes was compared using our small globular protein (SGP) dataset as a standard reference by Kullback-Leibler divergence [
15], which measures the extent of the dissimilarity between two probability distributions (
). MSF heterodimers show about the same similarity to MSF homodimers (D = 1.257) and small globular proteins (D = 1.879), while MSF homodimers are more similar to small globular proteins (D = 0.442). This result is in line with the observation that heterodimeric complexes from MFIB look much more disordered (~20%) than MFIB homodimers (MFHO) (~10%) [
7] based on MoRFpred [
16] and IUPred [
17] results. Some regions of the heterodimeric MFIB complexes are also capable of folding on the surface of a globular protein. Most of these can be found in the DIBS (Disordered Binding Site) database [
18]. It is rather rare, but it also shows the elevation of the group inhomogeneity. For example, the cellular tumor antigen p53 protein (UniProt: P04637) is able to establish a coactivator binding domain complex (MFIB: MF2201002, PDB: 2l14) with the CREB-binding MSF protein, although at the same part of the p53 capable to form a transactivator domain complex (DIBS: DI1000009, PDB: 2ly4) with the highly mobile folded B1 protein. We have also found examples of disordered proteins from UniProt (e.g., ID: Q9Y6Q9, Nuclear receptor coactivator 3) which are able to establish an MSF interaction (MF2201001, PDB: 1kbh), and another region is able to form a DIBS interaction (DI1000313, PDB: 3l3x), forming two different types of disordered protein complexes.
It is interesting to note, that a few MFIB homodimers occur in DIBS as ordered interaction partners. For example, the dynein light chain (Tctex-type) protein (UniProt: Q94524), which is disordered in monomeric form based on MFIB (MFIB: MF2110016, PDB: 1ygt), while this homodimeric complex is the ordered part of a DIBS-interaction complex (Cytosolic dynein intermediate chain bound to Tctex-type dynein light chain, DIBS: DI2100002, PDB: 3fm7). An additional example of these multiple structure organizations is the homodimeric S100BEF-hand calcium-binding protein superfamily (MFIB: MF2100013, PDB: 1uwo), which is the ordered component of a DIBS-interaction (RSK1 bound to S100B dimer, DIBS: DI2000012, PDB: 5csf).
Besides the amino acid compositions, other sequential parameters also display differences between GLHE and MFHE. Based on cleverMachine [
19] calculations (
p-value < 0.0001: 56 scale of all 80) and grouped properties results, membrane proteins (
p-value < 0.0001: 7 scale of 10), nucleic acid binding (
p-value < 0.0001: 3 scale of 10), disorder propensity (
p-value < 0.0001: 8 scale of 10), α-helix (
p-value < 0.0001: 9 scale of 10), β-sheet (
p-value < 0.0001: 9 scale of 10), aggregation (
p-value < 0.0001: 8 scale of 10), burial propensity (
p-value < 0.0001: 10 scale of 10), and hydrophobicity (
p-value < 0.0001: 2 scale of 10) properties in MFHE are in general stronger than in globular heterodimers (Reference number of the dataset: 196154). While there is no significant difference between the sequences of MFIB homodimers and globular homodimers (GLHO) in most of the properties (exception of some membrane proteins and aggregation scales;
p-value < 0.0001: 8 scale of all 80) (Reference number of the dataset: 199533).
We analyzed the Pfam database in conjunction with the intermolecular stabilization centers (SCs, see Chapter 2.2. Structure-based analysis) [
20] on MFIB heterodimeric and globular heterodimeric complexes (for detailed results, see
Table S2). In the MFHE have found 59 Pfam domains in a total of 19 families, while the GLHE have 64 Pfam domains in a total of 37 families. In the case of globular heterodimers, 3 of the 30 complexes have interactions and SCs between the Pfam domains of the monomers, whereas, for MFIB heterodimers much more, at least 15 of the complexes have Pfam domains in which monomers interactions and intermolecular SCs were found. This result confirms that the folding of the MSF proteins is related to their functional role since, in many cases, the two subunits form the biologically relevant unit.
2.2. Structure-based Analysis
In our recent analysis of MSF homodimeric proteins, we found differences in several structural parameters between our dataset and a globular reference dataset. These structural features were investigated including solvent accessibility, hydrogen bonds, stabilization center content, and ion-pairs with an additional investigation of the buried structural core size.
