Genome-Wide Identification and Characterization of the Vacuolar H+-ATPase Subunit H Gene Family in Crop Plants

 ,
, 
Abstract
:
1. Introduction
2. Results
2.1. Identification of VHA-H Gene Family Members in the Main Crops
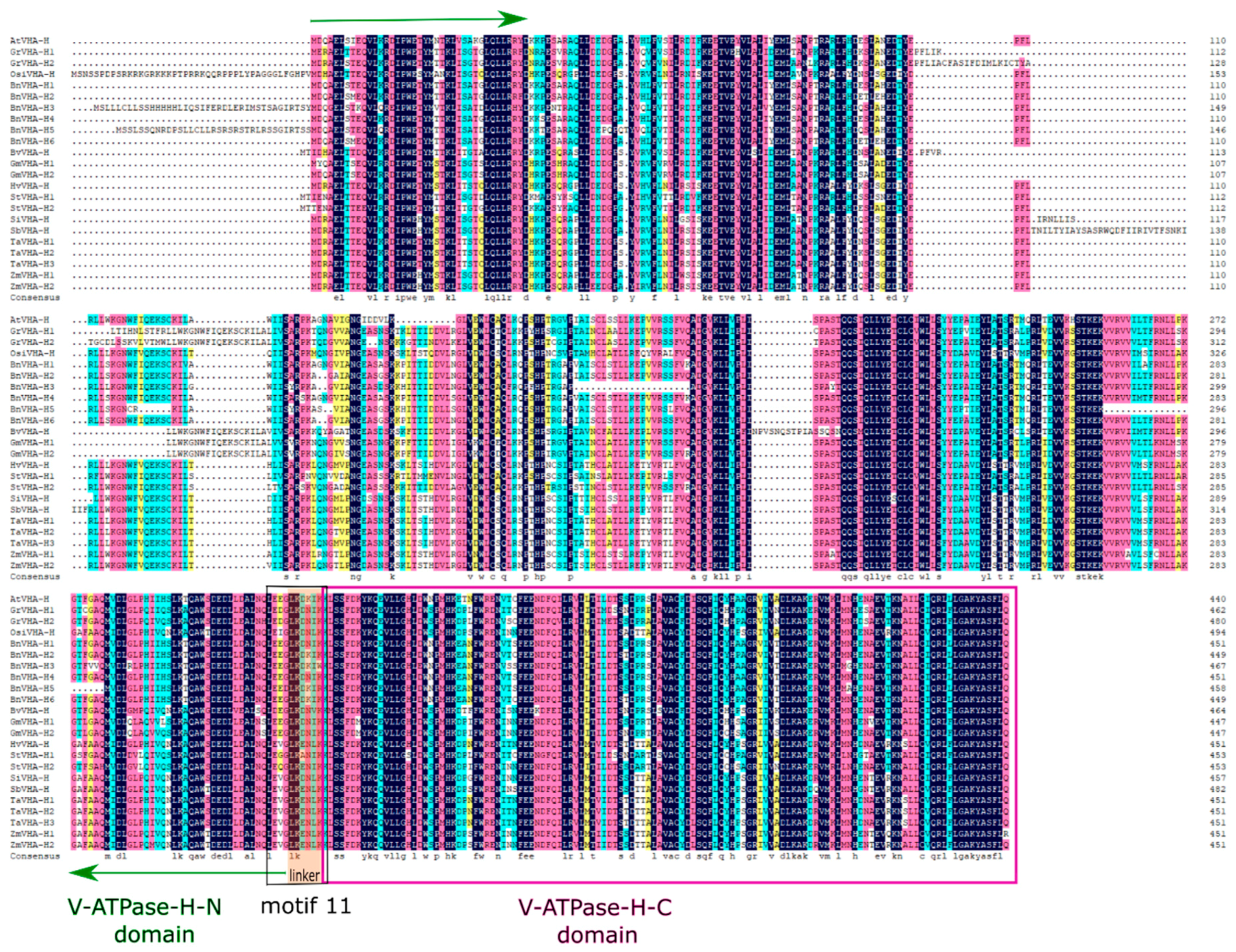
2.2. Protein Sequence Alignments and Phylogenetic and Motif Analyses
2.3. Structures of the VHA-H Genes
2.4. Splice Variants of VHA-H Genes
2.5. Analysis of Cis-acting Elements of VHA-H Promoters
2.6. Tissue-Specific Expression Patterns of VHA-H Genes
3. Discussion
3.1. Identification of V-ATPase Subunit H Genes in the Main Crops
3.2. Protein Sequence Alignments and Phylogenetic and Motif Analyses
3.3. Gene Structural Diversity
3.4. Splice Variants
3.5. Cis-Acting Elements of the VHA-H Promoters
3.6. Tissue-Specific Expression Patterns of VHA-H Genes
4. Materials and Methods
4.1. Identification of VHA-H Genes
4.2. Phylogenetic and Protein Motif Analyses
4.3. Gene Structure of VHA-H Genes
4.4. Splice Variants of VHA-H Genes
4.5. Analysis of cis-acting Elements of VHA-H Promoters
4.6. Tissue-Specific Expression Patterns of VHA-H Genes
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| V-ATPase | vacuolar H+-ATPase |
| VHA-H | V-ATPase subunit H |
| UTRs | Untranslated regions |
| ABA | Abscisic acid |
| OPR | 12-oxo-phytodienoic acid reductase |
| ORF | open reading frame |
| aa | amino acid |
| GA | gibberellin |
| SA | salicylic acid |
| MeJA | methyl jasmonate |
References
- Eisenach, C.; Francisco, R.; Martinoia, E. Plant vacuoles. Curr. Biol. 2015, 25, R136–R137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinoia, E.; Meyer, S.; De Angeli, A.; Nagy, R. Vacuolar transporters in their physiological context. Annu. Rev. Plant Biol. 2012, 63, 183–213. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.; Beyhl, D.; Gorlich, E.; Al-Rasheid, K.A.S.; Marten, I.; Stierhof, Y.D.; Hedrich, R.; Schumacher, K. Arabidopsis v-atpase activity at the tonoplast is required for efficient nutrient storage but not for sodium accumulation. Proc. Natl. Acad. Sci. USA 2010, 107, 3251–3256. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, H.; Brown, D.S.; Ehrenfeld, J.; Harvey, W. Animal plasma membrane energization by proton-motive v-atpases. BioEssays 2015, 21, 637–648. [Google Scholar] [CrossRef]
- Nishi, T.; Forgac, M. The vacuolar (H+)-ATPases-nature’s most versatile proton pumps. Nat. Rev. Mol. Cell Biol. 2002, 3, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Maxson, M.E.; Grinstein, S. The vacuolar-type H+-ATPase at a glance - more than a proton pump. J. Cell Sci. 2014, 127, 2367–2378. [Google Scholar] [CrossRef]
- Schumacher, K. Ph in the plant endomembrane system—An import and export business. Curr. Opin. Plant Biol. 2014, 22, 71–76. [Google Scholar] [CrossRef]
