1. Introduction
Notable progress in recent years has been made in biopolymers, synthetic polymers, and advanced composite materials. The exploitation of chemical, biological, and engineering methods has led to the generation of new polymeric productions by combining the polymer synthesis and polymer chemical modification for advanced applications in relevant industrial environments.
The use of bio-based polymers, such as poly(hydroxyalkanoate)s, has increased dramatically in various production fields including packaging, automotive, civil engineering, cosmetic, and medicine [
1]. An interesting new development considers the modification of polymer surfaces and interfaces, generating, for example, hybrid coatings using organic or inorganic nanostructures to improve the surface properties of traditional polymeric materials [
2,
3]. A central goal is to obtain a deeper understanding of the relationship between the microstructure and macrostructure of composites, leading to the development of modified and new polymers with unique and specific material properties.
Research in biopolymers and synthetic polymers has developed in the biomedical field, contributing to the production of new biocompatible materials. In this general context, there is a large interest in the development of implantable robotic devices to allow patients with severe hormonal alterations or chronic pain to benefit from an automatic and effective administration of specific drugs [
4]. However, long-term electronic implants present specific challenges, including stable performance and materials having poor bio- and cytocompatibility, resulting in immune reactions and infections [
5]. For implantable materials, it is important to address the interface between the device and human body. To allow a better interaction of the medical device with the biological system surrounding it, often, polymeric coatings are applied [
6].
A successful coating must be biocompatible, nontoxic and sterilizable, have sufficient mechanical properties, be durable under conditions of use, and adhere to the device. Hydrophilic coatings, prepared using polymers with functional groups able to absorb water, such as amino, hydroxyl, or carboxyl groups, are widely used to produce coatings on metallic or plastic substrates [
7]. Interestingly, coatings based on hydrogels can provide additional advantages such as good biocompatibility, good wetting, and low friction, making them excellent candidates as coating for soft tissue implants [
8].
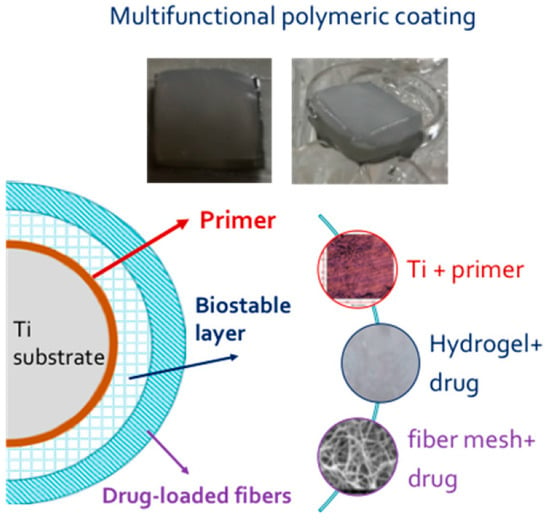
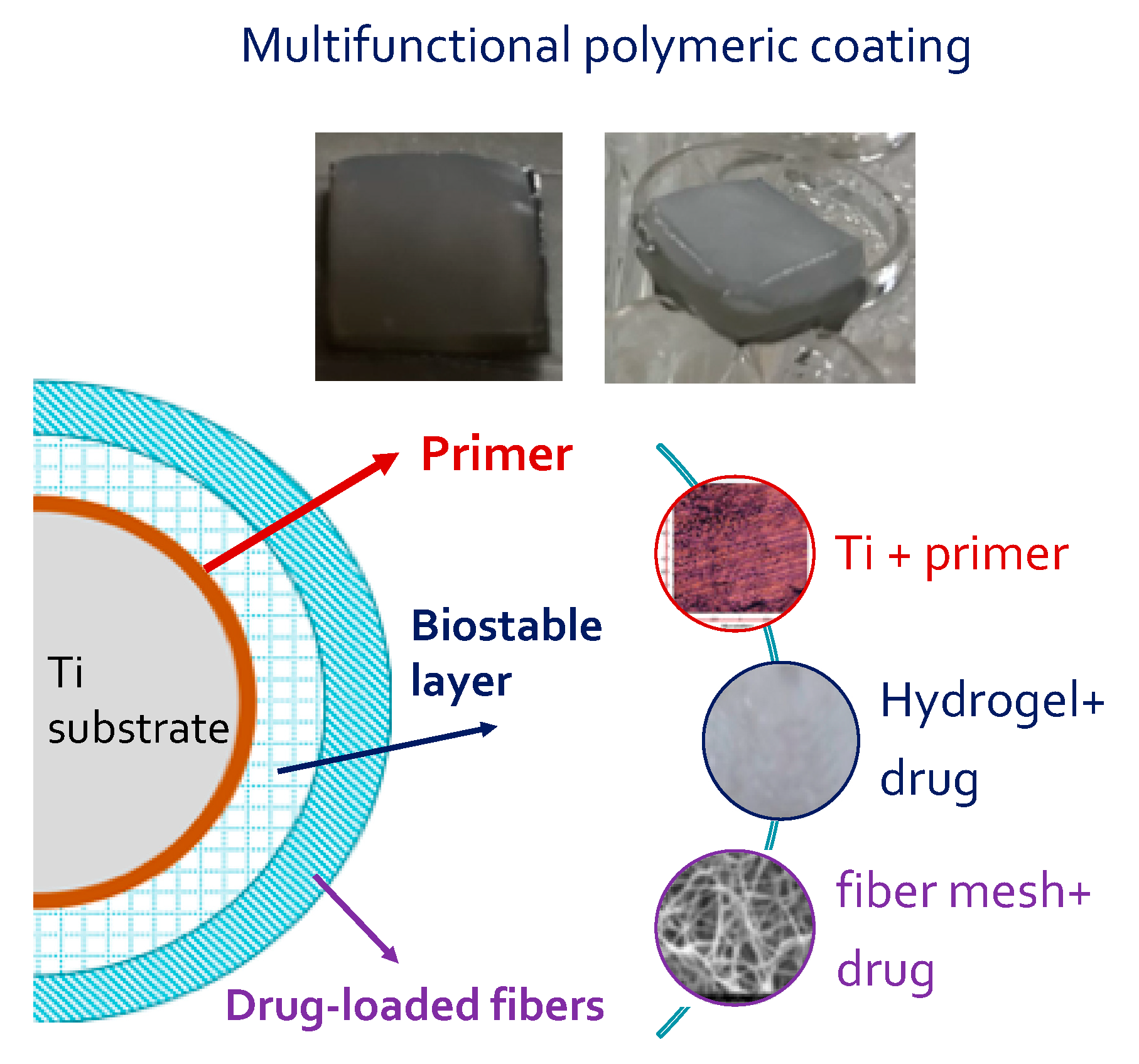
The objective of this study was to obtain a multifunctional coating for an innovative, fully implantable device. The coating was designed to have three fundamental properties: adhesion to device, close mechanical resemblance to human soft tissues, and control of the inflammatory response and tissue repair process.
