Autophagy, Metabolism, and Alcohol-Related Liver Disease: Novel Modulators and Functions



Abstract
:1. Introduction
2. General Interactions between Autophagy and Liver Diseases
2.1. Autophagy in Hepatic Lipid Metabolism
2.1.1. Autophagy as A Key Executor in Hepatic Steatosis
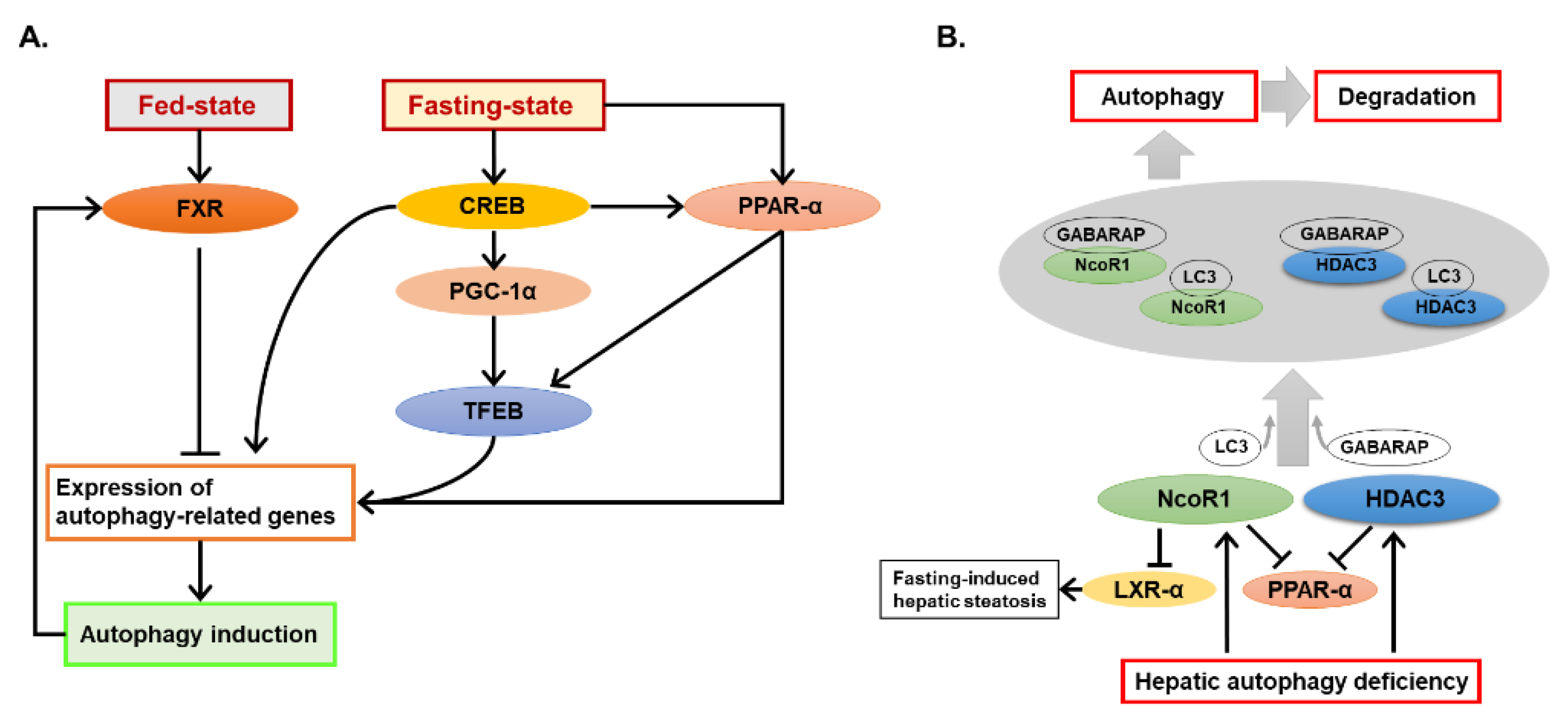
2.1.2. Crosstalk of Autophagy and the Nutrient-Sensing Nuclear Receptors
2.2. Regulation of Autophagy by Non-Coding RNAs in the Liver
2.3. Effects of Autophagy in Non-Parenchymal Hepatic Cells on Liver Diseases
3. Alcohol Modulates Autophagy in the Liver via Multiple Pathways
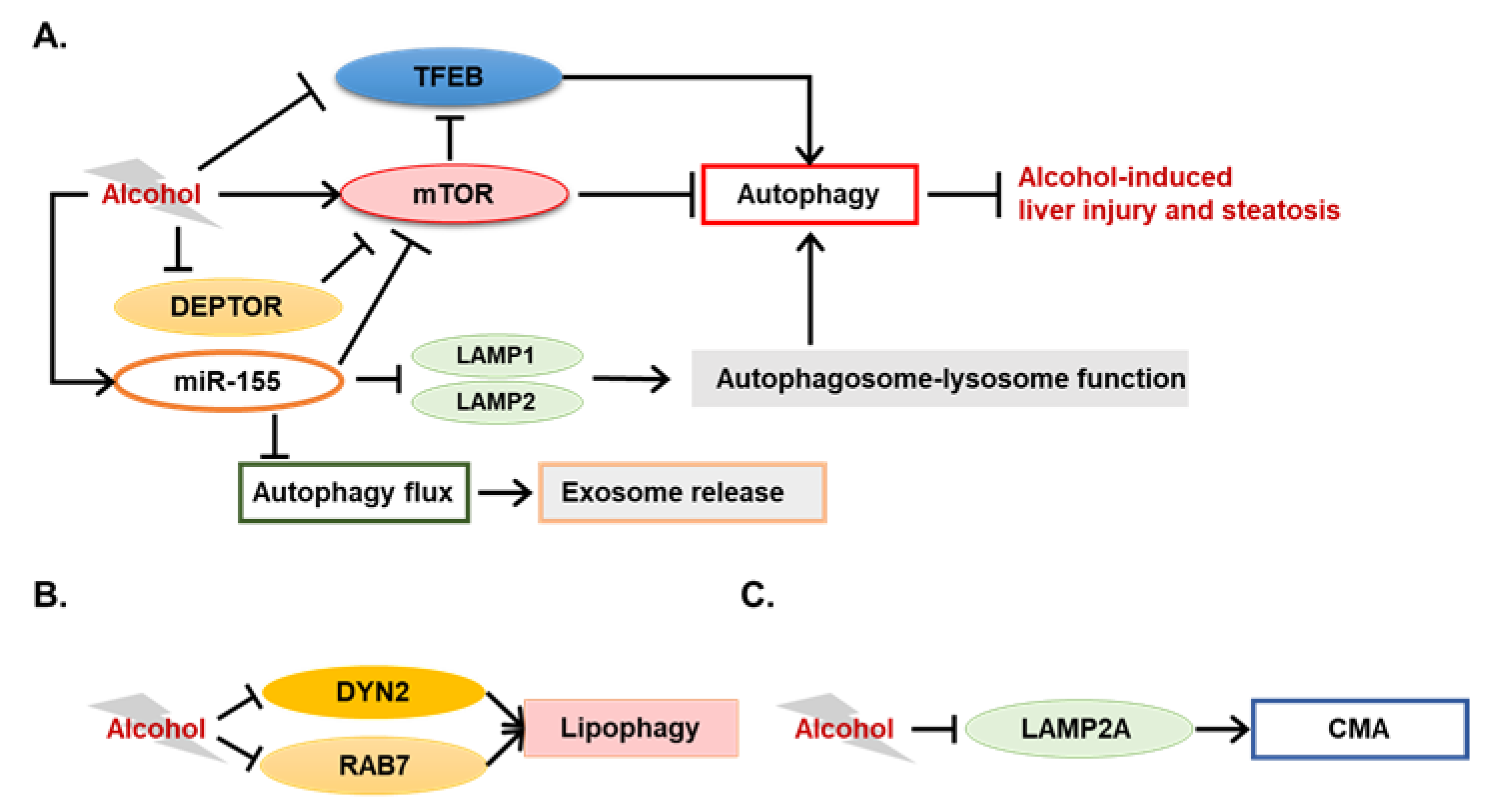
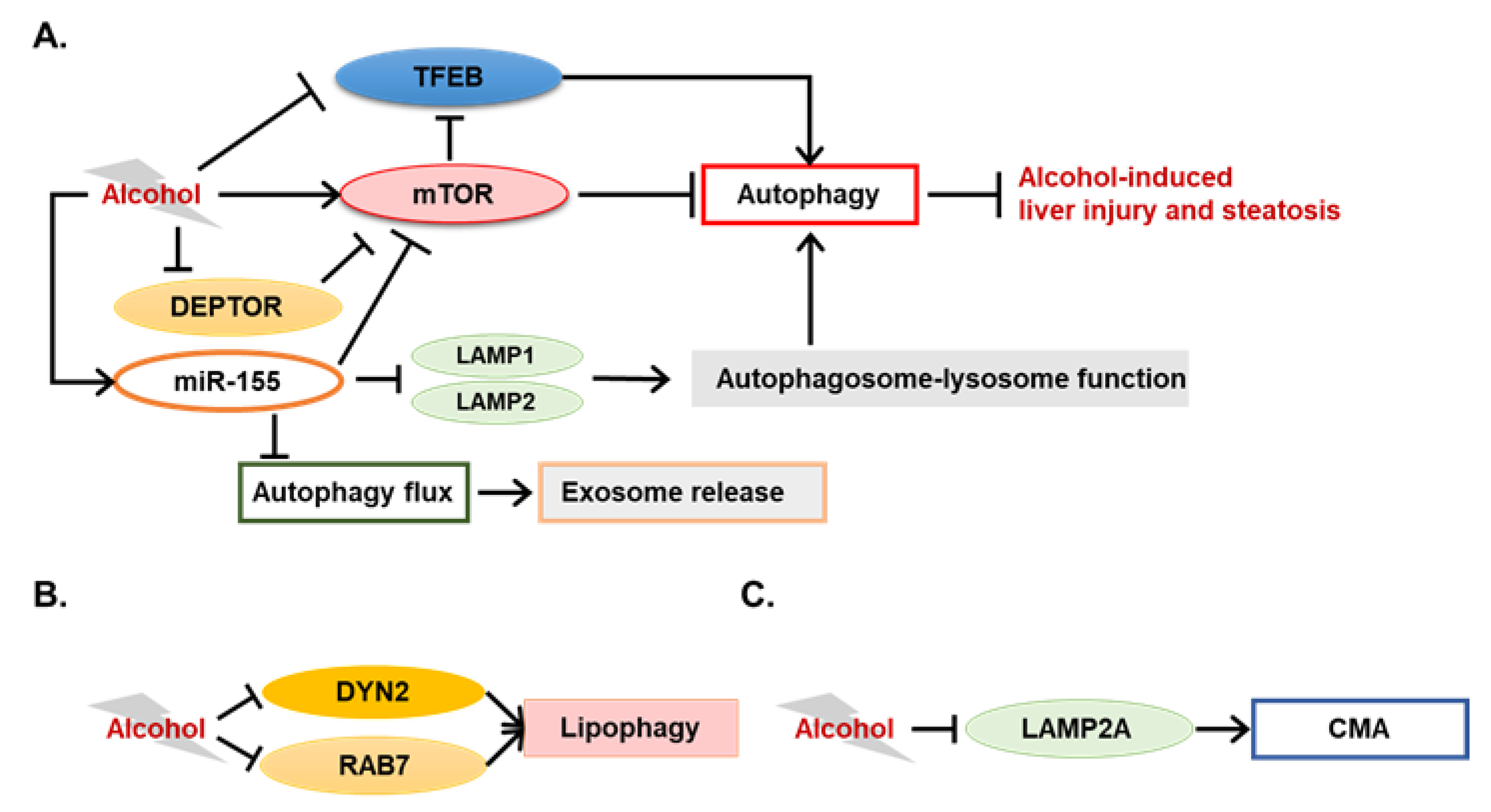
3.1. Novel Autophagy-Mediating Molecules in ALD
3.1.1. Novel Modulators of Alcohol-Induced Lipophagy
3.1.2. TFEB in ALD
3.1.3. Potential Involvement of Chaperone-Mediated Autophagy in ALD
3.2. Autophagy-Mediating miRNAs in ALD
4. Roles of Autophagy during Pathogenesis of ALD
4.1. Hepatocytes
4.2. Macrophages
4.3. Hepatic Stellate Cells
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 4E-BP1 | Eukaryotic translation initiation factor 4E-binding protein 1 |
| ACC1 | Acetyl-CoA carboxylase 1 |
| ADH | Alcohol dehydrogenase |
| AKT | Protein kinase B |
| ALD | Alcohol-related liver disease |
| ALDH2 | Aldehyde dedydrogenase-2 |
| ALR | Augmenter of liver regeneration |
| AMPK | 5′ AMP-activated protein kinase |
| ATGL | Adipose triglyceride lipase |
| CAT | Catalase |
| CCL4 | Carbon tetrachloride |
| CD36 | Cluster of differentiation 36 |
| CEBPβ | CCAAT/enhancer-binding protein beta |
| CMA | Chaperone-mediated autophagy |
| CREB | cAMP response element-binding protein |
| CTSA | Cathepsin A |
| Cyp2E1 | Cytochrome P450 family 2 subfamily E member 1 |
| DEPTOR | DEP domain-containing mTOR-interacting protein |
| DYN2 | Dynamin2 |
| ER | Endoplasmic reticulum |
| FXR | Farnesoid X receptor |
| G6PC | Glucose-6-phosphatase α |
| GABARAP | GABA type A receptor-associated protein |
| GalN | D-galactosamine |
| GSDIa | Glycogen storage diseases Ia |
| HCC | Hepatocellular carcinoma |
| HDAC3 | Histone deacetylase 3 |
| HMGB1 | High mobility group box 1 |
| HNF1A-AS1 | HNF1A antisense RNA 1 |
| HOTAIR | HOX transcript antisense RNA |
| HSC | Hepatic stellate cells |
| HSC70 | Heat shock-cognate protein of 70 kDa |
| HSL | Hormone-sensitive lipase |
| HULC | Hepatocellular carcinoma up-regulated long non-coding RNA |
| IL | Interleukin |
| IRF1 | Interferon regulatory factor1 |
| KC | Kupffer cells |
| LAMP | Lysosome-associated membrane protein |
| LC3 | Microtubule-associated proteins 1A/1B light chain 3 |
| LDs | Lipid droplets |
| lncRNA | Long non-coding RNAs |
| LPS | Lipopolysaccharide |
| LXRα | Liver X receptor alpha |
| miRNA | MicroRNAs |
| Mtor | The mammalian target of rapamycin |
| NAD | Nicotinamide adenine dinucleotide |