The inter-subunit interface was identified based on the solvent accessible surface area (SASA) calculations. However, an MSF protein subunit in itself does not have an ordered structure, structural properties were also calculated for their monomeric forms, which were created by deleting a polypeptide chain from the heterodimeric PDB structures. This is referred to as their “monomeric structure” hereafter. The all-atom SASA values were calculated for all residues from the heterodimeric and monomeric structures. If the dimeric SASA value was below 20% of the monomeric value, the residue was identified as an interface residue. In the case of the MFIB heterodimeric dataset, 908 interface residues were identified out of the 4615 residues, that is 19.7% of all residues participate in the formation of the interface. In the globular reference heterodimeric dataset 470 interface residues were identified out of the 5155 total residues, i.e., 9.1% of all residues are forming the interface. As a different measure of the interface region, all-atom SASA values were also compared. In MFHE, 27.3% of the total surface area becomes buried upon dimerization, while in GLHE, only 11.6%. This result is in agreement with the finding of Gunasekaran et al., that the per residue interface area is higher in disordered complexes [
3] In MSF proteins, the larger interface contact area underlines the importance of inter-subunit interactions, thus inter-subunit interactions were considered hereafter.
Completely buried residues were identified in the MSF and the globular reference heterodimeric datasets using a stricter definition of burial, defining the core of the protein structure shielded from the solvent. We identified all residues, which have less than 10% relative all-atom solvent accessibility in the heterodimeric and monomeric structures, respectively. In MFHE, 10.8% of all residues are buried in monomeric form, while in GLHE this value is 20.9%. If the dimeric structures were analyzed, the values change to 27.7% and 26.3%, respectively. There are significantly fewer residues buried in the monomeric forms of MSF proteins when compared to globular ones. In the dimeric forms, the ratio of buried residues is similar in both cases.
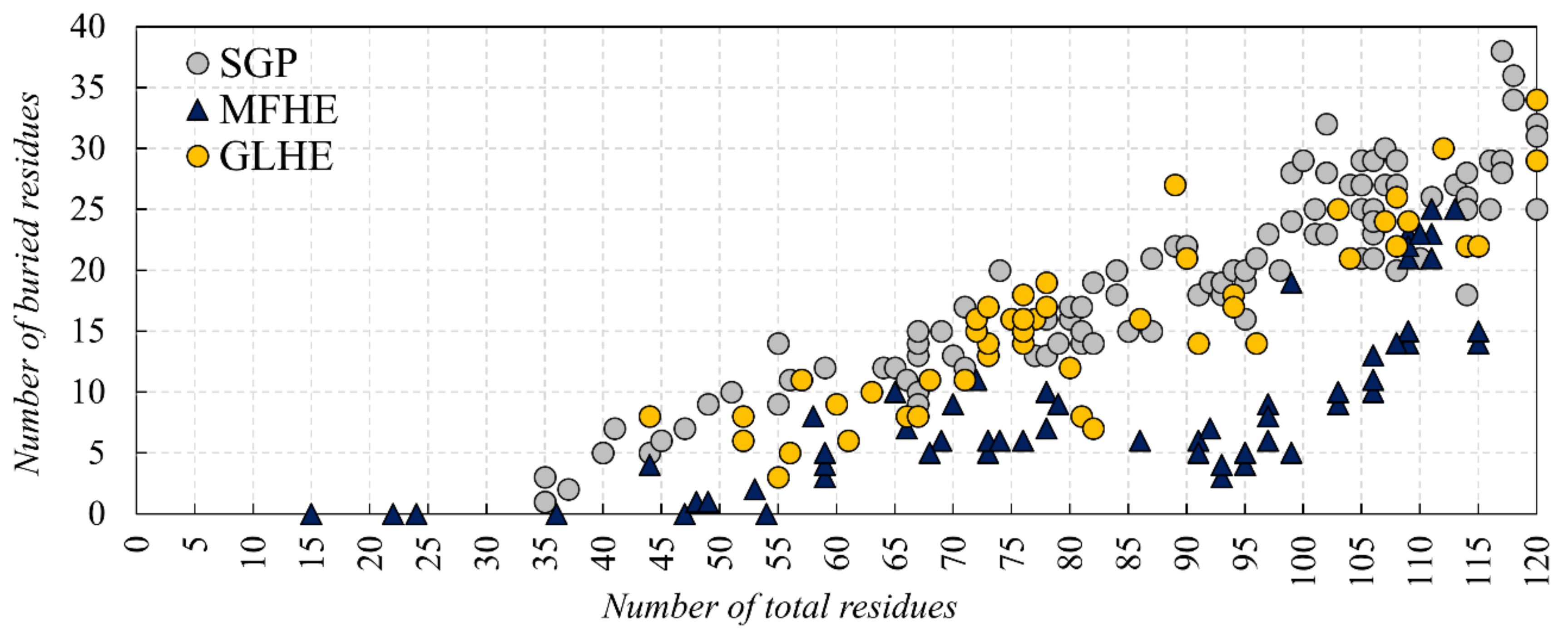
Figure 3 shows the number of buried residues in MSF (see
Figure 3A) and globular heterodimeric complexes (see
Figure 3B).
