Neuroimmunology: What Role for Autoimmunity, Neuroinflammation, and Small Fiber Neuropathy in Fibromyalgia, Chronic Fatigue Syndrome, and Adverse Events after Human Papillomavirus Vaccination?




{kind=link}
{kind=link}
Abstract
1. Introduction
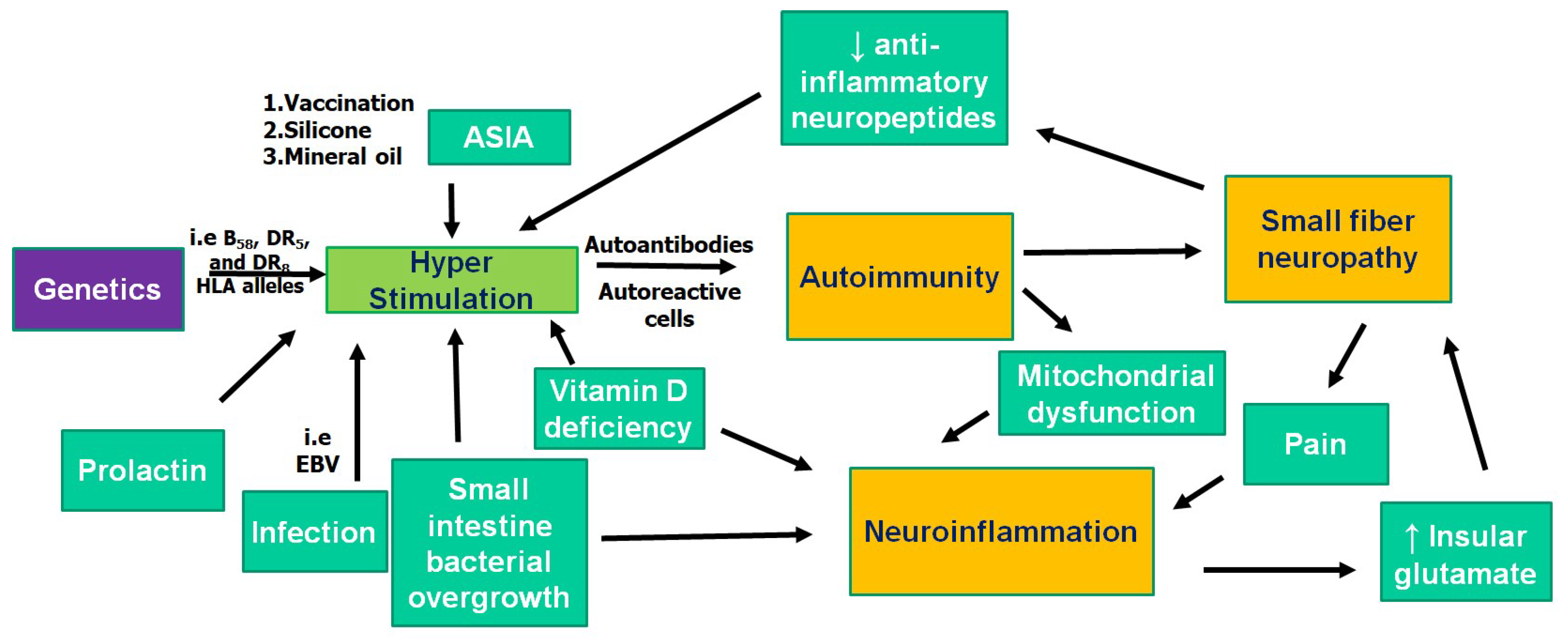
2. Pathogenesis of FM: Autoimmunity
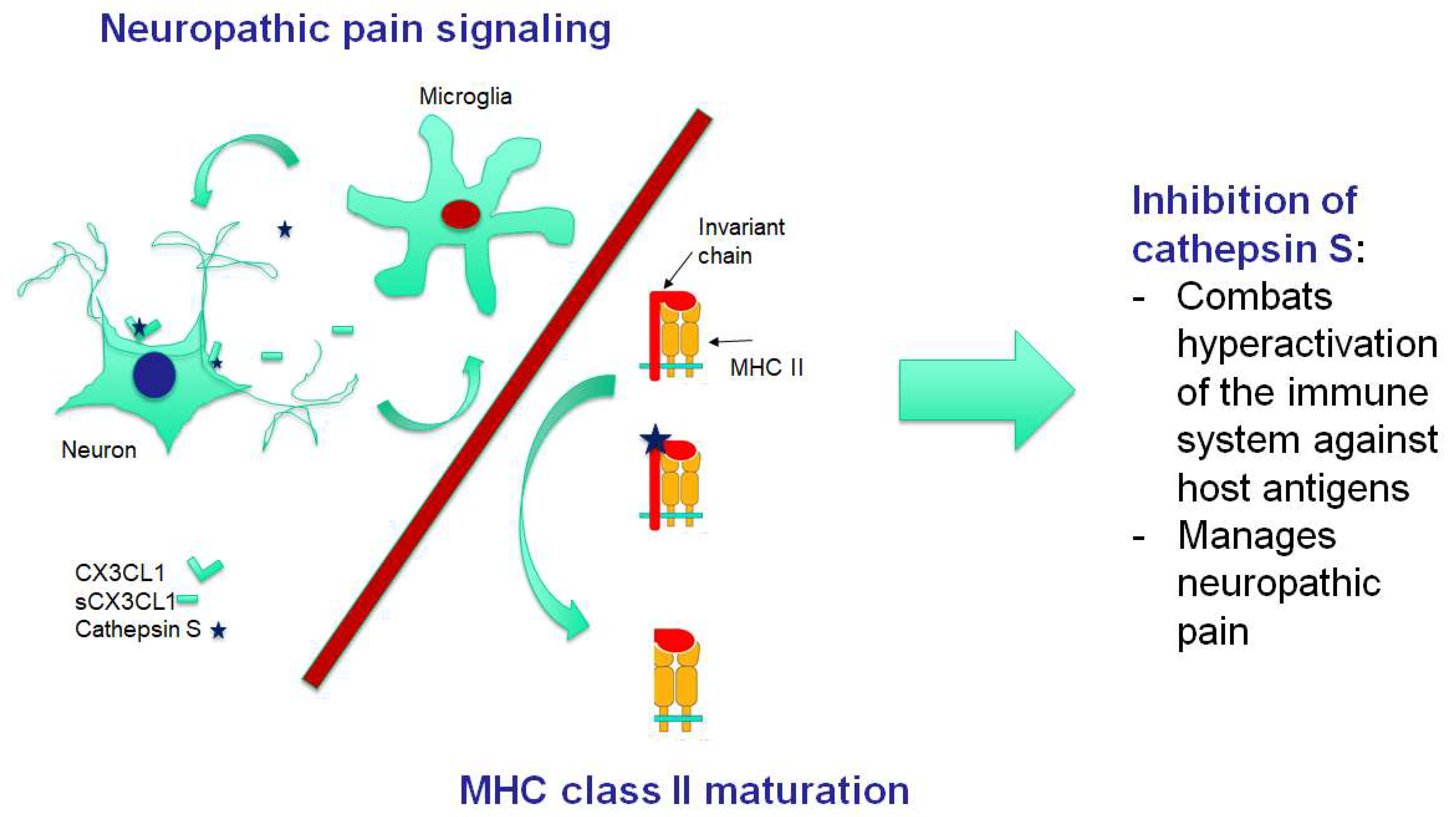
3. Pathogenesis of FM: Neuroinflammation
4. Pathogenesis of FM: Small Fiber Neuropathy
5. Chronic Fatigue Syndrome and FM
6. Human Papillomavirus Vaccination and FM
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviation
| AAb | autoantibodies |
| AChR | acetylcholine receptor |
| ACR | American College of Rheumatology |
| AdR | adrenergic receptor |
| CFS | chronic fatigue syndrome |
| CSF | cerebrospinal fluid |
| FM | fibromyalgia |
| HPA | hypothalamic-pituitary-adrenal |
| HPV | human papillomavirus |
| IENFD | intraepidermal nerve fiber density |
| LHVS | morpholinurea-leucine-homophenylalanine-vinyl sulfone-phenyl |
| MHC | major histocompatibility complex |
| SFN | small fiber neuropathy |
| TPO | thyroperoxidase |
References
- Boerma, T.; Harrison, J.; Jakob, R.; Mathers, C.; Schmider, A.; Weber, S. Revising the ICD: Explaining the WHO approach. Lancet 2016, 388, 2476–2477. [Google Scholar] [CrossRef]
- Wolfe, F.; Clauw, D.J.; Fitzcharles, M.-A.; Goldenberg, D.L.; Katz, R.S.; Mease, P.; Russell, A.S.; Russell, I.J.; Winfield, J.B.; Yunus, M.B. The American College of Rheumatology Preliminary Diagnostic Criteria for Fibromyalgia and Measurement of Symptom Severity. Arthritis Rheum. 2010, 62, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, F.; Smythe, H.A.; Yunus, M.B.; Bennett, R.M.; Bombardier, C.; Goldenberg, D.L.; Tugwell, P.; Campbell, S.M.; Abeles, M.; Clark, P. The American College of Rheumatology 1990 Criteria for the Classification of Fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum. 1990, 33, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Sarzi-Puttini, P.; Atzeni, F.; Masala, I.F.; Salaffi, F.; Chapman, J.; Choy, E. Are the ACR 2010 diagnostic criteria for fibromyalgia better than the 1990 criteria? Autoimmun. Rev. 2018, 17, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.D.; Hoffman, J.H. Fibromyalgia; Greenwood Press: Westport, CT, USA; ABC-CLIO: Santa Barbara, CA, USA, 2009. [Google Scholar]
- Amital, H.; Agmon-Levin, N.; Shoenfeld, N.; Arnson, Y.; Amital, D.; Langevitz, P.; Gurman, A.B.; Shoenfeld, Y. Olfactory impairment in patients with the fibromyalgia syndrome and systemic sclerosis. Immunol. Res. 2014, 60, 201–207. [Google Scholar] [CrossRef]
- Lichtenstein, A.; Tiosano, S.; Amital, H. The complexities of fibromyalgia and its comorbidities. Curr. Opin. Rheumatol. 2018, 30, 94–100. [Google Scholar] [CrossRef]
- Wolfe, F.; Clauw, D.J.; Fitzcharles, M.-A.; Goldenberg, D.L.; Häuser, W.; Katz, R.S.; Mease, P.; Russell, A.S.; Russell, I.J.; Winfield, J.B. Fibromyalgia Criteria and Severity Scales for Clinical and Epidemiological Studies: A Modification of the ACR Preliminary Diagnostic Criteria for Fibromyalgia. J. Rheumatol. 2011, 38, 1113–1122. [Google Scholar] [CrossRef]
- Fitzcharles, M.-A.; Ste-Marie, P.A.; Goldenberg, D.L.; Pereira, J.X.; Abbey, S.; Choinière, M. 2012 Canadian Guidelines for the Diagnosis and Management of Fibromyalgia Syndrome: Executive Summary. Pain Res. Manag. 2013, 18, 119–126. [Google Scholar] [CrossRef]
- Arnold, L.M.; Bennett, R.M.; Crofford, L.J.; Dean, L.E.; Clauw, D.J.; Goldenberg, D.L. AAPT Diagnostic Criteria for Fibromyalgia. J. Pain 2019, 20, 611–628. [Google Scholar] [CrossRef]