| NAFLD | Non-alcoholic fatty liver disease |
| NCoR1 | Nuclear receptor corepressor 1 |
| ncRNAs | Non-coding RNAs |
| Nogo-B | Reticulon 4B |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| oxLDL | Oxidized low-density lipoprotein |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PHLPP | PH domain and leucine rich repeat protein phosphatase |
| PI3KC3 | Class III phosphatidylinositol-3 kinase |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| PTEN | Phosphatase and tensin homolog |
| PTENP1 | Phosphatase and tensin homolog pseudogene 1 |
| RHEB | Ras homolog enriched in brain |
| ROS | Reactive oxidative species |
| S6 | Ribosomal protein S6 |
| SIRT1 | Sirtuin 1 |
| SNX | Sorting nexin |
| SQSTM1 | Sequestosome-1 |
| TFE3 | Transcription factor E3 |
| TFEB | Transcription factor EB |
| Vps34 | Vacuolar protein sorting 34 |
| vtRNA1-1 | Vault RNA1-1 |
| YAP | Yes-associated protein |
References
- Szabo, G.; Kamath, P.S.; Shah, V.H.; Thursz, M.; Mathurin, P.; Meeting, E.-A.J. Alcohol-related liver disease: Areas of consensus, unmet needs and opportunities for further study. Hepatology 2019, 69, 2271–2283. [Google Scholar] [CrossRef] [PubMed]
- Donohue, T.M., Jr.; Thomes, P.G. Ethanol-induced oxidant stress modulates hepatic autophagy and proteasome activity. Redox. Biol. 2014, 3, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Bailey, S.M.; Cunningham, C.C. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic. Biol. Med. 2002, 32, 11–16. [Google Scholar] [CrossRef]
- Demeilliers, C.; Maisonneuve, C.; Grodet, A.; Mansouri, A.; Nguyen, R.; Tinel, M.; Letteron, P.; Degott, C.; Feldmann, G.; Pessayre, D.; et al. Impaired adaptive resynthesis and prolonged depletion of hepatic mitochondrial DNA after repeated alcohol binges in mice. Gastroenterology 2002, 123, 1278–1290. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Demeilliers, C.; Amsellem, S.; Pessayre, D.; Fromenty, B. Acute ethanol administration oxidatively damages and depletes mitochondrial dna in mouse liver, brain, heart, and skeletal muscles: Protective effects of antioxidants. J. Pharmacol. Exp. Ther. 2001, 298, 737–743. [Google Scholar] [PubMed]
- Mansouri, A.; Gaou, I.; De Kerguenec, C.; Amsellem, S.; Haouzi, D.; Berson, A.; Moreau, A.; Feldmann, G.; Letteron, P.; Pessayre, D.; et al. An alcoholic binge causes massive degradation of hepatic mitochondrial DNA in mice. Gastroenterology 1999, 117, 181–190. [Google Scholar] [CrossRef]
- Cahill, A.; Cunningham, C.C.; Adachi, M.; Ishii, H.; Bailey, S.M.; Fromenty, B.; Davies, A. Effects of alcohol and oxidative stress on liver pathology: The role of the mitochondrion. Alcohol. Clin. Exp. Res. 2002, 26, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Cahill, A.; Stabley, G.J.; Wang, X.; Hoek, J.B. Chronic ethanol consumption causes alterations in the structural integrity of mitochondrial DNA in aged rats. Hepatology 1999, 30, 881–888. [Google Scholar] [CrossRef]