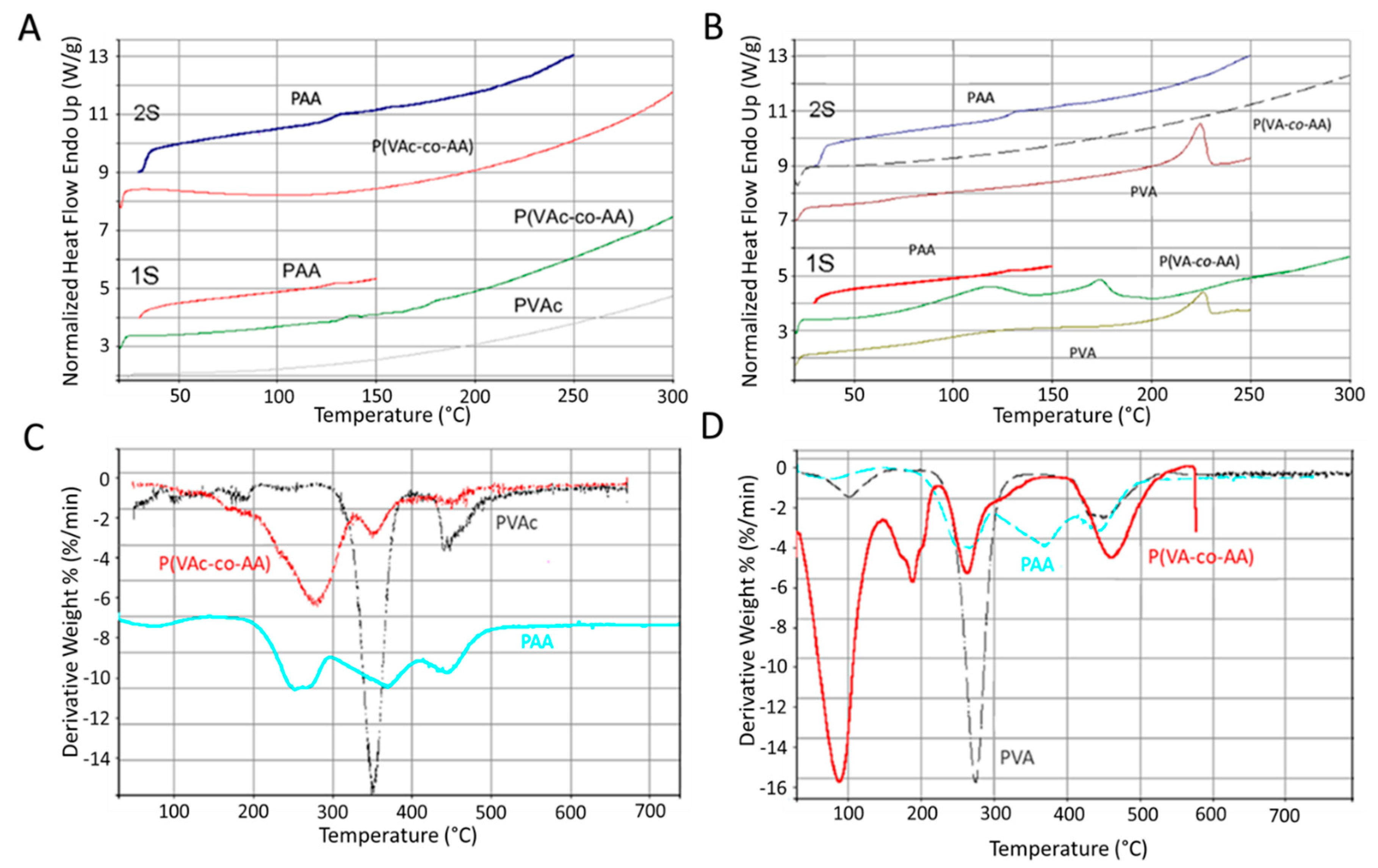
To achieve an effective adhesion to the device, two acrylic primers in the form of thin films were studied and characterized. Poly(vinyl acetate-acrylic acid) P(VAc-
co-AA) copolymer was synthetized by radical polymerization, starting from vinyl acetate (VAc) and acrylic acid (AA); a successive hydrolysis allowed the synthesis of poly(vinyl alcohol-acrylic acid) P(VA-
co-AA) copolymer [
9,
10]. A thin layer of P(VA-
co-AA) was dehydro-thermally cross-linked [
11]. The primers act as intermediates to improve the adhesion force between the device substrate, metallic or polymeric, and the real coating; a second layer of poly(vinyl alcohol) (PVA) hydrogel was prepared by repetitive freezing and thawing of aqueous PVA solutions [
12] and loaded with dexamethasone (Dexa). A third layer of biodegradable drug-loaded ultrafine fibers was obtained by electrospinning of PHBHV solutions containing Dexa. The copolymer formation was characterized and evaluated by FT-IR, differential scanning calorimetry (DSC), and thermal gravimetric analysis (TGA) analysis. The components of the coating were characterized from morphological (scanning electron microscopy, SEM), mechanical (dynamic mechanical analysis, DMA), chemical (FT-IR chemical imaging), biological (cytotoxicity test), and functional (high permeation liquid chromatography, HPLC, adhesion tests) properties.
3. Materials and Methods
3.1. Preparation of Titanium Substrate
NiTi (titanium alloy) was cut into 10 × 10 × 1 mm plates. The plates were washed in water and dish soap and then rinsed in deionized water and dried at 40 °C. They were then oxidized in NaOH 3 M for 1 h to promote the metal–polymer bond.
3.2. Preparation of Acrylic Primers
3.2.1. Synthesis of P(VAc-co-AA)
P(VAc-co-AA) copolymer was synthetized starting from vinyl acetate (VAc), potassium persulphate (KPS), and acrylic acid (AA). Preweighed amounts of PVA (1 g, 80% hydrolyzed, Sigma Aldrich, Milan, Italy) were put into a round-bottomed flask with 150 mL of water. The flask was stirred and heated at 60 °C in an oil bath. As the water began to steam, 45.5 mL of vinyl acetate monomer (VAc) were added. The monomer was treated with resin to eliminate the inhibitor. A 5% solution of K2S2O8 was added. The reaction was performed under nitrogen atmosphere stirring at 200 rpm with a reflux condenser for 15 min. A mixture of AA (21 mL), water (50 mL), and 5% K2S2O8 solution (5 mL) was prepared and 15 mL of that were added to the reaction. The remaining solution was dripped in the flask within 10 min. The reaction was carried out for 3 h under nitrogen atmosphere and then stopped with hydroquinone. The obtained polymer was purified in chloroform.
3.2.2. Preparation of P(VA-co-AA) through Hydrolysis of P(VAc-co-AA)
The polymer (10 g) was put into a round-bottomed flask and a solvent mixture composed by 200 mL of methanol (MeOH) and 150 mL of tetrahydrofurane (THF) was added. The flask was put under stirring and heated with a reflux condenser for 4–5 h. The solution was left under stirring at room temperature overnight. The hydrolysis was made with sodium metoxide (MeONa) in MeOH. MeONa (5 g) was added to 50 mL of MeOH into a beaker (the dissolution process was very exothermic). Then, 15 mL of this solution were added to the polymer solution and a white precipitate was immediately formed. After 30 min, the remaining methoxide solution (35 mL) was put into the flask. The precipitate was filtered and washed with MeOH. The hydrolized polymer was soluble in hot water.
3.3. Preparation of PVA Hydrogel
Commercial powder poly(vinyl alcohol) (PVA, Mw 115000, Sigma Aldrich) was dissolved in deionized water in autoclave to obtain a 8% w/v solution. Drug-loaded hydrogels of PVA were produced by adding dexamethasone (Sigma Aldrich) (0.01% w/v) and performing repetitive freezing and thawing of the PVA-Dexa solutions (8 cycles at least).
3.4. Preparation of PHBHV Electrospun Fiber Meshes
Poly (3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBHV, Sigma Aldrich) was dissolved in chloroform/methanol (9:1 v/v) mixture at a polymer/solvent concentration of 15% and stirred at 300 rpm for 12 h at room temperature (RT). For production of dexamethasone-loaded PHBHV fibers, 10wt% of Dexa was added to the solution and stirred at 300 rpm for 12 h at room temperature (RT). Each polymer solution was loaded into a 10 mL glass syringe, fitted with a blunt tip stainless steel needle (21G 3/4”), and placed into a syringe pump (NE-300, New Era Pump Systems, Inc. Farmingdale, NY, USA). The ground terminal of high voltage supply (S1600079 Linari High Voltage, Linari Engineering s.r.l, Pisa, Italy) was connected to the metal needle, while the positive terminal was connected to the collector; 30 kV potential was applied. A static collector made of a plastic plate covered with an aluminum foil, or a cylindrical collector (diameter 8 cm, Linari Engineering s.r.l), were placed at a distance of 15 cm from the tip of the needle. The polymer solution was injected from the needle in the presence of the electric field at a constant flow rate of 0.001 mL/min. All the fabrication steps were performed at RT with relative humidity (RH) of about 46% (if not differently specified). The fiber meshes were kept in an oven at 60 °C overnight to remove solvent traces.