- Buskila, D.; Atzeni, F.; Sarzi-Puttini, P. Etiology of fibromyalgia: The possible role of infection and vaccination. Autoimmun. Rev. 2008, 8, 41–43. [Google Scholar] [CrossRef]
- Pridgen, W.L.; Duffy, C.; Gendreau, J.F.; Gendreau, R.M. A famciclovir + celecoxib combination treatment is safe and efficacious in the treatment of fibromyalgia. J. Pain Res. 2017, 10, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Reshkova, V.; Kalinova, D.; Milanov, I. Evaluation of antiviral antibodies against Epstein-Barr Virus and neurotransmitters in patients with fibromyalgia. J. Neurol Neurosci. 2015, 6. [Google Scholar] [CrossRef][Green Version]
- Vera-Lastra, O.; Hernadez, P.B.; Sánchez-Rodríguez, A.; Jara, L. AB0991 Prevalence of fibromyalgia and depression in patients with autoimmune /inflammatory syndrome induced by adjuvants compared to patients with systemic sclerosis. Annu. Eur. Congr. Rheumatol. 2017, 76, 1401. [Google Scholar]
- Khoo, T.; Proudman, S.; Limaye, V. Silicone breast implants and depression, fibromyalgia and chronic fatigue syndrome in a rheumatology clinic population. Clin. Rheumatol. 2019, 38, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Agmon-Levin, N.; Zafrir, Y.; Kivity, S.; Balofsky, A.; Amital, H.; Shoenfeld, Y. Chronic fatigue syndrome and fibromyalgia following immunization with the hepatitis B vaccine: Another angle of the ‘autoimmune (auto-inflammatory) syndrome induced by adjuvants’ (ASIA). Immunol. Res. 2014, 60, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, F.; Walitt, B.; Perrot, S.; Rasker, J.J.; Häuser, W. Fibromyalgia diagnosis and biased assessment: Sex, prevalence and bias. PLoS ONE 2018, 13, e0203755. [Google Scholar] [CrossRef] [PubMed]
- Kilic Baygutalp, N.; Seferoglu, B.; Baygutalp, F.; Senel, K. The correlation of serum prolactin levels and clinical parameters in the patients with fibromyalgia syndrome. Fiz. Tip. Rehabil. Bilim. Derg. 2013, 16, 83–87. [Google Scholar]
- Branco, J.C.; Tavares, V.; Abreu, I.; Correia, M.M.; Caetano, J.A.M. HLA Studies in Fibromyalgia. J. Musculoskelet. Pain 1996, 4, 21–27. [Google Scholar] [CrossRef]
- Carvalho, L.S.C.; Correa, H.; Silva, G.C.; Campos, F.S.; Baião, F.R.; Ribeiro, L.S.; Faria, A.M.; Reis, D.D. May genetic factors in fibromyalgia help to identify patients with differentially altered frequencies of immune cells? Clin. Exp. Immunol. 2008, 154, 346–352. [Google Scholar] [CrossRef]
- Klein, R.; Berg, P.A. High incidence of antibodies to 5-hydroxytryptamine, gangliosides and phospholipids in patients with chronic fatigue and fibromyalgia syndrome and their relatives: Evidence for a clinical entity of both disorders. Eur. J. Med. Res. 1995, 1, 21–26. [Google Scholar]
- Dadabhoy, D.; Crofford, L.J.; Spaeth, M.; Russell, I.J.; Clauw, D.J. Biology and therapy of fibromyalgia. Evidence-based biomarkers for fibromyalgia syndrome. Arthritis Res. Ther. 2008, 10, 211. [Google Scholar] [CrossRef]
- Jacobsen, S.; Wiik, A.; Høyer-Madsen, M.; Danneskiold-Samsøe, B.; Høyer-Madsen, M.; Danneskiold-Samsøe, B. Screening for autoantibodies in patients with primary fibromyalgia syndrome and a matched control group. APMIS 1990, 98, 655–658. [Google Scholar] [CrossRef]
- Ribeiro, L.S.; Proietti, F.A. Interrelations between fibromyalgia, thyroid autoantibodies, and depression. J. Rheumatol. 2004, 31, 2036–2040. [Google Scholar]