- Wang, L.; Khambu, B.; Zhang, H.; Yin, X.M. Autophagy in alcoholic liver disease, self-eating triggered by drinking. Clin. Res. Hepatol. Gastroenterol. 2015, 39, S2–S6. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Allaire, M.; Rautou, P.E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Green, D.R. Autophagy-independent functions of the autophagy machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Li, M.; Chen, X.; Ni, H.M.; Lin, C.W.; Gao, W.; Lu, B.; Stolz, D.B.; Clemens, D.L.; Yin, X.M. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010, 139, 1740–1752. [Google Scholar] [CrossRef] [PubMed]
- Thomes, P.G.; Ehlers, R.A.; Trambly, C.S.; Clemens, D.L.; Fox, H.S.; Tuma, D.J.; Donohue, T.M. Multilevel regulation of autophagosome content by ethanol oxidation in HepG2 cells. Autophagy 2013, 9, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Wang, X.; Zhou, R.; Yang, L.; Cederbaum, A.I. Alcohol steatosis and cytotoxicity: The role of cytochrome P4502E1 and autophagy. Free Radic. Biol. Med. 2012, 53, 1346–1357. [Google Scholar] [CrossRef] [Green Version]
- Sid, B.; Verrax, J.; Calderon, P.B. Role of AMPK activation in oxidative cell damage: Implications for alcohol-induced liver disease. Biochem. Pharmacol. 2013, 86, 200–209. [Google Scholar] [CrossRef]
- Ding, W.X.; Li, M.; Yin, X.M. Selective taste of ethanol-induced autophagy for mitochondria and lipid droplets. Autophagy 2011, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Ni, H.M.; Du, K.; You, M.; Ding, W.X. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am. J. Pathol. 2013, 183, 1815–1825. [Google Scholar] [CrossRef]
- Ni, H.M.; Bhakta, A.; Wang, S.; Li, Z.; Manley, S.; Huang, H.; Copple, B.; Ding, W.X. Role of hypoxia inducing factor-1beta in alcohol-induced autophagy, steatosis and liver injury in mice. PLoS ONE 2014, 9, e115849. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy 2008, 4, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Ni, H.M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Yoo, C. Role of zinc in the regulation of autophagy during ethanol exposure in human hepatoma cells. Biol. Trace Elem. Res. 2013, 156, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Zhang, H.; Li, M.; Xiong, X.; Chen, X.; Chen, X.; Dong, X.C.; Yin, X.M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J. Hepatol. 