- Lüttge, U.; Ratajczak, R.; Rausch, T.; Rockel, B. Stress responses of tonoplast proteins: An example for molecular ecophysiology and the search for eco-enzymes. Acta Bot. Neerl. 1995, 44, 343–362. [Google Scholar] [CrossRef]
- He, H.L.; Huang, X.; Shen, Y.Z.; Huang, Z.J. Wheat V-H+-ATPase subunit genes significantly affect salt tolerance in Arabidopsis thaliana. PLoS ONE 2014, 9, e86982. [Google Scholar] [CrossRef]
- Shi, C.Y.; Hussain, S.B.; Guo, L.X.; Yang, H.; Ning, D.Y.; Liu, Y.Z. Genome-wide identification and transcript analysis of vacuolar-ATPase genes in citrus reveal their possible involvement in citrate accumulation. Phytochemistry 2018, 155, 147–154. [Google Scholar] [CrossRef]
- Nakanishi-Matsui, M.; Sekiya, M.; Nakamoto, R.K.; Futai, M. The mechanism of rotating proton pumping ATPases. Biochim. Et Biophys. Acta (BBa)/Bioenerg. 2010, 1797, 1343–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, K.; Krebs, M. The V-ATPase: Small cargo, large effects. Curr. Opin. Plant Biol. 2010, 13, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, R. Structure, function and regulation of the plant vacuolar H+-translocating ATPase. Biochim. Et Biophys. Acta 2000, 1465, 17–36. [Google Scholar] [CrossRef]
- Zhu, J.; Zhu, X.J.; Ying, S.H.; Feng, M.G. Effect of vacuolar ATPase subunit H (Vmah) on cellular pH, asexual cycle, stress tolerance and virulence in Beauveria bassiana. Fungal Genet. Biol. 2017, 1465, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, H.; Grber, G.; Harvey, W.R.; Huss, M.; Merzendorfer, H.; Zeiske, W. Structure and regulation of insect plasma membrane H(+)V-ATPase. J. Exp. Biol. 2000, 203, 127–135. [Google Scholar] [PubMed]
- Zhao, W.; Zhang, Y.; Yang, S.; Hao, Y.; Wang, Z.; Duan, X. Analysis of two transcript isoforms of vacuolar ATPase subunit H in mouse and zebrafish. Gene 2017, 638, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Jaquinod, M.; Villiers, F.; Kieffer-Jaquinod, S.; Hugouvieux, V.; Bruley, C.; Garin, J.; Bourguignon, J. A proteomics dissection of Arabidopsis thaliana vacuoles isolated from cell culture. Mol. Cell. Proteom. 2006, 6, 394–412. [Google Scholar] [CrossRef]
- Carter, C.; Pan, S.; Zouhar, J.; Avila, E.L.; Girke, T.; Raikhel, N.V. The vegetative vacuole proteome of Arabidopsis thaliana reveals predicted and unexpected proteins. Plant Cell 2004, 16, 3285–3303. [Google Scholar] [CrossRef]
- Kane, P.M.; Smardon, A.M. Assembly and Regulation of the Yeast Vacuolar H+-ATPase. J. Bioenerg. Biomembr. 2003, 35, 313–321. [Google Scholar] [CrossRef]
- Sambade, M.; Kane, P.M. The yeast vacuolar proton-translocating ATPase contains a subunit homologous to the manduca sexta and bovine e subunits that is essential for function. J. Biol. Chem. 2004, 279, 17361–17365. [Google Scholar] [CrossRef]