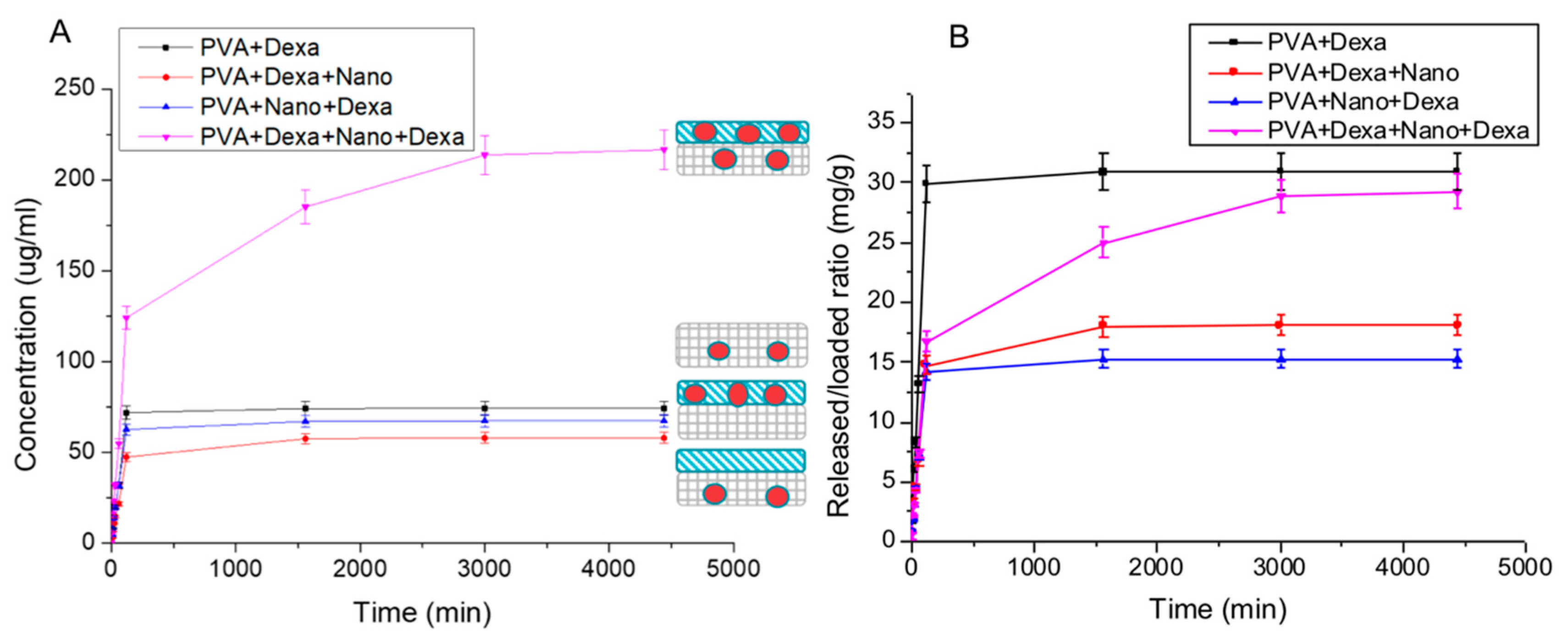
3.5. Final Assembly of the Multifunctional Composite System
P(VAc-co-AA) was dissolved in THF to prepare a 1%
w/
v solution and P(VA-co-AA) was dissolved in water to prepare a 0.3% w/v solution. Two layers of 250 µL of P(VAc-co-AA) solution or four layers of 250 µL of P(VA-co-AA) solution were stacked up on the oxidized titanium platelets to act as primer between the metal and the PVA hydrogel. The P(VA-co-AA) primer was cross-linked by dehydro-thermal method in a controlled temperature ramp under vacuum conditions: from RT to 50 °C for 2 h, 90 °C overnight, 120 °C for 3 h, and then cooling to 50 °C. The hydrogel layer was produced by deposition of 250 µL Dexa-PVA solution on the selected type of primer. Dexa-PHBHV fiber mesh (1 cm
2 squares) were put over the hydrogel before the freezing and thawing procedure. The photos of the multifunctional coating before and after immersion in incubation medium and a schematic representation of final assembly are reported in
Figure 11.
3.6. Morphological, Thermal, Physico-Chemical, and Mechanical Characterization
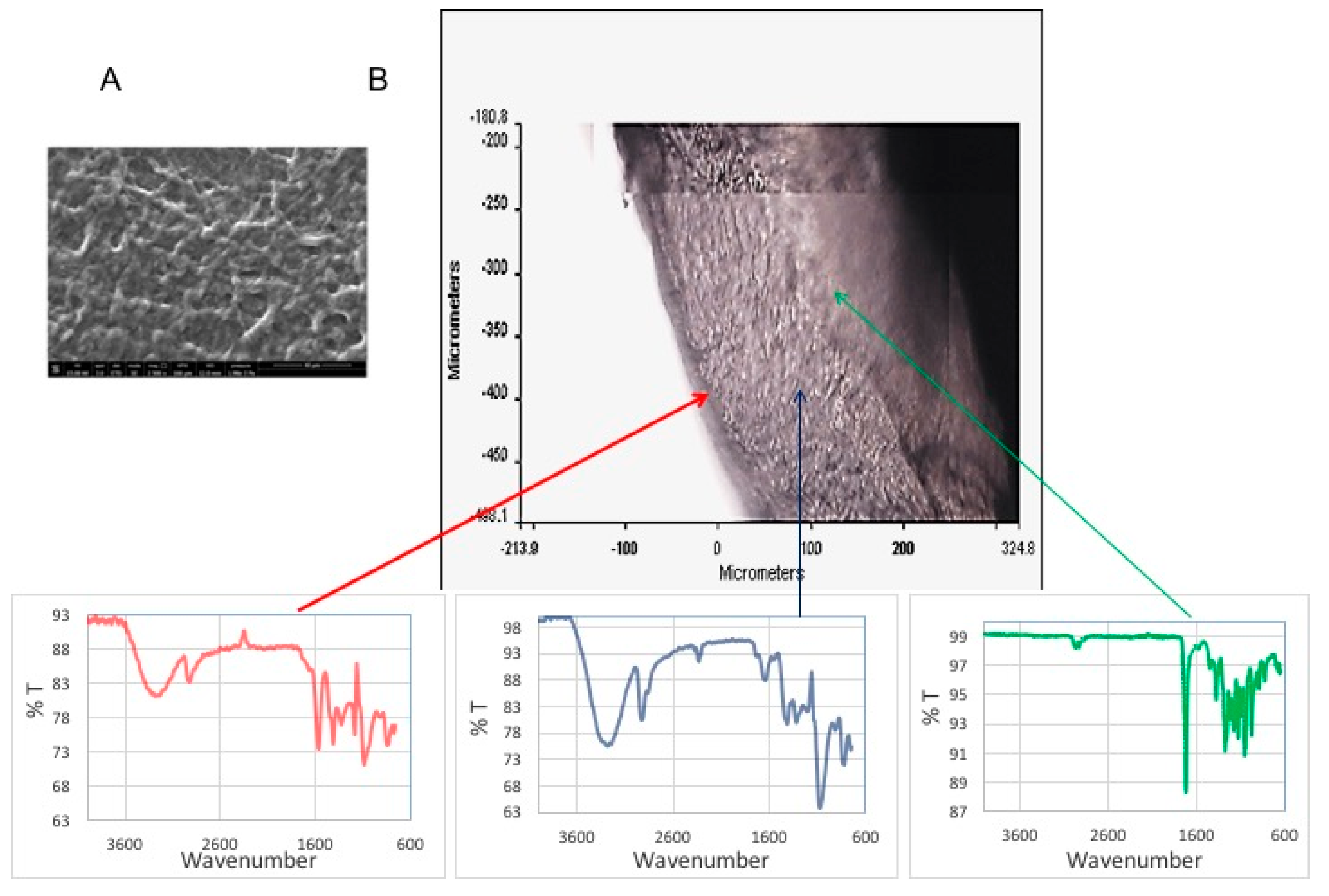
Scanning electron microscopy (SEM) was employed to analyze fibers and fibers–hydrogel coating, using a FEI Quanta™ 450 FEG instrument. SEM analysis was done at 15 kV in high-vacuum mode, with manual aperture and 2.5 beam spot size. The samples were sputtered with gold to improve the quality of the analyses.