2013, 58, 993–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khambu, B.; Yan, S.; Huda, N.; Liu, G.; Yin, X.M. Autophagy in non-alcoholic fatty liver disease and alcoholic liver disease. Liver Res. 2018, 2, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Huda, N.; Khambu, B.; Yin, X.M. Relevance of autophagy to fatty liver diseases and potential therapeutic applications. Amino Acids 2017, 49, 1965–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khambu, B.; Yan, S.; Huda, N.; Liu, G.; Yin, X.M. Homeostatic Role of Autophagy in Hepatocytes. Semin. Liver Dis. 2018, 38, 308–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of Liver Metabolism by Autophagy. Gastroenterology 2016, 150, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Khambu, B.; Huda, N.; Chen, X.; Antoine, D.J.; Li, Y.; Dai, G.; Kohler, U.A.; Zong, W.X.; Waguri, S.; Werner, S.; et al. HMGB1 promotes ductular reaction and tumorigenesis in autophagy-deficient livers. J. Clin. Investig. 2018, 128, 2419–2435. [Google Scholar] [CrossRef]
- Lee, Y.A.; Noon, L.A.; Akat, K.M.; Ybanez, M.D.; Lee, T.F.; Berres, M.L.; Fujiwara, N.; Goossens, N.; Chou, H.I.; Parvin-Nejad, F.P.; et al. Autophagy is a gatekeeper of hepatic differentiation and carcinogenesis by controlling the degradation of Yap. Nat. Commun. 2018, 9, 4962. [Google Scholar] [CrossRef]
- Ni, H.M.; Chao, X.; Yang, H.; Deng, F.; Wang, S.; Bai, Q.; Qian, H.; Cui, Y.; Cui, W.; Shi, Y.; et al. Dual Roles of Mammalian Target of Rapamycin in Regulating Liver Injury and Tumorigenesis in Autophagy-Defective Mouse Liver. Hepatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Misih, S.R.; Bloomston, M. Liver anatomy. Surg. Clin. North Am. 2010, 90, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farah, B.L.; Landau, D.J.; Sinha, R.A.; Brooks, E.D.; Wu, Y.; Fung, S.Y.S.; Tanaka, T.; Hirayama, M.; Bay, B.H.; Koeberl, D.D.; et al. Induction of autophagy improves hepatic lipid metabolism in glucose-6-phosphatase deficiency. J. Hepatol. 2016, 64, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Sathyanarayan, A.; Mashek, M.T.; Mashek, D.G. ATGL promotes autophagy/Lipophagy via SIRT1 to control hepatic lipid droplet catabolism. Cell Rep. 2017, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Yang, B.; Qiu, W.; Hao, Y.; Zhang, Z.; Yang, B.; Li, N.; Cheng, S.; Lin, Z.; Rui, Y.C.; et al. ER-residential Nogo-B accelerates NAFLD-associated HCC mediated by metabolic reprogramming of oxLDL lipophagy. Nat. Commun. 2019, 10, 3391. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.S.; Zimmermann, A.; Pendl, T.; Schroeder, S.; Schoenlechner, H.; Knittelfelder, O.; Lamplmayr, L.; Santiso, A.; Aufschnaiter, A.; Waltenstorfer, D.; et al. Acetyl-CoA carboxylase 1-dependent lipogenesis promotes autophagy downstream of AMPK. J. Biol. Chem. 2019, 294, 12020–12039. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Hikita, H.; Tatsumi, T.; Sakamori, R.; Nozaki, Y.; Sakane, S.; Shiode, Y.; Nakabori, T.; Saito, Y.; Hiramatsu, N.; et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology 2016, 64, 1994–2014. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Seok, S.; Fu, T.; Choi, S.E.; Li, Y.; Zhu, R.; Kumar, S.; Sun, X.; Yoon, G.; Kang, Y.; Zhong, W.; et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 2014, 516, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Williams, D.; Qiu, Y.; Song, Z.; Yang, Z.; Kimler, V.; Goldberg, A.; Zhang, R.; Yang, Z.; Chen, X.; et al. Regulation of hepatic autophagy by stress-sensing transcription factor CREBH. FASEB J. 2019, 33, 7896–7914. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Iershov, A.; Nemazanyy, I.; Alkhoury, C.; Girard, M.; Barth, E.; Cagnard, N.; Montagner, A.; Chretien, D.; Rugarli, E.I.; Guillou, H.; et al. The class 3 PI3K coordinates autophagy and mitochondrial lipid catabolism by controlling nuclear receptor PPARalpha. Nat. Commun. 2019, 10, 1566. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Kuma, A.; Sugiura, Y.; Ichimura, Y.; Obata, M.; Kitamura, H.; Okuda, S.; Lee, H.C.; Ikeda, K.; Kanegae, Y.; et al. Autophagy regulates lipid metabolism through selective turnover of NCoR1. Nat. Commun. 2019, 10, 1567. [Google Scholar] [CrossRef]
- Takahashi, S.-S.; Sou, Y.-S.; Saito, T.; Kuma, A.; Yabe, T.; Koike, M.; Terai, S.; Mizushima, N.; Waguri, S.; Komatsu, M. Autophagy controls lipid droplet formation by fine-tuning NCoR1 levels. BioRxiv 2019, 722686. [Google Scholar] [CrossRef]
- Li, Y.; Chao, X.; Yang, L.; Lu, Q.; Li, T.; Ding, W.X.; Ni, H.M. Impaired Fasting-Induced Adaptive Lipid Droplet Biogenesis in Liver-Specific Atg5-Deficient Mouse Liver Is Mediated by Persistent Nuclear Factor-Like 2 Activation. Am. J. Pathol. 2018, 188, 1833–1846. [Google Scholar] [CrossRef]
- Khambu, B.; Li, T.; Yan, S.; Yu, C.; Chen, X.; Goheen, M.; Li, Y.; Lin, J.; Cummings, O.W.; Lee, Y.A.; et al. Hepatic autophagy deficiency compromises farnesoid X receptor functionality and causes cholestatic injury. Hepatology 2019, 69, 2196–2213. [Google Scholar] [CrossRef]
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef]
- Frankel, L.B.; Lund, A.H. MicroRNA regulation of autophagy. Carcinogenesis 2012, 33, 2018–2025. [Google Scholar] [CrossRef] [PubMed]
- Gozuacik, D.; Akkoc, Y.; Ozturk, D.G.; Kocak, M. Autophagy-regulating microRNAs and cancer. Front. Oncol. 2017, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Z.; Zhang, W.; Zhang, L. MicroRNAs play an essential role in autophagy regulation in various disease phenotypes. BioFactors 2019. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Wang, Y.; Li, M.; Zhu, Y.; Liang, H.; Wang, C.; Wang, F.; Zhang, C.Y.; Zen, K.; Li, L. MiR-26 enhances chemosensitivity and promotes apoptosis of hepatocellular carcinoma cells through inhibiting autophagy. Cell Death Dis. 2017, 8, e2540. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhang, Q.; Cai, X.; Li, F.; Ma, Z.; Xu, M.; Lu, L. Exosomes derived from miR-181-5p-modified adipose-derived mesenchymal stem cells prevent liver fibrosis via autophagy activation. J. Cell. Mol. Med. 2017, 21, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- Islam Khan, M.Z.; Tam, S.Y.; Law, H.K.W. Autophagy-Modulating Long Non-coding RNAs (LncRNAs) and Their Molecular Events in Cancer. Front. Genet. 