- Suk, J.; Lee, J.; Kim, J. Association between Thyroid Autoimmunity and Fibromyalgia. Exp. Clin. Endocrinol. Diabetes 2012, 120, 401–404. [Google Scholar] [CrossRef]
- Ekinci, B.; Uzkeser, H.; Sevimli, H.; Macit, P.M.; Haliloglu, S.; Carlioglu, A. Fibromyalgia in patients with thyroid autoimmunity: Prevalence and relationship with disease activity. Clin. Rheumatol. 2017, 36, 1617–1621. [Google Scholar]
- Applbaum, E.; Lichtbroun, A. Novel Sjögren’s autoantibodies found in fibromyalgia patients with sicca and/or xerostomia. Autoimmun. Rev. 2019, 18, 199–202. [Google Scholar] [CrossRef]
- Bäckryd, E.; Tanum, L.; Lind, A.-L.; Larsson, A.; Gordh, T. Evidence of both systemic inflammation and neuroinflammation in fibromyalgia patients, as assessed by a multiplex protein panel applied to the cerebrospinal fluid and to plasma. J. Pain Res. 2017, 10, 515–525. [Google Scholar] [CrossRef]
- Clark, A.K.; Yip, P.K.; Grist, J.; Gentry, C.; Staniland, A.A.; Marchand, F.; Dehvari, M.; Wotherspoon, G.; Winter, J.; Ullah, J.; et al. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc. Natl. Acad. Sci. USA 2007, 104, 10655–10660. [Google Scholar] [CrossRef]
- Flight, M.H. CatS relief. Nat. Rev. Drug Discov. 2007, 6, 604. [Google Scholar] [CrossRef]
- Clark, A.K.; Yip, P.K.; Malcangio, M. The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J. Neurosci. 2009, 29, 6945–6954. [Google Scholar] [CrossRef]
- Allan, E.R.O.; Yates, R.M. Redundancy between Cysteine Cathepsins in Murine Experimental Autoimmune Encephalomyelitis. PLoS ONE 2015, 10, e0128945. [Google Scholar] [CrossRef]
- Clark, A.K.; Grist, J.; Al-Kashi, A.; Perretti, M.; Malcangio, M.; Al-Kashi, A. Spinal cathepsin S and fractalkine contribute to chronic pain in the collagen-induced arthritis model. Arthritis Rheum. 2012, 64, 2038–2047. [Google Scholar] [CrossRef]
- Nieto, F.R.; Clark, A.K.; Grist, J.; Hathway, G.J.; Chapman, V.; Malcangio, M. Neuron-immune mechanisms contribute to pain in early stages of arthritis. J. Neuroinflamm. 2016, 13, 96. [Google Scholar] [CrossRef]
- Suzuki, F.; Nanki, T.; Imai, T.; Kikuchi, H.; Hirohata, S.; Kohsaka, H.; Miyasaka, N. Inhibition of CX3CL1 (fractalkine) improves experimental autoimmune myositis in SJL/J mice. J. Immunol. 2005, 175, 6987–6996. [Google Scholar] [CrossRef]
- Xu, J.; Wang, H.; Ding, K.; Lu, X.; Li, T.; Wang, J.; Wang, C.; Wang, J. Inhibition of Cathepsin S Produces Neuroprotective Effects after Traumatic Brain Injury in Mice. Mediat. Inflamm. 2013, 2013, 1–11. [Google Scholar] [CrossRef]
- Grandhi, R.; Tavakoli, S.; Ortega, C.; Simmonds, M.J. A Review of Chronic Pain and Cognitive, Mood, and Motor Dysfunction Following Mild Traumatic Brain Injury: Complex, Comorbid, and/or Overlapping Conditions? Brain Sci. 2017, 7, 160. [Google Scholar] [CrossRef]
- Allen, E.M.; Vitali, N.; Underwood, S.; Sweeney, D.; Wheeler, D.; Lawrence, C. Reversible cathepsin S (CATS) inhibitors block invariant chain degradation both in vitro and in vivo. Inflamm. Res. 2001, 50 (Suppl. 3), 159. [Google Scholar]
- Fissolo, N.M.; Kraus, M.; Reich, M.; Ayturan, M.; Overkleeft, H.; Driessen, C.; Weissert, R. Dual inhibition of proteasomal and lysosomal proteolysis ameliorates autoimmune central nervous system inflammation. Eur. J. Immunol. 2008, 38, 2401–2411. [Google Scholar] [CrossRef]
- Baugh, M.; Black, D.; Westwood, P.; Kinghorn, E.; McGregor, K.; Bruin, J.; Hamilton, W.; Dempster, M.; Claxton, C.; Cai, J.; et al. Therapeutic dosing of an orally active, selective cathepsin S inhibitor suppresses disease in models of autoimmunity. J. Autoimmun. 2011, 36, 201–209. [Google Scholar] [CrossRef]