It can be seen that in the case of MSF heterodimers, there is a more considerable difference between the number of buried residues in the dimeric and monomeric forms, than in the case of globular heterodimers. In the case of globular heterodimers (see
Figure 3B), the sum of the number of buried residues in the two monomeric subunits is close to the number of buried residues in the dimeric form. These subunits are ordered by themselves, and they do not need another subunit to help to order their structures. In the case of MSF heterodimers (see
Figure 3A), the sum of the number of buried residues in the monomeric forms is lower than in the case of the globular heterodimers and, more importantly, they are much smaller than the number of buried residues in the dimeric form. These polypeptide chains are disordered by themselves, they need the presence of an interacting partner to help in ordering their structures. These protein chains need each other to form a reasonably sized core, needed for a stable, ordered structure.
The secondary structural element content was determined in the heterodimeric structures using the DSSP program [
21]. We found that in the MFHE dataset, 43.6% of the residues have the α-helical conformation and only 16.1% of the residues belonged to β-sheets, in the globular heterodimeric dataset, these values were 21.5% and 27.5%, respectively. In the MSF, heterodimeric dataset β-sheets were less abundant than in globular heterodimeric proteins. This will have some consequences in the interpretation of our later results.
We counted the number of inter-subunit ion-pairs. While there is only a small difference in the number of charged residues between MFHE and GLHE (1224 vs. 1380), the total charge is +320 for all 30 MFHE proteins and –91 for all 30 GLHE proteins. We found only 16 charged residues participating in 8 strong ion-pairs in the MFHE, while 28 residues are participating in 15 ion-pairs in the GLHE dataset. If we also consider weak ion-pairs, these values change to 73 residues participating in 42 ion-pairs for MFHE and 59 residues in 35 ion-pairs for GLHE. This is a 5.25-fold increase for MFHE and only a 2.33-fold increase for GLHE, respectively. Weak ion-pairs, presumably do not contribute to the enthalpic stabilization of the dimers, but probably play a role in the formation of electrostatic complementarity, already observed by Wong et al. in the case of complexes containing IDPs [
22] This behavior was unexpected, and further investigation of the role of electrostatic interactions in the stabilization of MSF dimers is planned.
In the case of the MSF homodimers, we found that the main-chain solvent accessibility may play an important role in the stabilization of homodimer structures [
8]. We identified residues with solvent accessible main-chain patches (RSAMPs). We have found a total of 161 RSAMPs in the MFHE dataset, and 90 RSAMPs in the GLHE dataset, respectively. There are 2 out of the 30 proteins in the MFHE dataset, which does not contain an RSAMP residue, while there are four such entries in the GLHE dataset. The average RSAMP content was 5.4 per heterodimeric complexes; thus, 17.7% of the interface residues are RSAMPs. In 26 of the 30 globular heterodimeric complexes, the average RSAMP content was 3, thus 19.1% of the interface residues are RSAMPs.
On the one hand, the composition of the RSAMPs of MFIB heterodimers suggested that five types of amino acids (glycine, alanine, isoleucine, leucine, and valine) play a major role in these interactions (see
Figure 4). These RSAMP contributing amino acids are mainly hydrophobic, are exposed to the inter-subunit interface. These residues do not contribute to the stabilization of the monomeric form since exposed hydrophobic surfaces are energetically not favorable. However, next to the favorable burial of their main-chain, they might help the formation of the tertiary structure by building sticky hydrophobic patches at the inter-subunit interface. On the other hand, in the case of the globular heterodimer dataset, the two amino acids with the smallest side-chains, glycine and alanine are the most abundant residues under RSAMPs. We investigated the secondary structural distribution of RSAMP, as well. We found that 33.5% of RSAMPs are located in β-sheets and 44.7% in α-helices. We checked the secondary structural composition of the interface residues, from which RSAMPs are selected. We found that 19.5% of interface residues have β-sheet and 63.9% have α-helical secondary structure. Considering the 3.3-fold higher occurrence of helical secondary structure at the interface, we can conclude that RSAMPs are more abundantly found in β structures, which can be easily broken by disturbing their hydrogen bonding network through interactions with accessible solvent molecules.
We counted the number of inter-subunit hydrogen bonds. We found a total number of 181 H-bonds in the MFHE and only 67 in the GLHE dataset, respectively. This is in agreement with our observation that inter-subunit interactions are of high importance in MSF heterodimers. We calculated the average wrapping of hydrogen bonds [
10]. Hydrogen bonds with a low wrapping (dehydrons) are less shielded from the solvent. The average value was 13.8 for the MFHE and 14.6 for the GLHE. Inter-subunit hydrogen bonds are slightly less wrapped in the MSF heterodimers, which also indicates the importance of solvent accessibility.