The thermal behavior of the P(VAc-co-AA) and P(VA-co-AA) were studied by a differential scanning calorimeter (DSC 7; Perkin Elmer Inc., Waltham, MA, USA), using aluminum pans. Two consecutive scans were carried out on each sample, at scan rate of 10 and 20 °C/min respectively.
Thermogravimetric analysis (TGA-6, Perkin Elmer) was carried out on samples of ca. 10 mg, heated from 25 to 700 °C at a rate of 10 °C/min with a nitrogen purge and ceramic pans.
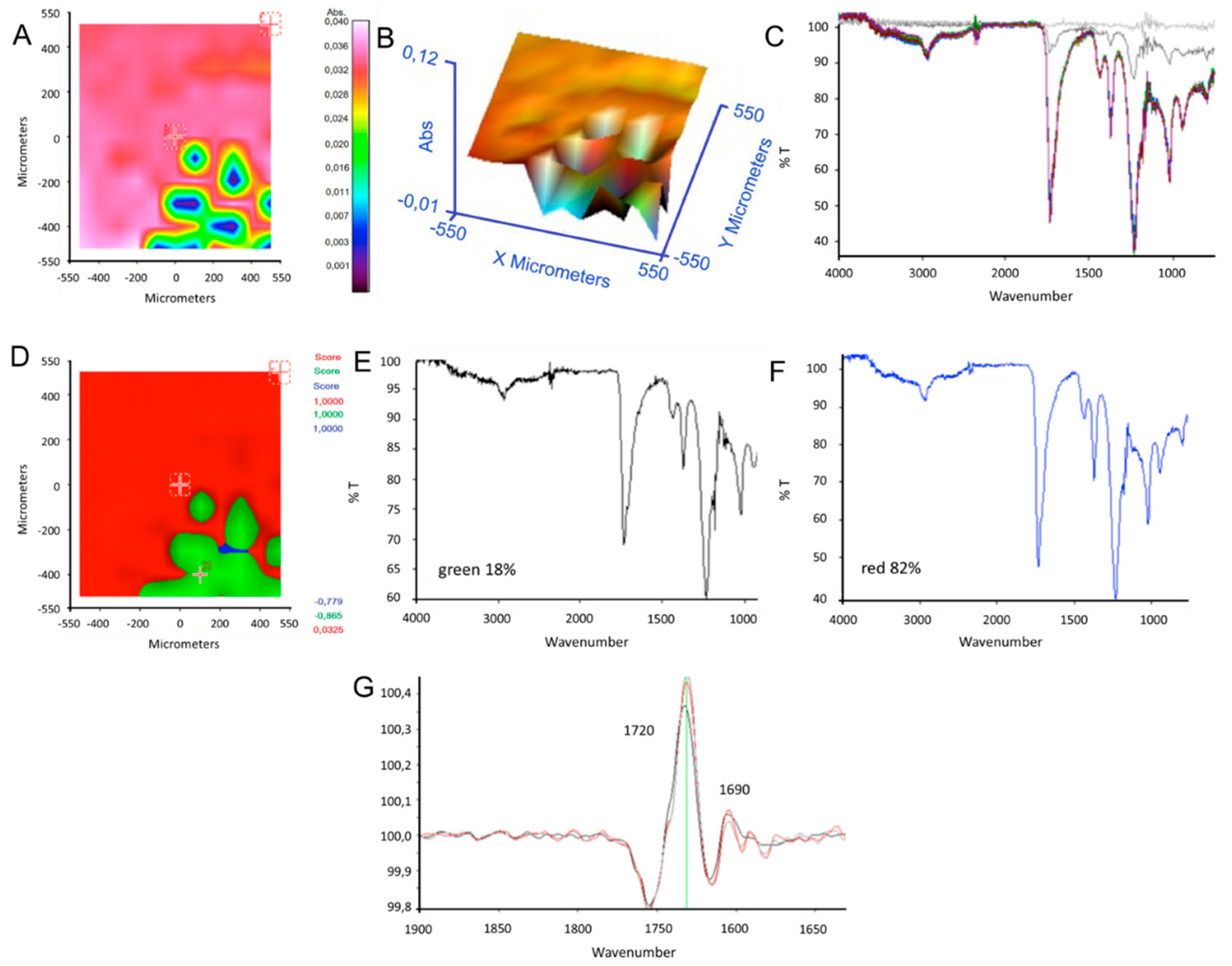
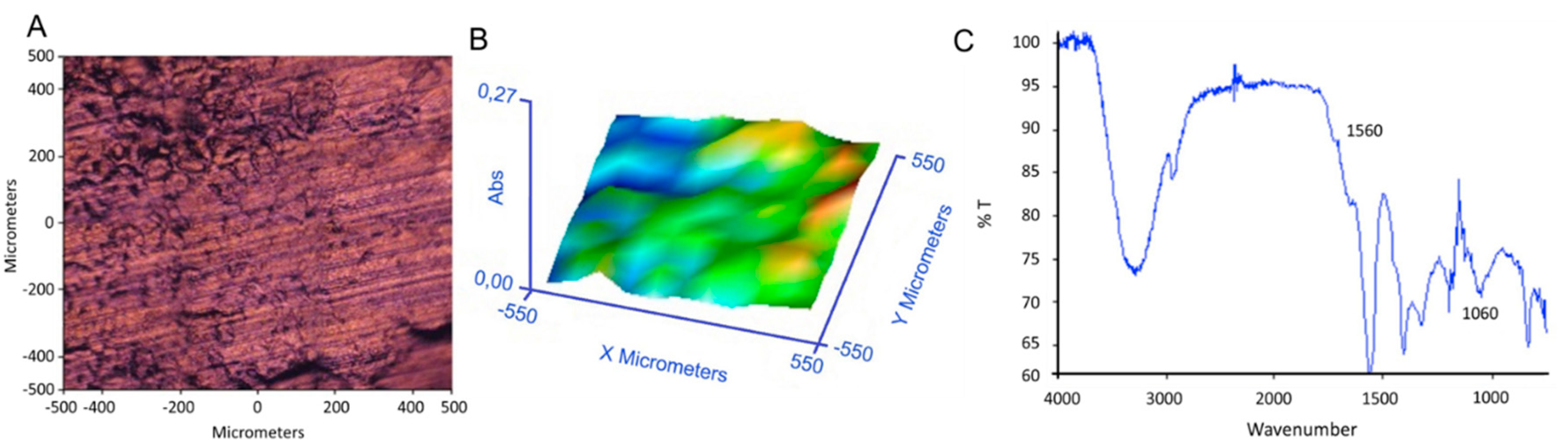
FTIR analysis in ATR mode (Spectrum 400, Perkin Elmer) was performed on each layer and on the complete system. Infrared spectra were acquired with a Perkin Elmer Spectrum One FT-IR Spectrometer, equipped with ATR objective lens with a penetration depth of less than 1 μm. All spectra were obtained in the middle range (4000–720 cm−1) with a resolution of 4 cm−1, representing an average of 16 scans. Spectral images were acquired in transmission and in μATR mode using the infrared imaging system Spotlight 300 (Perkin Elmer). The spectral resolution was 4 cm−1. The spatial resolution was 100 × 100 μm2 in μATR mode and 6.25 μm in transmission. Background scans were obtained from a region of no sample. IR images were acquired with a liquid nitrogen-cooled mercury cadmium telluride line detector composed of 16 pixel elements. Each absorbance spectrum composing the IR images, 16 scans, was recorded for each pixel in the μATR mode using the Spotlight software. Spectra were collected by touching the ATR objective on the sample and collecting the spectrum generated from the surface layer of the sample. The Spotlight software used for the acquisition was also used to preprocess the spectra. IR spectral images were produced by using the absorbance in a given frequency range, 4000–720 cm−1. Spectra contained in the spectral images were analyzed using a compare correlation image. The obtained correlation map indicates the areas of an image where the spectra are most similar to a reference spectrum.