- Albrecht, D.S.; Forsberg, A.; Sandström, A.; Bergan, C.; Kadetoff, D.; Protsenko, E. Brain glial activation in fibromyalgia—A multi-site positron emission tomography investigation. Brain Behav. Immun. 2019, 75, 72–83. [Google Scholar] [CrossRef]
- Littlejohn, G.; Guymer, E. Neurogenic inflammation in fibromyalgia. Semin. Immunopathol. 2018, 40, 291–300. [Google Scholar] [CrossRef]
- Vasquez, A. Neuroinflammation in fibromyalgia and CRPS is multifactorial. Nat. Rev. Rheumatol. 2016, 12, 242. [Google Scholar] [CrossRef]
- Perricone, C.; Shoenfeld, Y. Mosaic of Autoimmunity: The Novel Factors of Autoimmune Diseases, 1st ed.; Academic Press: New York, NY, USA, 2019. [Google Scholar]
- Polkowska-Pruszyńska, B.; Gerkowicz, A.; Szczepanik-Kułak, P.; Krasowska, D. Small intestinal bacterial overgrowth in systemic sclerosis: A review of the literature. Arch. Dermatol. Res. 2019, 311, 1–8. [Google Scholar] [CrossRef]
- Konrad, P.; Chojnacki, J.; Kaczka, A.; Pawłowicz, M.; Rudnicki, C.; Chojnacki, C. [Thyroid dysfunction in patients with small intestinal bacterial overgrowth]. Polski Merkur. Lek. Organ. Polskiego Towar. Lek. 2018, 44, 15–18. [Google Scholar]
- Losurdo, G.; Marra, A.M.; Shahini, E.; Girardi, B.; Giorgio, F.; Amoruso, A.; Pisani, A.; Piscitelli, D.; Barone, M.; Principi, M.; et al. Small intestinal bacterial overgrowth and celiac disease: A systematic review with pooled-data analysis. Neurogastroenterol. Motil. 2017, 29, e13028. [Google Scholar] [CrossRef]
- Li, X.; Xiao-Feng, L.; Gao, C.; Dong, S.; Zhang, S.-X.; Zhao-Hua, L. ab0533 positive rate of small intestinal bacterial overgrowth test (sibo) was significant correlations to disease activity of primary sjogren’s syndrome (pss). Abstr. Accept. Publ. 2019, 78, 1728. [Google Scholar]
- Rosen, Y.; Daich, J.; Soliman, I.; Brathwaite, E.; Shoenfeld, Y. Vitamin D and autoimmunity. Scand. J. Rheumatol. 2016, 45, 439–447. [Google Scholar] [CrossRef]
- Morris, G.; Berk, M.; Walder, K.; Maes, M. Central pathways causing fatigue in neuro-inflammatory and autoimmune illnesses. BMC Med. 2015, 13, 28. [Google Scholar] [CrossRef]
- Platt, M.P.; Agalliu, D.; Cutforth, T. Hello from the Other Side: How Autoantibodies Circumvent the Blood–Brain Barrier in Autoimmune Encephalitis. Front. Immunol. 2017, 8, 299. [Google Scholar] [CrossRef]
- Al-Nimer, M.S.M.; Mohammad, T.A.M.; Alsakeni, R.A. Serum levels of serotonin as a biomarker of newly diagnosed fibromyalgia in women: Its relation to the platelet indices. J. Res. Med. Sci. 2018, 23, 71. [Google Scholar] [CrossRef]
- Carta, M.G.; Loviselli, A.; Hardoy, M.C.; Massa, S.; Cadeddu, M.; Sardu, C.; Carpiniello, B.; Dell’Osso, L.; Mariotti, S. The link between thyroid autoimmunity (antithyroid peroxidase autoantibodies) with anxiety and mood disorders in the community: A field of interest for public health in the future. BMC Psychiatry 2004, 4, 25. [Google Scholar] [CrossRef]
- Harte, S.E.; Clauw, D.J.; Hayes, J.M.; Feldman, E.L.; St Charles, I.C.; Watson, C.J. Reduced intraepidermal nerve fiber density after a sustained increase in insular glutamate: A proof-of-concept study examining the pathogenesis of small fiber pathology in fibromyalgia. Pain Rep. 2017, 2, e590. [Google Scholar] [CrossRef]
- Basantsova, N.Y.; Starshinova, A.A.; Dori, A.; Zinchenko, Y.S.; Yablonskiy, P.K.; Shoenfeld, Y. Small-fiber neuropathy definition, diagnosis, and treatment. Neurol. Sci. 2019, 40, 1343–1350. [Google Scholar] [CrossRef]