- Cipriano, D.J.; Wang, Y.; Bond, S.; Hinton, A.; Jefferies, K.C.; Qi, J.; Forgac, M. Structure and regulation of the vacuolar ATPases. Biochim. Et Biophys. Acta (BBA)/Bioenerg. 2008, 1777, 599–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, M.N.; Hirata, R.; Umemoto, N.; Ohya, Y.; Anraku, Y. VIMA13 encodes a 54-kda vacuolar H-ATPase subunit required for activity but not assembly of the enzyme complex in Saccharomyces cerevisiae. J. Biol. Chem. 1993, 268, 18286–18292. [Google Scholar] [PubMed]
- Xie, X.S.; Crider, B.P.; Ma, Y.M.; Stone, D.K. Role of a 50–57-kda polypeptide heterodimer in the function of the clathrin-coated vesicle proton pump. J. Biol. Chem. 1994, 269, 25809–25815. [Google Scholar] [PubMed]
- Sagermann, M.; Stevens, T.H.; Matthews, B.W. Crystal structure of the regulatory subunit H of the v-type ATPase of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2001, 98, 7134–7139. [Google Scholar] [CrossRef]
- Liu, M.; Tarsio, M.; Charsky, C.M.; Kane, P.M. Structural and functional separation of the N- and C-terminal domains of the yeast V-ATPase subunit H. J. Biol. Chem. 2005, 280, 36978–36985. [Google Scholar] [CrossRef]
- Flannery, A.R.; Stevens, T.H. Functional characterization of the n-terminal domain of subunit H (vma13p) of the yeast vacuolar ATPase. J. Biol. Chem. 2008, 283, 29099–29108. [Google Scholar] [CrossRef]
- Wang, F.W.; Wang, C.; Sun, Y.; Wang, N.; Li, X.W.; Dong, Y.Y.; Yao, N.; Liu, X.M.; Chen, H.; Chen, X.F.; et al. Overexpression of vacuolar proton pump ATPase (V-H+-ATPase) subunits B, C and H confers tolerance to salt and saline-alkali stresses in transgenic alfalfa (Medicago sativa L.). J. Integr. Agric. 2016, 15, 2279–2289. [Google Scholar] [CrossRef]
- Zhang, Q.; Maroof, M.A.; Lu, T.Y.; Shen, B.Z. Genetic diversity and differentiation of indica and japonica rice detected by RFLP analysis. Theor. Appl. Genet. 1992, 83, 495–499. [Google Scholar] [CrossRef]
- Gogarten, J.P. Evolution and isoforms of V-ATPase subunits. J. Exp. Biol. 1992, 172, 137–147. [Google Scholar]
- Nelson, N.; Perzov, N.; Cohen, A.; Hagai, K.; Padler, V.; Nelson, H. The cellular biology of proton-motive force generation by V-ATPases. J. Exp. Biol. 2000, 203, 89–95. [Google Scholar]
- Lu, L.; Qi, Z.; Wu, W. Cloning, expression and purification of subunit H of vacuolar H+-ATPase from Mythimna separata Walker (Lepidoptera: Noctuidae). Int. J. Mol. Sci. 2014, 15, 15443–15455. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Y.; Wang, X.; Li, R.; Li, W.Q.; Chen, K.M. Genome-wide analysis of the NADK gene family in plants. PLoS ONE 2014, 9, e101051. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, B.; Yu, L.; Feng, D.; Wang, H.; Wang, J. Phylogenetic analysis, structural evolution and functional divergence of the 12-oxo-phytodienoate acid reductase gene family in plants. BMC Evol. Biol. 2009, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Yoshihama, M.; Kenmochi, N. Phase distribution of spliceosomal introns: Implications for intron origin. BMC Evol. Biol. 2006, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, E.M.; Casano, L.M.; Barreno, E. Evolutionary implications of intron-exon distribution and the properties and sequences of the RPL10A gene in eukaryotes. Mol. Phylogenetics Evol. 2013, 66, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Rogozin, I.B.; Carmel, L.; Csuros, M.; Koonin, E.V. Origin and evolution of spliceosomal introns. Biol. Direct 2012, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Roos, A.D.D. Conserved intron positions in ancient protein modules. Biol. Direct 2007, 2, 7. [Google Scholar] [CrossRef]
- Min, X.J.; Powell, B.; Braessler, J.; Meinken, J.; Yu, F.; Sablok, G. Genome-wide cataloging and analysis of alternatively spliced genes in cereal crops. BMC Genom. 2015, 16, 721. [Google Scholar] [CrossRef]
- Ding, F.; Cui, P.; Wang, Z.; Zhang, S.; Ali, S.; Xiang, L. Genome-wide analysis of alternative splicing of pre-mRNA under salt stress in Arabidopsis. BMC Genom. 2014, 15, 431. [Google Scholar] [CrossRef]
- Reddy, A.S.N.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef]
- Staiger, D.; Brown, J.W.S. Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell 2013, 25, 3640–3656. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, A.M.; Marone, D.; Laidò, G.; De Leonardis, A.M.; De Vita, P. Alternative splicing: Enhancing ability to cope with stress via transcriptome plasticity. Plant Sci. 2012, 185, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Aw, V.D.V.; Thomas, A.A. The role of the 5′ untranslated region of an mRNA in translation regulation during development. Int. J. Biochem. Cell Biol. 1999, 31, 87. [Google Scholar]
- Bashirullah, A.; Cooperstock, R.L.; Lipshitz, H.D. Spatial and temporal control of RNA stability. Proc. Natl. Acad. Sci. USA 2001, 98, 7025–7028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poursani, E.M.; Mehravar, M.; Shahryari, A.; Mowla, S.J.; Soltani, B.M. Alternative splicing generates different 5′ UTRs in OCT4B variants. Avicenna J. Med. Biotechnol. 2017, 9, 201–204. [Google Scholar] [PubMed]
- Fetherson, R.A.; Strock, S.B.; White, K.N.; Vaughn, J.C. Alternative pre-mRNA splicing in Drosophila spliceosomal assembly factor RNP-4F during development. Gene 2006, 371, 234–245. [Google Scholar] [CrossRef]
- Thiele, A.; Nagamine, Y.; Hauschildt, S.; Clevers, H. AU-rich elements and alternative splicing in the β-catenin 3′ UTR can influence the human β-catenin mRNA stability. Exp. Cell Res. 2006, 312, 2367–2378. [Google Scholar] [CrossRef]