DMA analysis was performed with a GABO Eplexor 150 N equipped with compression clamps. The test was conducted at room temperature with a frequency of 1 Hz. Dimension of the specimens used were 17.5 × 15 × 10 mm. PVA hydrogels were tested in two different conditions: static strain of 1%, dynamic strain of 0.5% (initial clamp distance = 10mm) and static strain of 5%, dynamic strain of 1% (initial clamp distance = 10 mm).
3.7. Biological Assay
3.7.1. Materials
CaCl2 l-glutamine and penicillin/streptomycin, EDTA, trypan blue, and resazurin were purchased from Sigma Aldrich (Milan, Italy). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS) and phosphate-buffered saline 1×, (PBS), Calcein AM were bought from Gibco by Life Technologies—Thermo Fisher Scientific (Waltham, MA, USA). Diflucan was supplied by Pfizer (New York, NY, USA) and Levoflaxcin (500 mg/100 mL) was bought from Fresenius Kabi (Verona, Italy). Collagenase I was obtained from Worthington Biochemical Corp. (Lakewood, NJ, USA).
3.7.2. Preparation of PHBHV fiber/PVA hydrogel (F/H) composite for cell culture experiments
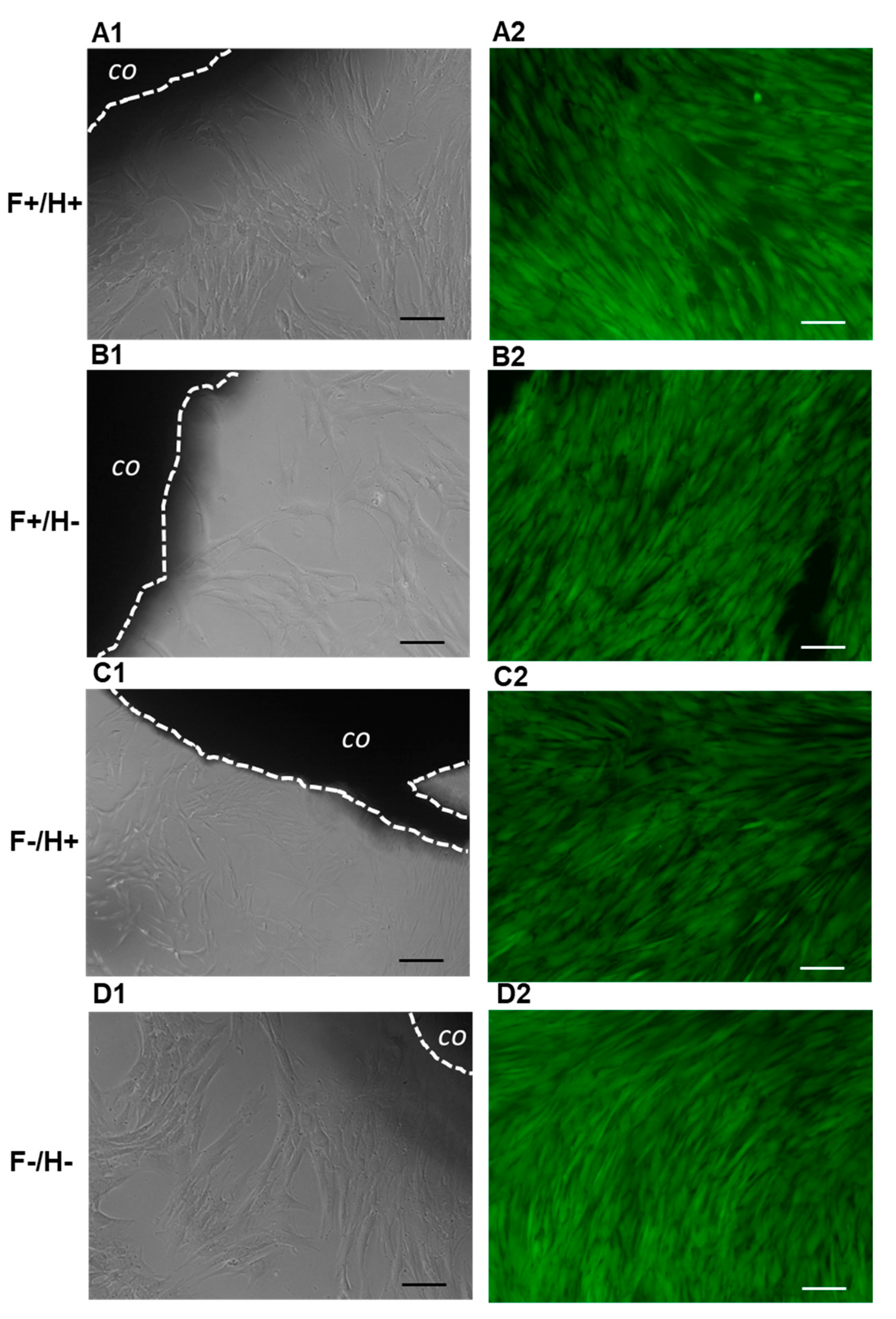
For cell culture experiments, the composite samples were prepared and handled in sterile conditions. PHBHV fibers, with and without Dexa samples, were cut into 3 cm diameter discs by using a circle stencil template and diamond tip pen, placed at the bottom of sterile 6-well culture plates, and exposed to UV radiation overnight to provide sterilization. The A 10% (w/v%) PVA solution in double-distilled water was prepared via autoclaving at 121 °C for 1 h (total volume 80 mL) and cooled under a biohood. At the same time, a 0.05% (w/v%) Dexa solution of D-MEM (Sigma Aldrich, Milan Italy) was prepared and sterile-filtered. D-MEM, either with (Dex+) or without Dexa (Dex-) was added to the PVA solution at 1:4 volume ratio and gently stirred, thus obtaining PVA(Dex+) and PVA(Dex−) solutions, respectively. Each solution was therefore added to each type of fiber samples at 5 mL/well, thus obtaining 4 types of composites, namely PHBHV(Dex-)/PVA(Dex−) (F−/H−), PHBHV(Dex−)/PVA(Dex+) (F−/H+), PHBHV(Dex+)/PVA(Dex−) (F+/H−), and PVA(Dex+)/PHBHV(Dex+) (F+/H+). The 6-well plates were sealed with parafilm and 8 freeze-thawing cycles were performed, consisting of an initial freezing at −20 °C overnight, followed by 7 cycles, each one consisting of thawing at room temperature (RT) for 1 h and freezing at −20 °C for 1 h. Afterwards, the samples were cut into cylinders using a sterile 5 mm diameter puncher.
3.7.3. Ethical Statement
Adult dermal fibroblasts (hDFs) were isolated from waste samples of normal skin derived from contralateral mastectomy to be discarded after surgery. The samples were treated anonymously and in conformity with the Declaration of Helsinki.
3.7.4. Cell Cultures
Skin specimens were washed with sterile PBS added with antibiotics, cut into 2–3 mm pieces, and washed again. Skin pieces were then treated over night with DMEM, CaCl2 5 mM, 0.25% Collagenase I. After digestion, collagenase was neutralized by adding EDTA and washing twice with DMEM mixed with 10% FBS. Cell suspension was cultured in complete medium and hDFs were used for all the experiments at 4–7 passages. Characterization of hDFs was performed by FACS analysis, checking the expression of fibroblastic markers, such as cadherin-11 and CD-90. Cell culture was carried out in a growth culture medium (CM), consisting of DMEM supplemented with 10% (v%) FBS, 2 mM L-glutamine, 100 IU/mL penicillin, and 100 mg/mL streptomycin. Once the 80% confluence was reached, hDFs were trypsinized and viable cells were counted with a hemocytometer, using trypan blue exclusion dye. All the cell culture experiments were performed in a humidified incubator at 37 °C and in 95%/5% air/CO2 environment.