- Ramírez, M.; Martínez-Martínez, L.-A.; Hernández-Quintela, E.; Velazco-Casapía, J.; Vargas, A.; Martínez-Lavín, M. Small fiber neuropathy in women with fibromyalgia. An in vivo assessment using corneal confocal bio-microscopy. Semin. Arthritis Rheum. 2015, 45, 214–219. [Google Scholar] [CrossRef]
- Caro, X.J.; Winter, E.F. The Role and Importance of Small Fiber Neuropathy in Fibromyalgia Pain. Curr. Pain Headache Rep. 2015, 19, 55. [Google Scholar] [CrossRef]
- Oaklander, A.L.; Herzog, Z.D.; Downs, H.M.; Klein, M.M. Objective evidence that small-fiber polyneuropathy underlies some illnesses currently labeled as fibromyalgia. Pain 2013, 154, 2310–2316. [Google Scholar] [CrossRef]
- Kosmidis, M.L.; Koutsogeorgopoulou, L.; Alexopoulos, H.; Mamali, I.; Vlachoyiannopoulos, P.G.; Voulgarelis, M.; Moutsopoulos, H.M.; Tzioufas, A.G.; Dalakas, M.C. Reduction of Intraepidermal Nerve Fiber Density (IENFD) in the skin biopsies of patients with fibromyalgia: A controlled study. J. Neurol. Sci. 2014, 347, 143–147. [Google Scholar] [CrossRef]
- Giannoccaro, M.P.; Donadio, V.; Incensi, A.; Avoni, P.; Liguori, R. Small nerve fiber involvement in patients referred for fibromyalgia. Muscle Nerve 2014, 49, 757–759. [Google Scholar] [CrossRef]
- de Tommaso, M.; Nolano, M.; Iannone, F.; Vecchio, E.; Ricci, K.; Lorenzo, M. Update on laser-evoked potential findings in fibromyalgia patients in light of clinical and skin biopsy features. J. Neurol. 2014, 261, 461–472. [Google Scholar] [CrossRef]
- Üçeyler, N.; Zeller, D.; Kahn, A.-K.; Kewenig, S.; Kittel-Schneider, S.; Schmid, A.; Casanova-Molla, J.; Reiners, K.; Sommer, C. Small fibre pathology in patients with fibromyalgia syndrome. Brain 2013, 136, 1857–1867. [Google Scholar] [CrossRef]
- Caro, X.J.; Winter, E.F. Evidence of Abnormal Epidermal Nerve Fiber Density in Fibromyalgia: Clinical and Immunologic Implications. Arthritis Rheumatol. 2014, 66, 1945–1954. [Google Scholar] [CrossRef]
- Varela, N.; Chorny, A.; Gonzalez-Rey, E.; Delgado, M. Tuning inflammation with anti-inflammatory neuropeptides. Expert Opin. Boil. Ther. 2007, 7, 461–478. [Google Scholar] [CrossRef]
- Metyas, S.; Youssef, H.; Che, C.; Quismorio, A.; Bui, J. Improvement in Fibromyalgia Symptoms and Skin Biopsy Results in Patients with Fibromyalgia and Small Fiber Neuropathy Treated with Intravenous Immune Globulin Infusion( IVIG) [abstract]. In Arthritis Rheumatology; Wiley: New Jersey, NJ, USA, 2018. [Google Scholar]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2017, 42, 193–215. [Google Scholar] [CrossRef]
- Harris, R.E.; Sundgren, P.C.; Craig, A.; Kirshenbaum, E.; Sen, A.; Napadow, V.; Clauw, D.J. Elevated insular glutamate in fibromyalgia is associated with experimental pain. Arthritis Rheum. 2009, 60, 3146–3152. [Google Scholar] [CrossRef]
- Sharif, K.; Watad, A.; Bragazzi, N.L.; Lichtbroun, M.; Martini, M.; Perricone, C.; Amital, H.; Shoenfeld, Y. On chronic fatigue syndrome and nosological categories. Clin. Rheumatol. 2018, 37, 1161–1170. [Google Scholar] [CrossRef]
- Sotzny, F.; Blanco, J.; Capelli, E.; Castro-Marrero, J.; Steiner, S.; Murovska, M.; Scheibenbogen, C.; (Euromene), O.B.O.T.E.N.O.M. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome—Evidence for an autoimmune disease. Autoimmun. Rev. 2018, 17, 601–609. [Google Scholar] [CrossRef]
- Blomberg, J.; Gottfries, C.-G.; Elfaitouri, A.; Rizwan, M.; Rosén, A. Infection Elicited Autoimmunity and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: An Explanatory Model. Front. Immunol. 2018, 9, 229–249. [Google Scholar] [CrossRef]