- Kaur, S.; Gupta, A.K.; Kaur, N. Gibberellin A3 reverses the effect of salt stress in chickpea (Cicer arietinum L.) seedlings by enhancing amylase activity and mobilization of starch in cotyledons. Plant Growth Regul. 1998, 26, 85–90. [Google Scholar]
- Shi, Z.; Ji, G.; Jing, S. Mitigative effects of salicylic acid and aspirin on salt stress induced injuries in wheat (Triticum aestivum L.). Acta Phytophysiol. Sin. 1999, 25, 159–164. [Google Scholar]
- Battal, P.; Erez, M.E.; Turker, M.; Berber, I. Molecular and physiological changes in maize (Zea mays) induced by exogenous NAA, ABA and MeJa during cold stress. Ann. Bot. Fenn. 2008, 45, 173–185. [Google Scholar] [CrossRef]
- Nakashima, K.; Yamaguchi-Shinozaki, K. ABA signaling in stress-response and seed development. Plant Cell Rep. 2013, 32, 959–970. [Google Scholar] [CrossRef]
- Strizhov, N.; Abrahám, E.; Okrész, L.; Blickling, S.; Zilberstein, A.; Schell, J.; Koncz, C.; Szabados, L. Differential expression of two P5CS genes controlling proline accumulation during salt-stress requires ABA and is regulated by ABA1, ABI1 and AXR2 in Arabidopsis. Plant J. 2010, 12, 557–569. [Google Scholar]
- Jia, W. Salt-stress-induced ABA accumulation is more sensitively triggered in roots than in shoots. J. Exp. Bot. 2002, 53, 2201–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, I.S.; Momose, Y.; Yamamoto, A.; Kim, D.W.; Usui, K. Inhibition of catalase activity by oxidative stress and its relationship to salicylic acid accumulation in plants. Plant Growth Regul. 2003, 39, 285–292. [Google Scholar] [CrossRef]
- Hall, T. BioEdit: Biological sequence alignment editorfor Win95/98/NT/2K/XP. Nucleic Acids Symp. Series 1999, 41, 95–98. [Google Scholar]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; Salazar, G.A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2015, 44, D279–D285. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Guo, A.Y.; Zhu, H.Q.; Chen, X.; Luo, J.C. GSDS: A gene structure display server. Hereditas 2007, 29, 1023–1026. [Google Scholar] [CrossRef]
- Kaur, S.; Dhugga, K.S.; Beech, R.; Singh, J. Genome-wide analysis of the cellulose synthase-like (Csl) gene family in bread wheat (Triticum aestivum L.). BMC Plant Biol. 2017, 17, 193. [Google Scholar] [CrossRef]
- Sammeth, M.; Foissac, S.; Guigó, R. A general definition and nomenclature for alternative splicing events. PLoS Comput. Biol. 2008, 4, e1000147. [Google Scholar] [CrossRef]
- Lescot, M. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zeng, D.E.; Liao, J.; Cheng, C.; Sahito, Z.A.; Xiang, M.; Fu, M.; Chen, Y.; Wang, D. Genome-wide analysis of auxin receptor family genes in Brassica juncea var. tumida. Genes 2019, 10, 165. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Species | Ensembl ID | Chromosome: Locations | ORF Length (bp) | No. of Exon | Deduced Polypeptide | ||
|---|---|---|---|---|---|---|---|---|