2018, 9, 750. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wei, X.; Zhang, A.; Li, C.; Bai, J.; Dong, J. Long non-coding RNA HNF1A-AS1 functioned as an oncogene and autophagy promoter in hepatocellular carcinoma through sponging hsa-miR-30b-5p. Biochem. Biophys. Res. Commun. 2016, 473, 1268–1275. [Google Scholar] [CrossRef]
- Chen, C.L.; Tseng, Y.W.; Wu, J.C.; Chen, G.Y.; Lin, K.C.; Hwang, S.M.; Hu, Y.C. Suppression of hepatocellular carcinoma by baculovirus-mediated expression of long non-coding RNA PTENP1 and MicroRNA regulation. Biomaterials 2015, 44, 71–81. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, X.; Li, H.; Liu, J. The long noncoding RNA HOTAIR activates autophagy by upregulating ATG3 and ATG7 in hepatocellular carcinoma. Mol. Biosyst. 2016, 12, 2605–2612. [Google Scholar] [CrossRef]
- Panzitt, K.; Tschernatsch, M.M.; Guelly, C.; Moustafa, T.; Stradner, M.; Strohmaier, H.M.; Buck, C.R.; Denk, H.; Schroeder, R.; Trauner, M.; et al. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology 2007, 132, 330–342. [Google Scholar] [CrossRef]
- Xiong, H.; Ni, Z.; He, J.; Jiang, S.; Li, X.; He, J.; Gong, W.; Zheng, L.; Chen, S.; Li, B.; et al. LncRNA HULC triggers autophagy via stabilizing Sirt1 and attenuates the chemosensitivity of HCC cells. Oncogene 2017, 36, 3528–3540. [Google Scholar] [CrossRef] [PubMed]
- Horos, R.; Buscher, M.; Kleinendorst, R.; Alleaume, A.M.; Tarafder, A.K.; Schwarzl, T.; Dziuba, D.; Tischer, C.; Zielonka, E.M.; Adak, A.; et al. The small non-coding vault RNA1-1 acts as a riboregulator of autophagy. Cell 2019, 176, 1054–1067. [Google Scholar] [CrossRef] [PubMed]
- Kmiec, Z. Cooperation of liver cells in health and disease. Adv. Anat. Embryol. Cell Biol. 2001, 161, 1–151. [Google Scholar]
- Weiskirchen, R.; Tacke, F. Relevance of autophagy in parenchymal and non-parenchymal liver cells for health and disease. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Hilscher, M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 2013, 59, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Lodder, J.; Denaes, T.; Chobert, M.N.; Wan, J.; El-Benna, J.; Pawlotsky, J.M.; Lotersztajn, S.; Teixeira-Clerc, F. Macrophage autophagy protects against liver fibrosis in mice. Autophagy 2015, 11, 1280–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruart, M.; Chavarria, L.; Camprecios, G.; Suarez-Herrera, N.; Montironi, C.; Guixe-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagan, J.C.; Hernandez-Gea, V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J. Hepatol. 2019, 70, 458–469. [Google Scholar] [CrossRef]