- Perez, M.; Jaundoo, R.; Hilton, K.; Del Alamo, A.; Gemayel, K.; Klimas, N.G.; Craddock, T.J.A.; Nathanson, L. Genetic Predisposition for Immune System, Hormone, and Metabolic Dysfunction in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Pilot Study. Front. Pediatr. 2019, 7, 206. [Google Scholar] [CrossRef]
- Castro-Marrero, J.; Faro, M.; Aliste, L.; Sáez-Francàs, N.; Calvo, N.; Martinez-Martinez, A.; De Sevilla, T.F.; Alegre, J. Comorbidity in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: A Nationwide Population-Based Cohort Study. Psychosomatics 2017, 58, 533–543. [Google Scholar] [CrossRef]
- Loebel, M.; Grabowski, P.; Heidecke, H.; Bauer, S.; Hanitsch, L.G.; Wittke, K.; Meisel, C.; Reinke, P.; Volk, H.-D.; Fluge, Ø.; et al. Antibodies to β adrenergic and muscarinic cholinergic receptors in patients with Chronic Fatigue Syndrome. Brain Behav. Immun. 2016, 52, 32–39. [Google Scholar] [CrossRef]
- Scheibenbogen, C.; Loebel, M.; Freitag, H.; Krueger, A.; Bauer, S.; Antelmann, M.; Doehner, W.; Scherbakov, N.; Heidecke, H.; Reinke, P.; et al. Immunoadsorption to remove β2 adrenergic receptor antibodies in Chronic Fatigue Syndrome CFS/ME. PLoS ONE 2018, 13, e0193672. [Google Scholar] [CrossRef] [PubMed]
- Tomas, C.; Newton, J.; Watson, S. A Review of Hypothalamic-Pituitary-Adrenal Axis Function in Chronic Fatigue Syndrome. ISRN Neurosci. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Crofford, L.J. The Hypothalamic-Pituitary-Adrenal Axis in Fibromyalgia: Where Are We in 2001? J. Musculoskelet. Pain 2002, 10, 215–220. [Google Scholar] [CrossRef]
- Morris, G.; Anderson, G.; Maes, M. Hypothalamic-Pituitary-Adrenal Hypofunction in Myalgic Encephalomyelitis (ME)/Chronic Fatigue Syndrome (CFS) as a Consequence of Activated Immune-Inflammatory and Oxidative and Nitrosative Pathways. Mol. Neurobiol. 2017, 54, 6806–6819. [Google Scholar] [CrossRef]
- Nakatomi, Y.; Mizuno, K.; Ishii, A.; Wada, Y.; Tanaka, M.; Tazawa, S.; Onoe, K.; Fukuda, S.; Kawabe, J.; Takahashi, K.; et al. Neuroinflammation in Patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: An 11C-(R)-PK11195 PET Study. J. Nucl. Med. 2014, 55, 945–950. [Google Scholar] [CrossRef]
- Zaichik, A.S.; Churilov, L. P Fundamentals of General Pathology. Fundamentals of General Pathophysiology; ELBI Publishers: Saint Petersburg, Russia, 1999; Volume 1, p. 536. [Google Scholar]
- Noda, M.; Ifuku, M.; Hossain, M.S.; Katafuchi, T. Glial Activation and Expression of the Serotonin Transporter in Chronic Fatigue Syndrome. Front. Psychol. 2018, 9, 589–595. [Google Scholar] [CrossRef]
- Fomicheva, E.E.; Filatenkova, T.A.; Rybakina, E.G. Activity in the Hypothalamo-Hypophyseal-Adrenocortical System on Experimental Induction of Chronic Fatigue Syndrome. Neurosci. Behav. Physiol. 2010, 40, 245–250. [Google Scholar] [CrossRef]
- Giannoccaro, M.P.; Cossins, J.; Sørland, K.; Fluge, Ø.; Vincent, A. Searching for Serum Antibodies to Neuronal Proteins in Patients with Myalgic Encephalopathy/Chronic Fatigue Syndrome. Clin. Ther. 2019, 41, 836–847. [Google Scholar] [CrossRef]
- Churilov, L.P.; Danilenko, O.V. [Immunoreactivity in chronic fatigue syndrome during remission, exacerbation and virus carriage] In Russian. Clin. Pathophysiol. 2019, 25, 26–36. [Google Scholar]
- Hatziagelaki, E.; Adamaki, M.; Tsilioni, I.; Dimitriadis, G.; Theoharides, T.C. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome—Metabolic Disease or Disturbed Homeostasis due to Focal Inflammation in the Hypothalamus? J. Pharmacol. Exp. Ther. 2018, 367, 155–167. [Google Scholar] [CrossRef]