| Length (aa) | MW (Da) | PI | ||||||
| AtVHA-H | Arabidopsis thaliana | AT3G42050 | 3: 14,229,669−14,233,441 | 1326 | 11 | 441 | 50284.42 | 6.58 |
| GrVHA-H1 | Gossypium raimondii | B456_003G056300 | 3: 9,065,281−9,069,803 | 1392 | 11 | 463 | 52652.25 | 7.55 |
| GrVHA-H2 | B456_004G256400 | 4: 59,286,789−59,291,565 | 1446 | 12 | 481 | 54453.38 | 6.48 | |
| OsiVHA-H | Oryza sativa Indica Group | BGIOSGA025913 | 7: 19,885,495−19,891,284 | 1488 | 12 | 495 | 56165.18 | 9.07 |
| BnVHA-H1 | Brassica napus | BnaA01g02950D | A1: 1,436,042−1,439,972 | 1359 | 11 | 452 | 51168.20 | 6.40 |
| BnVHA-H2 | BnaA03g50930D | A3: 26,436,023−26,439,323 | 1353 | 11 | 450 | 51179.39 | 6.25 | |
| BnVHA-H3 | BnaA08g11340D | A8: 10,300,929−10,304,240 | 1407 | 13 | 468 | 54010.52 | 6.36 | |
| BnVHA-H4 | BnaC01g04210D | C1: 2,192,503−2,196,646 | 1359 | 11 | 452 | 51151.19 | 6.58 | |
| BnVHA-H5 | BnaC03g77060D | C3: 5,609,855−5,617,966 | 1380 | 14 | 459 | 52350.47 | 7.13 | |
| BnVHA-H6 | BnaC07g44770D | C7: 43,161,725−43,165,037 | 1353 | 11 | 450 | 51191.44 | 6.25 | |
| BvVHA-H | Beta vulgaris | BVRB_4g074640 | 4: 3,654,561−3,661,113 | 1398 | 11 | 465 | 52980.14 | 7.07 |
| GmVHA-H1 | Glycine max | GLYMA_02G059800 | 2: 5,381,068−5,387,111 | 1347 | 11 | 448 | 51058.20 | 6.53 |
| GmVHA-H2 | GLYMA_16G142600 | 16: 30,131,160−30,136,817 | 1347 | 11 | 448 | 51004.12 | 6.48 | |
| HvVHA-H | Hordeum vulgare | HORVU2Hr1G042700 | 2H: 214,952,610−214,958,367 | 1359 | 11 | 452 | 51420.65 | 7.58 |
| StVHA-H1 | Solanum tuberosum | PGSC0003DMG400007911 | 12: 2,431,205−2,438,621 | 1365 | 11 | 454 | 51347.52 | 6.36 |
| StVHA-H2 | PGSC0003DMG401011206 | 7: 1,259,585−1,265,564 | 1365 | 11 | 454 | 51545.58 | 6.43 | |
| SiVHA-H | Setaria italica | SETIT_029790mg | II: 42,735,808−42,741,948 | 1377 | 12 | 458 | 52115.37 | 6.76 |
| SbVHA-H | Sorghum bicolor | SORBI_3004G347600 | 4: 67,696,418−67,701,157 | 1452 | 12 | 483 | 55159.96 | 7.56 |
| TaVHA-H1 | Triticum aestivum | TraesCS2A02G212100 | 2A: 196,521,667−196,527,664 | 1359 | 11 | 452 | 51412.63 | 7.57 |
| TaVHA-H2 | TraesCS2B02G237200 | 2B: 237,967,455−237,974,014 | 1359 | 11 | 452 | 51382.58 | 7.98 | |
| TaVHA-H3 | TraesCS2D02G218000 | 2D: 181,306,271−181,312,853 | 1359 | 11 | 452 | 51426.61 | 7.57 | |
| ZmVHA-H1 | Zea mays | Zm00001d006565 | 2: 211,028,576−211,035,348 | 1359 | 11 | 452 | 51428.57 | 7.55 |
| ZmVHA-H2 | Zm00001d021721 | 7: 161,424,744−161,430,332 | 1359 | 11 | 452 | 51554.77 | 7.56 | |
| Number | Motif Logo | E-value | Site Count | Number of aa |
|---|---|---|---|---|
| Motif 1 |  | 5.6 × 10−1013 | 23 | 50 |
| Motif 2 |  | 2.1 × 10−981 | 24 | 50 |
| Motif 3 |  | 2.8 × 10−889 | 23 | 50 |
| Motif 4 |  | 4.8 × 10−939 | 24 | 50 |
| Motif 5 |  | 3.9 × 10−886 | 24 | 50 |
| Motif 6 |  | 3.7 × 10−823 | 22 | 50 |
| Motif 7 |  | 3.1 × 10−669 | 22 | 41 |