- Liu, K.; Zhao, E.; Ilyas, G.; Lalazar, G.; Lin, Y.; Haseeb, M.; Tanaka, K.E.; Czaja, M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015, 11, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Ilyas, G.; Zhao, E.; Liu, K.; Lin, Y.; Tesfa, L.; Tanaka, K.E.; Czaja, M.J. Macrophage autophagy limits acute toxic liver injury in mice through down regulation of interleukin-1beta. J. Hepatol. 2016, 64, 118–127. [Google Scholar] [CrossRef]
- Yan, S.; Zhou, J.; Chen, X.; Dong, Z.; Yin, X.M. Diverse consequences in liver injury in mice with different autophagy functional status treated with alcohol. Am. J. Pathol. 2019, 189, 1744–1762. [Google Scholar] [CrossRef] [PubMed]
- Chao, X.; Wang, S.; Zhao, K.; Li, Y.; Williams, J.A.; Li, T.; Chavan, H.; Krishnamurthy, P.; He, X.C.; Li, L.; et al. Impaired TFEB-mediated lysosome biogenesis and autophagy promote chronic ethanol-induced liver injury and steatosis in mice. Gastroenterology 2018, 155, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Denaes, T.; Lodder, J.; Chobert, M.N.; Ruiz, I.; Pawlotsky, J.M.; Lotersztajn, S.; Teixeira-Clerc, F. The cannabinoid receptor 2 protects against alcoholic liver disease via a macrophage autophagy-dependent pathway. Sci. Rep. 2016, 6, 28806. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, G.; Cingolani, F.; Zhao, E.; Tanaka, K.; Czaja, M.J. Decreased macrophage autophagy promotes liver injury and inflammation from alcohol. Alcohol. Clin. Exp. Res. 2019, 43, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Zhong, Z.; Kim, S.Y.; Uchiyama, R.; Roh, Y.S.; Matsushita, H.; Gottlieb, R.A.; Seki, E. Murine macrophage autophagy protects against alcohol-induced liver injury by degrading interferon regulatory factor 1 (IRF1) and removing damaged mitochondria. J. Biol. Chem. 2019, 294, 12359–12369. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.Y.; Xiao, Z.H.; Wang, F.F. Inhibition of autophagy reverses alcohol-induced hepatic stellate cells activation through activation of Nrf2-Keap1-ARE signaling pathway. Biochimie 2018, 147, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, C.; Thomes, P.G.; Kharbanda, K.K.; Casey, C.A.; McNiven, M.A.; Donohue, T.M., Jr. Lipophagy and Alcohol-Induced Fatty Liver. Front. Pharm. 2019, 10, 495. [Google Scholar] [CrossRef] [Green Version]
- Schott, M.B.; Rasineni, K.; Weller, S.G.; Schulze, R.J.; Sletten, A.C.; Casey, C.A.; McNiven, M.A. beta-Adrenergic induction of lipolysis in hepatocytes is inhibited by ethanol exposure. J. Biol. Chem. 2017, 292, 11815–11828. [Google Scholar] [CrossRef]
- Zhong, Z.; Ramshesh, V.K.; Rehman, H.; Liu, Q.; Theruvath, T.P.; Krishnasamy, Y.; Lemasters, J.J. Acute ethanol causes hepatic mitochondrial depolarization in mice: Role of ethanol metabolism. PLoS ONE 2014, 9, e91308. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, J.; Yan, S.; Lei, G.; Lee, C.H.; Yin, X.M. Ethanol-triggered Lipophagy requires SQSTM1 in AML12 hepatic cells. Sci. Rep. 2017, 7, 12307. [Google Scholar] [CrossRef]