- Mackay, A.; Tate, W.P. A compromised paraventricular nucleus within a dysfunctional hypothalamus: A novel neuroinflammatory paradigm for ME/CFS. Int. J. Immunopathol. Pharmacol. 2018, 32, 1–8. [Google Scholar] [CrossRef]
- Bugajski, A.; Gadek-Michalska, A.; Bugajski, J. The involvement of nitric oxide and prostaglandins in the cholinergic stimulation of hypothalamic-pituitary-adrenal response during crowding stress. J. Physiol. Pharmacol. Polish Physiol. Soc. 2006, 57, 463–477. [Google Scholar]
- Smail, M.A.; Soles, J.L.; Karwoski, T.E.; Rubin, R.T.; Rhodes, M.E. Sexually diergic hypothalamic-pituitary-adrenal axis responses to selective and non-selective muscarinic antagonists prior to cholinergic stimulation by physostigmine in rats. Brain Res. Bull. 2018, 137, 23–34. [Google Scholar] [CrossRef]
- Wang, D.; Feng, H.; Li, Y.-S.; Qiu, D.-L.; Jin, H.; Jin, Q.-H. β-Adrenoceptors in the hypothalamic paraventricular nucleus modulate the baroreflex in conscious rats. Neurosci. Lett. 2013, 551, 43–46. [Google Scholar] [CrossRef]
- Okada, S.; Yamaguchi, N. Possible role of adrenoceptors in the hypothalamic paraventricular nucleus in corticotropin-releasing factor-induced sympatho-adrenomedullary outflow in rats. Auton. Neurosci. 2017, 203, 74–80. [Google Scholar] [CrossRef]
- Hazell, G.G.J.; Hindmarch, C.C.; Pope, G.R.; Roper, J.A.; Lightman, S.L.; Murphy, D. G protein-coupled receptors in the hypothalamic paraventricular and supraoptic nuclei—serpentine gateways to neuroendocrine homeostasis. Front. Neuroendocrinol. 2012, 33, 45–66. [Google Scholar] [CrossRef]
- Yamamoto, S.; Ouchi, Y.; Nakatsuka, D.; Tahara, T.; Mizuno, K.; Tajima, S. Reduction of [11C](+)3-MPB binding in brain of chronic fatigue syndrome with serum autoantibody against muscarinic cholinergic receptor. PLoS ONE 2012, 7, e51515. [Google Scholar] [CrossRef]
- Ikeda, S.-I.; Hineno, A.; Scheibenbogen, C.; Heidecke, H.; Shulze-Forster, K.; Junker, J. Autoantibodies against autonomic nerve receptors in adolescent Japanese girls after immunization with human papillomavirus vaccine. Ann. Arthritis Clin. Rheumatol. 2019, 2, 1014. [Google Scholar]
- Pellegrino, P.; Perrone, V.; Pozzi, M.; Carnovale, C.; Perrotta, C.; Clementi, E.; Radice, S. The epidemiological profile of ASIA syndrome after HPV vaccination: An evaluation based on the Vaccine Adverse Event Reporting Systems. Immunol. Res. 2014, 61, 90–96. [Google Scholar] [CrossRef]
- Brinth, L.; Theibel, A.C.; Pors, K.; Mehlsen, J. Suspected side effects to the quadrivalent human papilloma vaccine. Dan. Med. J. 2015, 62, 5064. [Google Scholar]
- Kinoshita, T.; Abe, R.-T.; Hineno, A.; Tsunekawa, K.; Nakane, S.; Ikeda, S.-I. Peripheral Sympathetic Nerve Dysfunction in Adolescent Japanese Girls Following Immunization with the Human Papillomavirus Vaccine. Intern. Med. 2014, 53, 2185–2200. [Google Scholar] [CrossRef] [PubMed]
- Arata, H. Clinical analysis of neurological symptoms in the patients with HPV vaccination. J. Neurol. Sci. 2017, 381, 530. [Google Scholar] [CrossRef]
- Martínez-Lavín, M.; Martínez-Martínez, L.-A.; Reyes-Loyola, P. HPV vaccination syndrome. A questionnaire-based study. Clin. Rheumatol. 2015, 34, 1981–1983. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Assessment Report EMA/762033/2015. Available online: https://www.ema.europa.eu/en/documents/referral/hpv-vaccines-article-20-procedure-assessment-report_en.pdf (accessed on 17 October 2019).