3.7.5. Direct Cytotoxicity Tests
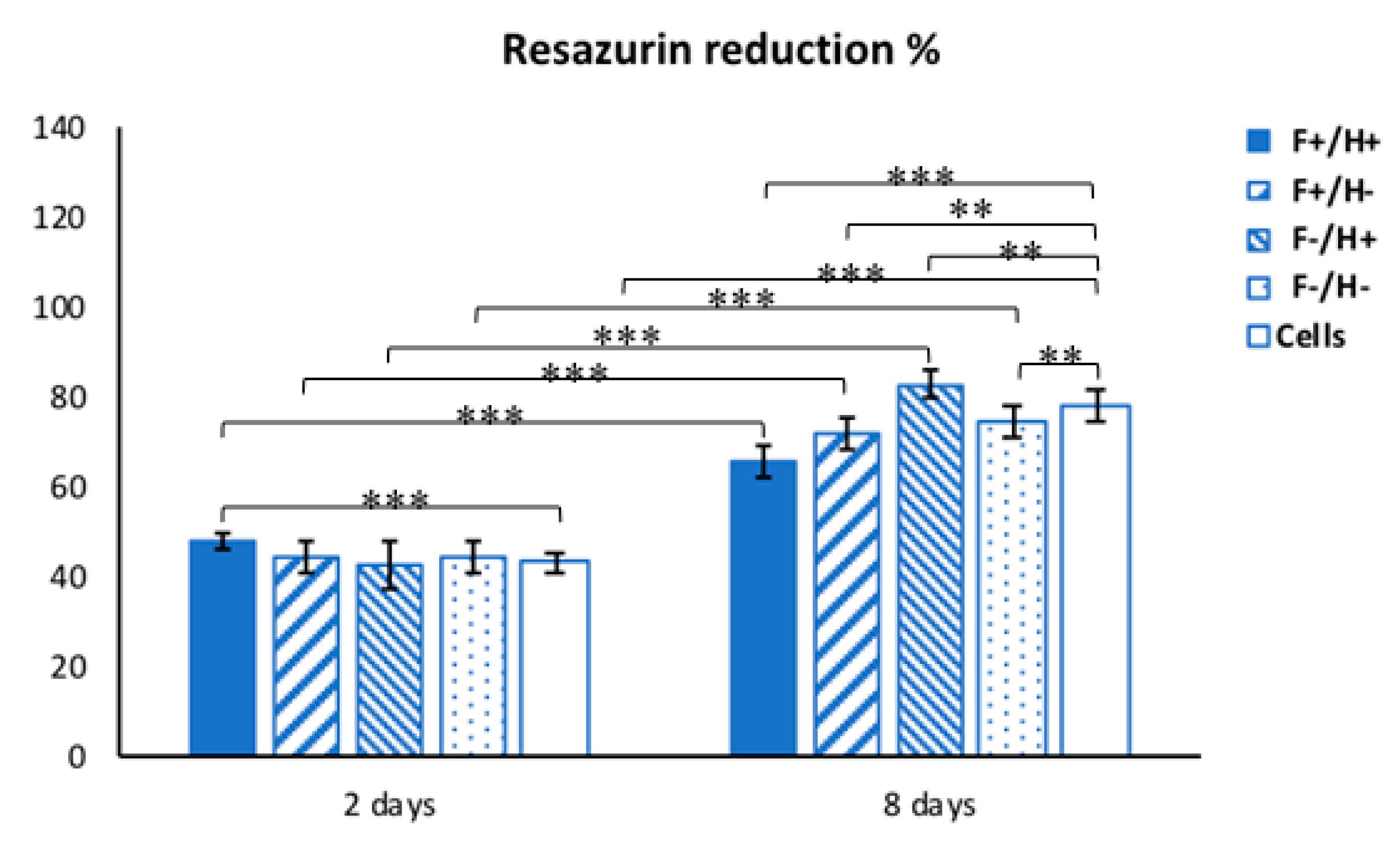
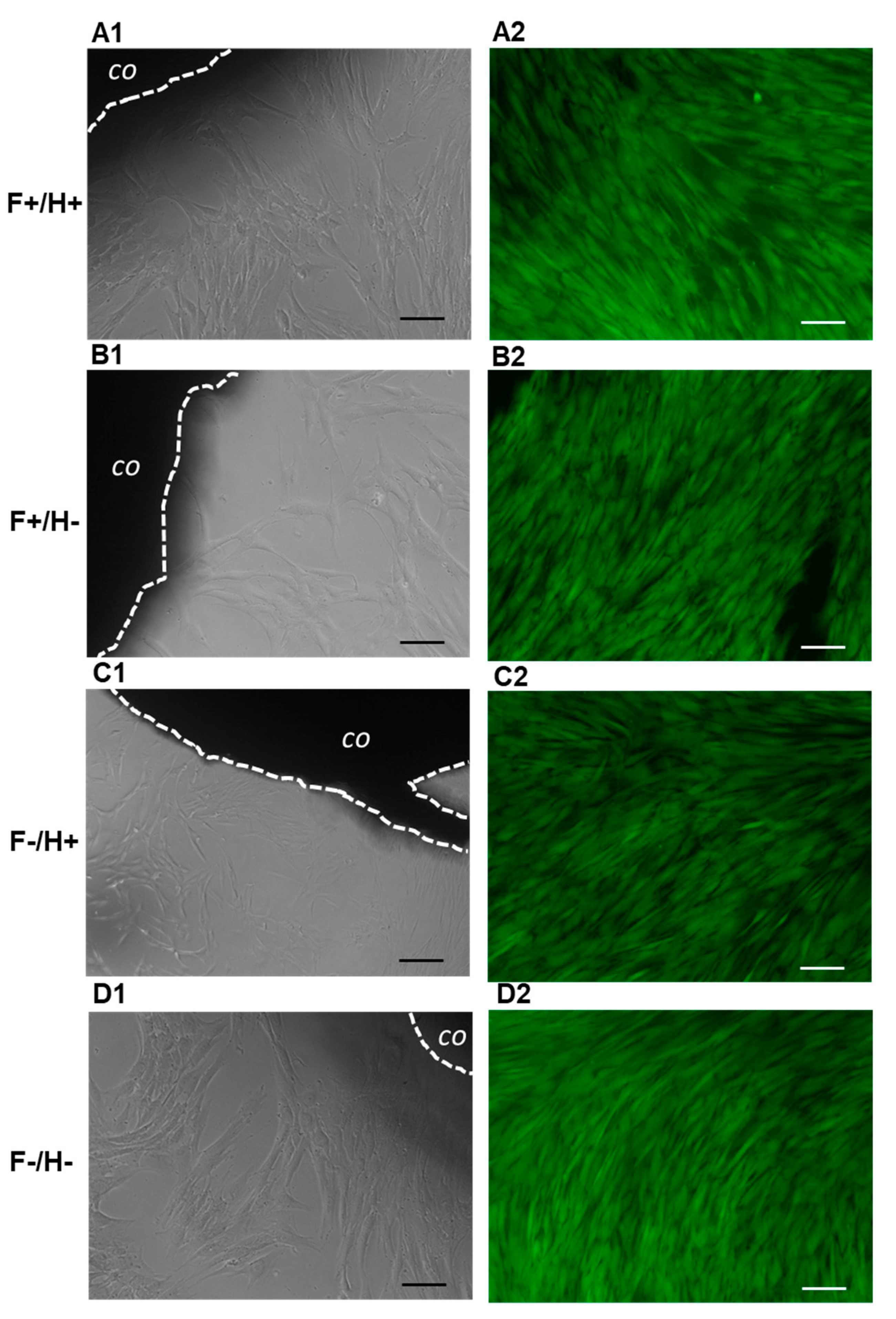
HDFs were seeded in 24-well plates at a density of 20,000 cells/well and 1 mL CM. The following day, F/H composites (5 mm diameter) of the 4 different types (n = 4) were placed in the wells containing hDFs, one sample per well, and cell culture was continued for 8 days by replacing the CM every 2–3 days. HDFs that were not exposed to the biomaterials were kept in the same culture conditions and used as controls. The samples were monitored daily until the endpoint by reverse microscopy observation and micrographs, which were acquired every 2–3 days to image cell morphology at the periphery of the F/H composites.