| Motif 8 |  | 3.4 × 10−475 | 22 | 29 |
| Motif 9 |  | 8.6 × 10−260 | 22 | 21 |
| Motif 10 |  | 5.6 × 10−179 | 23 | 11 |
| Motif 11 |  | 4.1 × 10−116 | 23 | 11 |
| Motif 12 |  | 5.3 × 10−105 | 22 | 8 |
| Motif 13 |  | 2.4 × 10−081 | 24 | 8 |
| Motif 14 |  | 8.6 × 10−061 | 23 | 8 |
| Motif 15 |  | 4.9 × 10002 | 2 | 12 |
| Gene | Transcript | Ensembl Transcript ID | Predicted Amino Acid Length (aa) | Spliced Exon | Status |
|---|---|---|---|---|---|
| GrVHA-H2 | GrVHA-H2.1 | KJB26715 | 481 | Wild type | |
| GrVHA-H2.2 | KJB26714 | 351 | Exon 1 | Alternative 5′ donor site | |
| Exon 3 | Alternative 3′ acceptor site | ||||
| Exons 4-7 | Exon skipping | ||||
| Exon 12 | Alternative 3′ acceptor site | ||||
| HvVHA-H | HvVHA-H.1 | HORVU2Hr1G042700.1 | 452 | Wild type | |
| HvVHA-H.2 | HORVU2Hr1G042700.2 | 452 | Exon 1 (5′ UTR) | Alternative 5′ donor site | |
| Exon 12 (3′ UTR) | Mutually exclusive exons | ||||
| HvVHA-H.3 | HORVU2Hr1G042700.3 | 452 | Exon 1 (5′ UTR) | Alternative 5′ donor site | |
| Exon 12 (3′ UTR) | Mutually exclusive exons | ||||
| HvVHA-H.4 | HORVU2Hr1G042700.4 | 450 | Exon 1 (5′ UTR) | Alternative 5′ donor site | |
| Exon 11 | Mutually exclusive exons | ||||
| Exon 12 (3′ UTR) | Exon skipping | ||||
| HvVHA-H.5 | HORVU2Hr1G042700.5 | 494 | Exon 1 (5′ UTR) | Alternative 5′ donor site | |
| Exon 10 | Alternative 3′ acceptor site | ||||
| Exons 11-12 (3′ UTR) | Exon skipping | ||||
| HvVHA-H.6 | HORVU2Hr1G042700.6 | 110 | Exon 1 (5′ UTR) | Exon skipping | |
| Exons 2-7 | Exon skipping | ||||
| Exons 10 | Alternative 3′ acceptor site | ||||
| Exons 11-12 (3′ UTR) | Exon skipping | ||||
| HvVHA-H.7 | HORVU2Hr1G042700.7 | 109 | Exon 1 (5′ UTR) | Exon skipping | |
| Exons 2-7 | Exon skipping | ||||
| Exon 11 (3′ UTR) | Mutually exclusive exons | ||||
| Exon 12 (3′ UTR) | Exon skipping | ||||
| StVHA-H2 | StVHA-H2.1 | PGSC0003DMT400029149 | 454 | Wild type | |
| StVHA-H2.2 | PGSC0003DMT400029148 | 454 | Exon 8 | Mutually exclusive exons | |
| Exon 12 (3′ UTR) | Exon skipping | ||||
| StVHA-H2.3 | PGSC0003DMT400029147 | 454 | Exon 12 (3′ UTR) | Exon skipping | |
| StVHA-H2.4 | PGSC0003DMT400029150 | 369 | Exon 9 (3′ UTR) | Mutually exclusive exons | |
| Exons 10-11 | Exon skipping | ||||
| Exons 12-13 (3′ UTR) | Exon skipping | ||||
| StVHA-H2.5 | PGSC0003DMT400029145 | 144 | Exons 1 (5′ UTR) | Exon skipping | |
| Exons 2-7 | Exon skipping | ||||
| Exon 8 | Alternative 5′ donor site | ||||
| Retained one exon between exons 8 and 9 | Exon skipping | ||||
| Exon 11 (3′ UTR) | Alternative 3′ acceptor site | ||||
| Exon 12, 13 (3′ UTR) | Exon skipping | ||||
| SiVHA-H | SiVHA-H.1 | KQL26169 | 458 | Wild type | |
| SiVHA-H.2 | KQL26168 | 404 | Exon 1 (5′ UTR) | Alternative 5′ donor site | |
| Exon 12 (3′ UTR) | Mutually exclusive exons | ||||