We also identified inter-subunit stabilization centers in both the MFHE and GLHE datasets. Stabilization centers are special residue pairs, which together with their sequential neighbors, participate in above than average long-range interactions and are believed to contribute to the stabilization of protein structures [
23]. The two residues that form a stabilization center are called stabilization center elements (SCEs). In MFHE, the average inter-subunit SCE content was 8.1, and we found at least one inter-subunit SC in 26 of the 30 heterodimers. In GLHE, the average SCE content was 0.5, and we found an inter-subunit SC is only 5 out of the 30 structures.
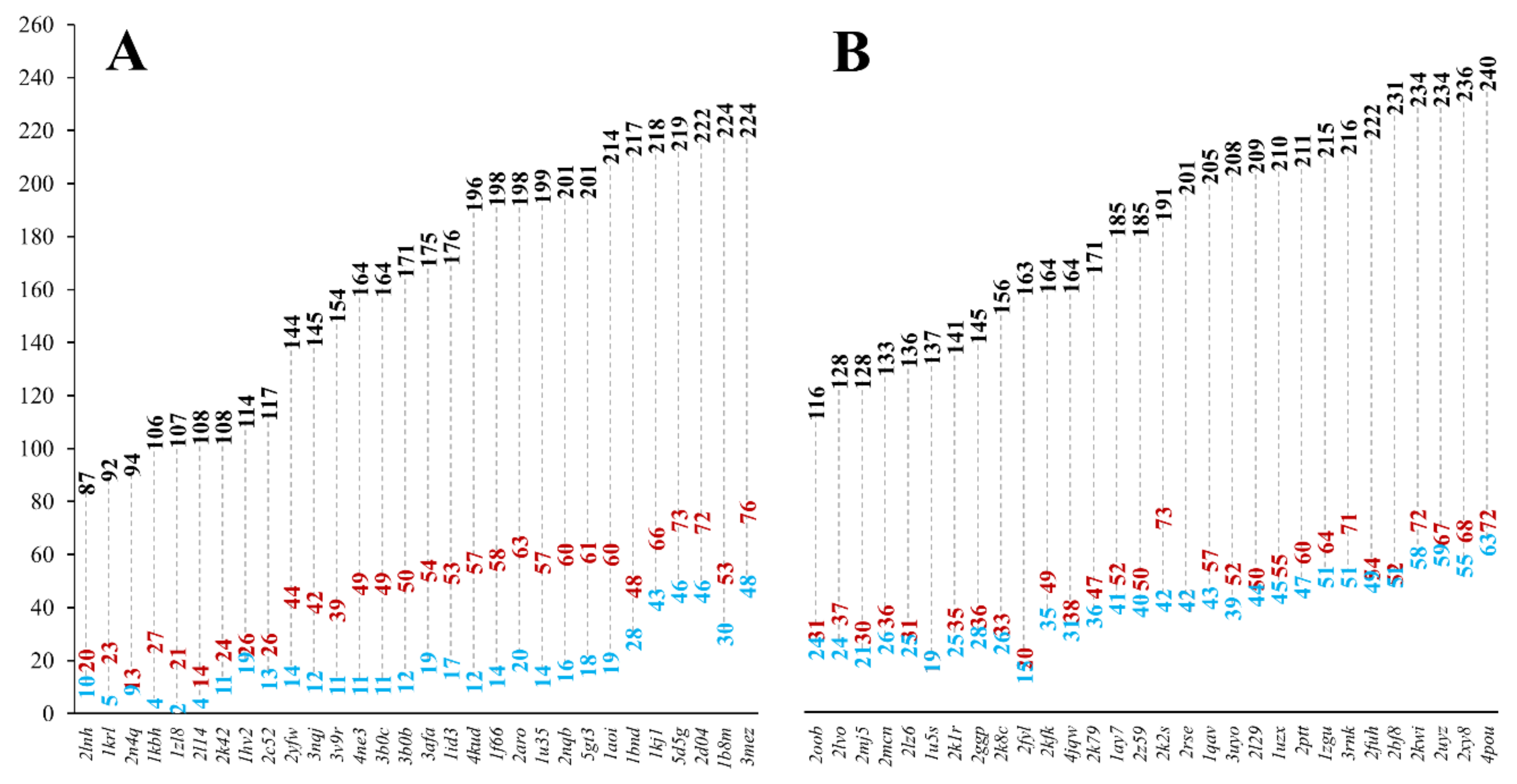
We investigated if there is a lower size limit for globular proteins, which already bear a buried core structure. Our analysis of monomeric, single-domain globular (SGP) dataset pointed out that proteins with 35 residues are already containing a buried structural core (see
Figure 5). Our results, regarding the buried core size of the MFIB heterodimers, indicate that although a couple of polypeptide chains are too small to contain a buried core, this is not a general trend for the MFHE dataset.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