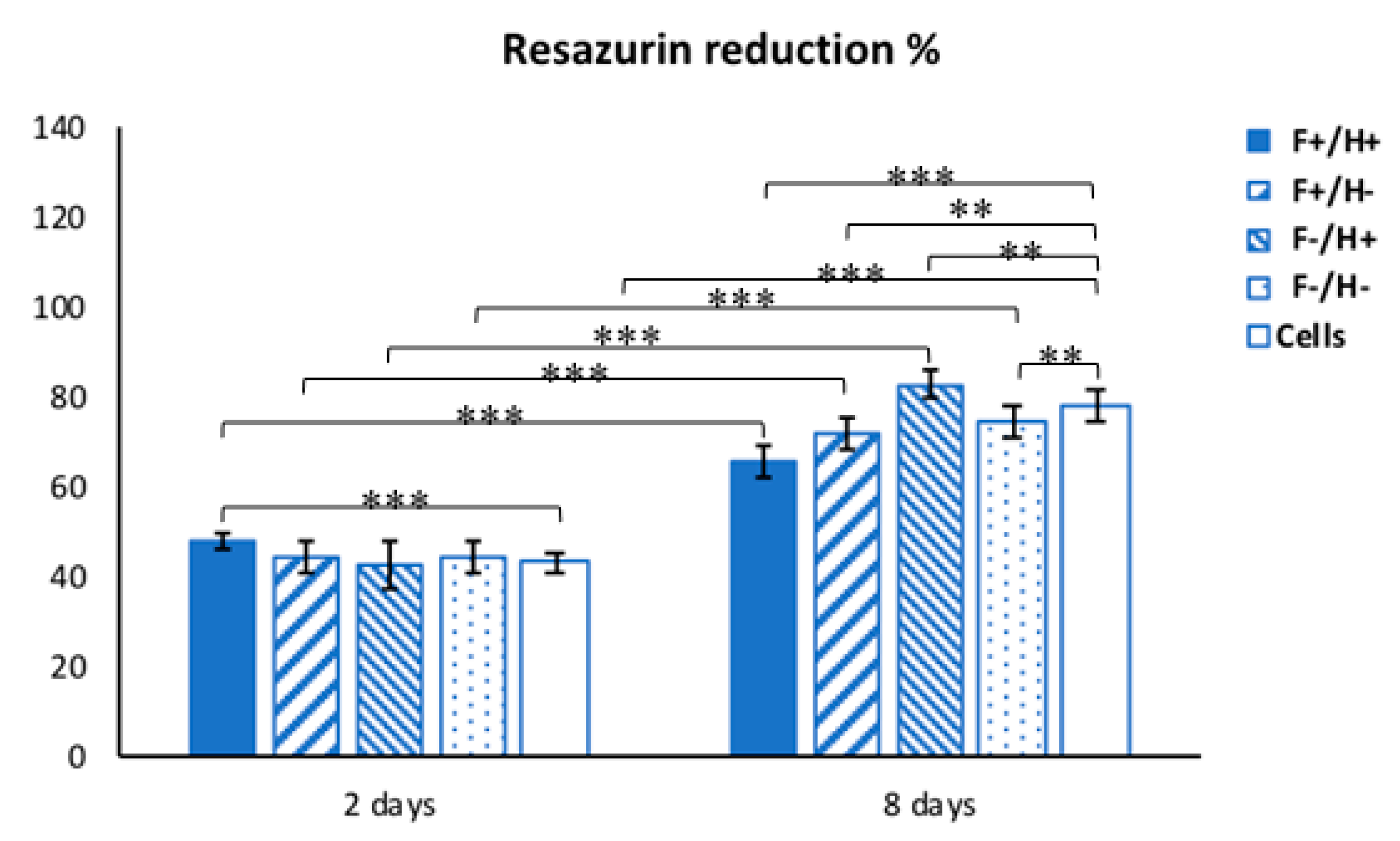
Along the culture time, resazurin dye assay, a nondisruptive metabolic activity test, was performed to check the viability of hDFs. Resazurin is a blue dye, which is irreversibly reduced to the pink colored fluorescent resorufin and used as a REDOX indicator to measure cell metabolic activity. A stock 5 mg/mL solution of resazurin in PBS was freshly prepared and sterile-filtered. At 2 time points, namely day 2 and day 8 after the hDFs were exposed to the composites, the CM was replaced with a 20 µL/mL resazurin working solution prepared in CM. Briefly, samples (
n = 3) and negative controls (
n = 3) were incubated for 3 h at 37 °C with the resazurin working solution. At each time point, 100 µL of supernatant from sample or control was loaded into 96-well plates; then, excess supernatant was removed from the cultures and replaced with fresh culture media. The absorbance (λ) of supernatants was measured with a spectrophotometer (Victor 3; PerkinElmer, Waltham, MA, USA) under double-wavelength reading (570 nm and 600 nm). Data were acquired and expressed as the percentage of reduced resazurin by correlating the absorbance values and the molar extinction coefficients of the dye at the selected wavelengths, as shown in the formula:
At the endpoint, the CM was removed and 1 mL of Calcein AM solution (1 µL/mL of in sterile PBS) was added to the wells and incubated in the dark for 20 min. Calcein AM is a cell-permeant fluorescent dye, which stains the cytoplasm of viable cells in green. Micrographs were acquired under fluorescence mode using a FITC fluorescein filter (Nikon Eclipse Ti, Nikon, Tokyo, Japan).