| TaVHA-H1 | TaVHA-H1.1 | TraesCS2A02G212100.2 | 452 | Wild type | |
| TaVHA-H1.2 | TraesCS2A02G212100.1 | 455 | Exon 12 | Mutually exclusive exons | |
| Exon 13 (3′ UTR) | Exon skipping | ||||
| ZmVHA-H1 | ZmVHA-H1.1 | Zm00001d006565_T002 | 452 | Wild type | |
| ZmVHA-H1.2 | Zm00001d006565_T001 | 379 | Exons 4-6 | Exon skipping | |
| Exon 7 | Alternative 5′ donor site | ||||
| ZmVHA-H1.3 | Zm00001d006565_T003 | 431 | Exons 1-2 (5′ UTR) | Exon skipping | |
| ZmVHA-H2 | ZmVHA-H2.1 | Zm00001d021721_T003 | 452 | Wild type | |
| ZmVHA-H2.2 | Zm00001d021721_T001 | 464 | Retained two exons between exons 3 and 4 | Exon skipping | |
| Exon 5 | Exon skipping | ||||
| ZmVHA-H2.3 | Zm00001d021721_T002 | 461 | Retained two exons between exons 3 and 4 | Exon skipping | |
| Exon 5 | Exon skipping | ||||
| Exon 6 | Alternative 3′ acceptor site | ||||
| Exon 7 | Alternative 5′ donor site | ||||
| ZmVHA-H2.4 | Zm00001d021721_T004 | 199 | Exons 1-2 (5′ UTR) | Exon skipping | |
| Retained exon between exons 2 and 3 (5′ UTR) | Exon skipping | ||||
| Introns 3-5 (5′ UTR) | Intron retention | ||||
| ZmVHA-H2.5 | Zm00001d021721_T005 | 431 | Exons 1-2 (5′ UTR) | Exon skipping |
| Species | VHA-H1 | VHA-H2 | VHA-H3 | VHA-H4 | VHA-H5 | VHA-H6 |
|---|---|---|---|---|---|---|
| Gossypium raimondii | 1 | 2 | ||||
| Oryza sativa (Indica Group) | 1 | |||||
| Brassica napus | 1 | 1 | 1 | 1 | 1 | 1 |
| Beta vulgaris | 1 | |||||
| Glycine max | 1 | 1 | ||||
| Hordeum vulgare | 6 | |||||
| Solanum tuberosum | 1 | 4 | ||||
| Setaria italica | 2 | |||||
| Sorghum bicolor | 1 | |||||
| Triticum aestivum | 2 | 1 | 1 | |||
| Zea mays | 3 | 5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, C.; Sun, F.; Yan, L.; Li, R.; Bai, J.; Caetano-Anollés, G. Genome-Wide Identification and Characterization of the Vacuolar H+-ATPase Subunit H Gene Family in Crop Plants. Int. J. Mol. Sci. 2019, 20, 5125. https://doi.org/10.3390/ijms20205125
Kang C, Sun F, Yan L, Li R, Bai J, Caetano-Anollés G. Genome-Wide Identification and Characterization of the Vacuolar H+-ATPase Subunit H Gene Family in Crop Plants. International Journal of Molecular Sciences. 2019; 20(20):5125. https://doi.org/10.3390/ijms20205125
Chicago/Turabian StyleKang, Chen, Fengjie Sun, Lei Yan, Rui Li, Jianrong Bai, and Gustavo Caetano-Anollés. 2019. "Genome-Wide Identification and Characterization of the Vacuolar H+-ATPase Subunit H Gene Family in Crop Plants" International Journal of Molecular Sciences 20, no. 20: 5125. https://doi.org/10.3390/ijms20205125
APA StyleKang, C., Sun, F., Yan, L., Li, R., Bai, J., & Caetano-Anollés, G. (2019). Genome-Wide Identification and Characterization of the Vacuolar H+-ATPase Subunit H Gene Family in Crop Plants. International Journal of Molecular Sciences, 20(20), 5125. https://doi.org/10.3390/ijms20205125