- Donegan, K.; Beau-Lejdstrom, R.; King, B.; Seabroke, S.; Thomson, A.; Bryan, P. Bivalent human papillomavirus vaccine and the risk of fatigue syndromes in girls in the UK. Vaccine 2013, 31, 4961–4967. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.C.; Bell, C.A.; Simmonds, K.A.; Svenson, L.W.; Russell, M.L. Adverse events following HPV vaccination, Alberta 2006–2014. Vaccine 2016, 34, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Galán, M.; Pérez-Vilar, S.; Díez-Domingo, J.; Tuells, J.; Gomar-Fayos, J.; Morales-Olivas, F.; Pastor-Villalba, E. Notificación de reacciones adversas a la vacuna frente al virus del papiloma humano en la Comunidad Valenciana (2007-2011). Anales Pediatría 2014, 81, 303–309. [Google Scholar] [CrossRef]
- Martínez-Lavín, M. HPV Vaccination Syndrome: A Clinical Mirage, or a New Tragic Fibromyalgia Model. Reumatol. Clín. 2018, 14, 211–214. [Google Scholar] [CrossRef]
- Martínez-Lavín, M.; Amezcua-Guerra, L. Serious adverse events after HPV vaccination: A critical review of randomized trials and post-marketing case series. Clin. Rheumatol. 2017, 36, 2169–2178. [Google Scholar] [CrossRef]
- Sekiguchi, K.; Yasui, N.; Kowa, H.; Kanda, F.; Toda, T. Two Cases of Acute Disseminated Encephalomyelitis Following Vaccination against Human Papilloma Virus. Intern. Med. 2016, 55, 3181–3184. [Google Scholar] [CrossRef][Green Version]
- Karussis, D.; Petrou, P. The spectrum of post-vaccination inflammatory CNS demyelinating syndromes. Autoimmun. Rev. 2014, 13, 215–224. [Google Scholar] [CrossRef]
- Hu, Y.; Tornes, L.; Lopez-Alberola, R. Two Cases of Pediatric Multiple Sclerosis after Human Papillomavirus Vacciation. Neurology 2018, 90 (Suppl. 15), P4-353. [Google Scholar]
- Mouchet, J.; Salvo, F.; Raschi, E.; Poluzzi, E.; Antonazzo, I.C.; De Ponti, F.; Bégaud, B. Human papillomavirus vaccine and demyelinating diseases—A systematic review and meta-analysis. Pharmacol. Res. 2018, 132, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Blitshteyn, S.; Brinth, L.; Hendrickson, J.E.; Martinez-Lavin, M.; Blitshetyn, S. Autonomic dysfunction and HPV immunization: An overview. Immunol. Res. 2018, 66, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, B.; Poddighe, D.; Vadalà, M.; Laurino, C.; Carnovale, C.; Clementi, E. Severe somatoform and dysautonomic syndromes after HPV vaccination: Case series and review of literature. Immunol. Res. 2017, 65, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, R.K.; Crépeaux, G.; Authier, F.-J. Myalgia and chronic fatigue syndrome following immunization: Macrophagic myofasciitis and animal studies support linkage to aluminum adjuvant persistency and diffusion in the immune system. Autoimmun. Rev. 2019, 18, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Combadière, C.; Authier, F.-J.; Itier, V.; Lux, F.; Exley, C. Slow CCL2-dependent translocation of biopersistent particles from muscle to brain. BMC Med. 2013, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef]
- Inbar, R.; Weiss, R.; Tomljenovic, L.; Arango, M.-T.; Deri, Y.; Shaw, C.A. Behavioral abnormalities in female mice following administration of aluminum adjuvants and the human papillomavirus (HPV) vaccine Gardasil. Immunol Res. 2017, 65, 136–149. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryabkova, V.A.; Churilov, L.P.; Shoenfeld, Y. Neuroimmunology: What Role for Autoimmunity, Neuroinflammation, and Small Fiber Neuropathy in Fibromyalgia, Chronic Fatigue Syndrome, and Adverse Events after Human Papillomavirus Vaccination? Int. J. Mol. Sci. 2019, 20, 5164. https://doi.org/10.3390/ijms20205164
Ryabkova VA, Churilov LP, Shoenfeld Y. Neuroimmunology: What Role for Autoimmunity, Neuroinflammation, and Small Fiber Neuropathy in Fibromyalgia, Chronic Fatigue Syndrome, and Adverse Events after Human Papillomavirus Vaccination? International Journal of Molecular Sciences. 2019; 20(20):5164. https://doi.org/10.3390/ijms20205164
Chicago/Turabian StyleRyabkova, Varvara A., Leonid P. Churilov, and Yehuda Shoenfeld. 2019. "Neuroimmunology: What Role for Autoimmunity, Neuroinflammation, and Small Fiber Neuropathy in Fibromyalgia, Chronic Fatigue Syndrome, and Adverse Events after Human Papillomavirus Vaccination?" International Journal of Molecular Sciences 20, no. 20: 5164. https://doi.org/10.3390/ijms20205164
APA StyleRyabkova, V. A., Churilov, L. P., & Shoenfeld, Y. (2019). Neuroimmunology: What Role for Autoimmunity, Neuroinflammation, and Small Fiber Neuropathy in Fibromyalgia, Chronic Fatigue Syndrome, and Adverse Events after Human Papillomavirus Vaccination? International Journal of Molecular Sciences, 20(20), 5164. https://doi.org/10.3390/ijms20205164