3.7.6. Indirect Cytotoxicity Tests on Primers
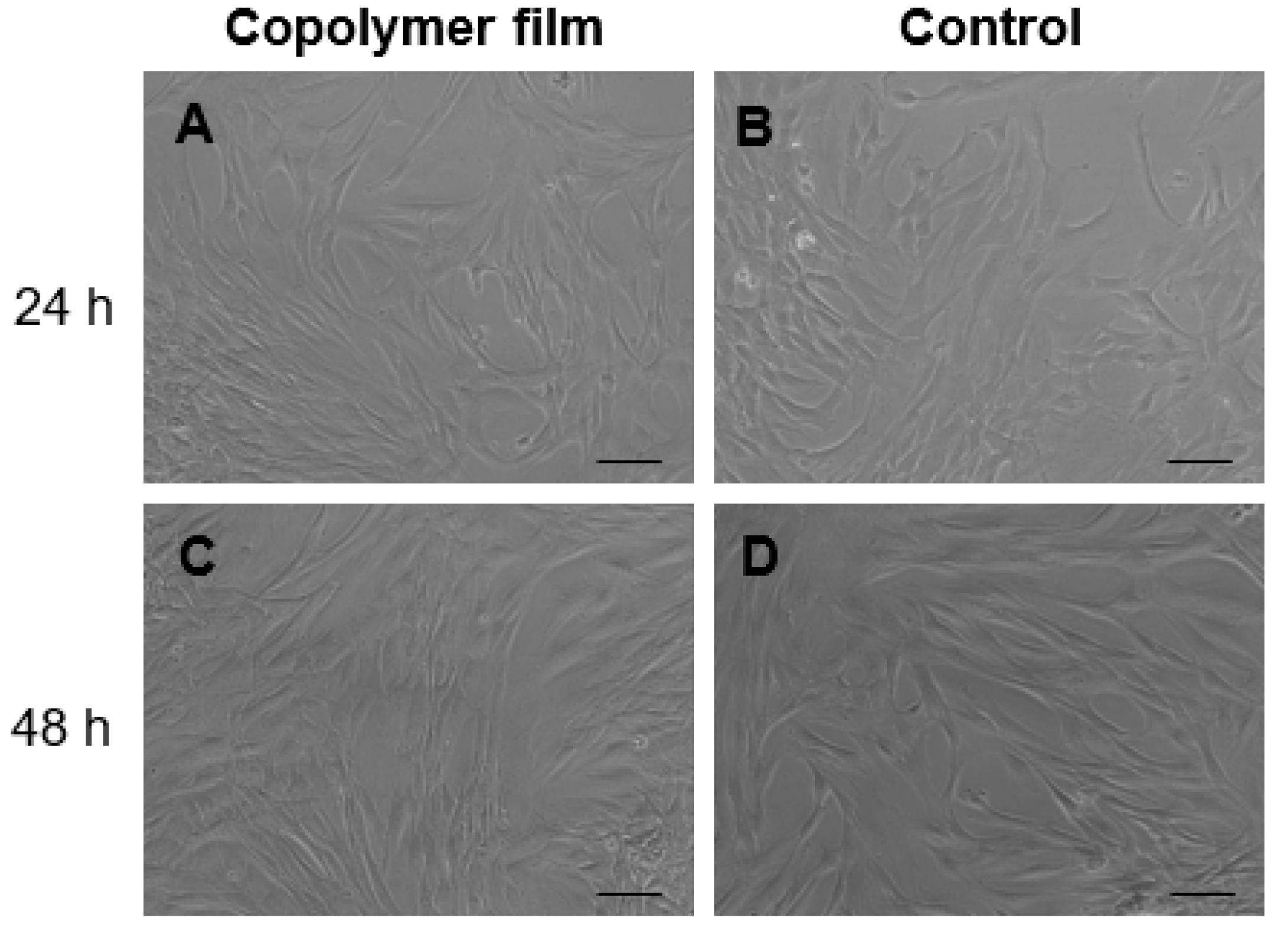
Films of synthesized copolymers P(VAc-co-AA) and P(VA-co-AA) were used to assess cytocompatibility with hDFs. Briefly, the material was soaked in complete CM for 24 h and sterile filtered. HDFs were seeded in 24-well plates at 2 × 104 cells/well and cultured with the conditioned CM and with regular CM as control. Microscopy observations and resazurin assay were performed as described above.
3.7.7. Statistical Analysis
Statistical analyses were carried out by SPSS (SPSS v.16.0; IBM) to specify the significant level of different processing parameters. All the data were analyzed using one-way analysis of variance and Duncan post hoc test for multiple comparisons. Probability (p) values <0.05 were considered as statistically significant.
3.8. Functional Analysis
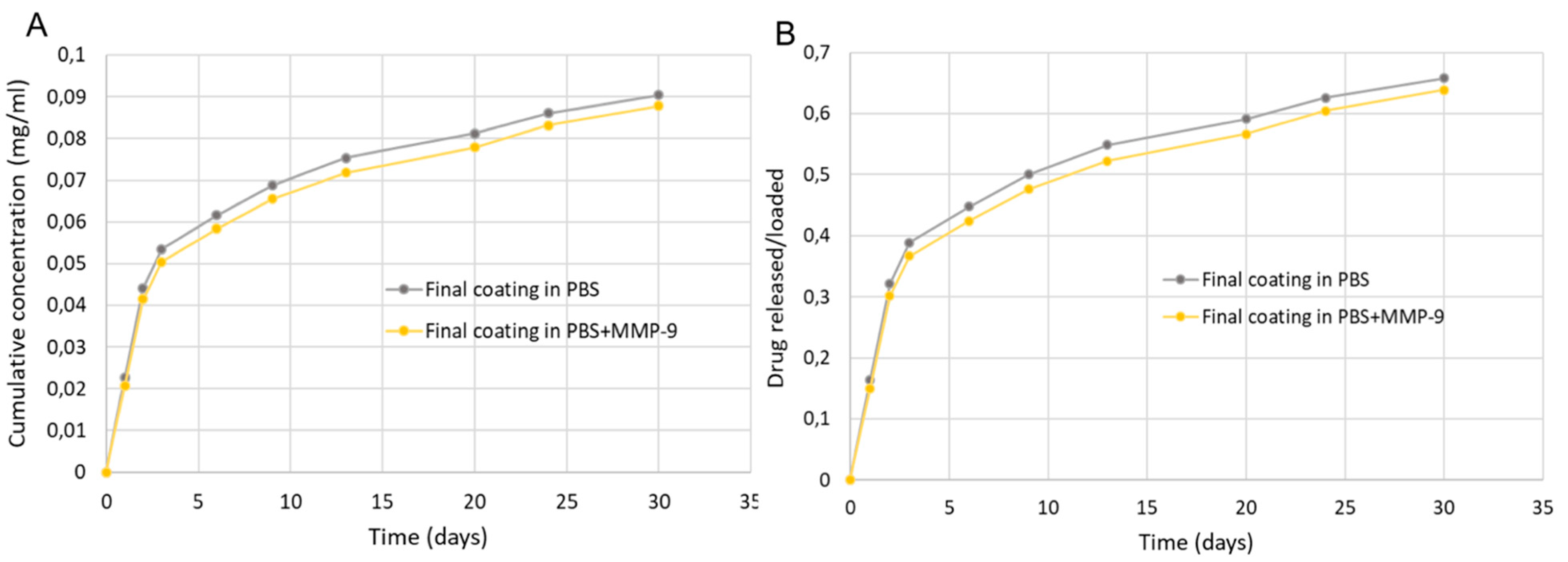
Release tests were performed using high-performance liquid chromatography (HPLC, Perkin Elmer Series 200). The systems were placed in tubes containing PBS at 37 °C, under constant stirring. For final coating the release test was carried out also in PBS, with the addition of MMP-9 solution (0.05 μg/mL). At predefined time intervals, the supernatant was collected and analyzed to evaluate the amount of Dexa released. For HPLC analysis the supernatants were placed into 1.5 mL HPLC vials. A 50 µL aliquot of each sample was then injected into the HPLC system. For HPLC analysis, a C18 Phenomenex column, acetonitrile/MilliQ water 70/30 as the mobile phase, UV detector set at 254 nm wavelength, and 1 mL/min eluent flow rate were used.
3.9. Adhesion Evaluation
The adhesion of the composite system to metallic device substrates is a fundamental requirement for the success of the implant. The adhesion test was carried out by immersing the samples (composite coatings on Ti platelets, in triplicate), in wells containing two different solutions: PBS at pH 7.4 and PBS with addition of MMP-9 solution (0.05 μg/mL). The addition of MMP-9 was set in order to simulate a pathological condition, considering that MMP-9 is one of the most frequent proteases associated with the inflammation process in fibrous tissue interfaces [
17]. Samples were maintained in a thermostatic bath at 37 °C under continuous stirring. The samples were checked daily over the whole period of analysis (70 days).
4. Conclusions
The realization of a suitable polymeric coating for an implantable device still represents one of main challenges for the success of a permanent implant. Two copolymers, P(Vac-co-AA) and P(VA-co-AA), were synthetized and characterized with the aim to act as a primer, a thin layer at the interface between the metallic surface of device and external coating, to improve the adhesion to device surface. The external layer of PVA hydrogel was produced as a coating of metallic substrate for soft tissue implant. PVA hydrogel was shown to be biocompatible, biostable, and mechanically compatible with soft tissues and able to incorporate and release Dexa. The third component is based on PHBHV; the biopolymer was processed to obtain electrospun fibers showing uniform dimension, smooth surfaces, and capability to incorporate the drug in a homogeneous way. The composite coating showed a good adhesion to titanium substrate, no in vitro cytotoxicity, and a prolonged and controlled drug release. The multifunctional composite coating offers the potential for a long-term interface, able to modulate the release of an anti-inflammatory drugs, thus contrasting acute and chronic inflammation responses following device implantation.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