- Schulze, R.J.; Weller, S.G.; Schroeder, B.; Krueger, E.W.; Chi, S.; Casey, C.A.; McNiven, M.A. Lipid droplet breakdown requires dynamin 2 for vesiculation of autolysosomal tubules in hepatocytes. J. Cell Biol. 2013, 203, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Rasineni, K.; Donohue, T.M., Jr.; Thomes, P.G.; Yang, L.; Tuma, D.J.; McNiven, M.A.; Casey, C.A. Ethanol-induced steatosis involves impairment of lipophagy, associated with reduced Dynamin2 activity. Hepatol. Commun. 2017, 1, 501–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, B.; Schulze, R.J.; Weller, S.G.; Sletten, A.C.; Casey, C.A.; McNiven, M.A. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 2015, 61, 1896–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, R.J.; Rasineni, K.; Weller, S.G.; Schott, M.B.; Schroeder, B.; Casey, C.A.; McNiven, M.A. Ethanol exposure inhibits hepatocyte lipophagy by inactivating the small guanosine triphosphatase Rab7. Hepatol. Commun. 2017, 1, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; McVicker, D.L.; Zetterman, R.K.; Donohue, T.M., Jr. Ethanol consumption alters trafficking of lysosomal enzymes and affects the processing of procathepsin L in rat liver. Biochim. Biophys. Acta 1996, 1291, 45–52. [Google Scholar] [CrossRef]
- Kharbanda, K.K.; McVicker, D.L.; Zetterman, R.K.; MacDonald, R.G.; Donohue, T.M., Jr. Flow cytometric analysis of vesicular pH in rat hepatocytes after ethanol administration. Hepatology 1997, 26, 929–934. [Google Scholar] [CrossRef]
- Dolganiuc, A.; Thomes, P.G.; Ding, W.X.; Lemasters, J.J.; Donohue, T.M., Jr. Autophagy in alcohol-induced liver diseases. Alcohol. Clin. Exp. Res. 2012, 36, 1301–1308. [Google Scholar] [CrossRef]
- Thomes, P.G.; Trambly, C.S.; Fox, H.S.; Tuma, D.J.; Donohue, T.M., Jr. Acute and Chronic Ethanol Administration Differentially Modulate Hepatic Autophagy and Transcription Factor EB. Alcohol. Clin. Exp. Res. 2015, 39, 2354–2363. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Xu, X.; Babcock, S.A.; Zhang, Y.; Ren, J. Aldehyde dedydrogenase-2 plays a beneficial role in ameliorating chronic alcohol-induced hepatic steatosis and inflammation through regulation of autophagy. J. Hepatol. 2015, 62, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shen, F.; Sherban, A.; Nocon, A.; Li, Y.; Wang, H.; Xu, M.J.; Rui, X.; Han, J.; Jiang, B.; et al. DEP domain-containing mTOR-interacting protein suppresses lipogenesis and ameliorates hepatic steatosis and acute-on-chronic liver injury in alcoholic liver disease. Hepatology 2018, 68, 496–514. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Li, W.Z.; Zhang, S.; Hu, B.; Li, Y.X.; Li, H.D.; Tang, H.H.; Li, Q.W.; Guan, Y.Y.; Liu, L.X.; et al. SNX10 mediates alcohol-induced liver injury and steatosis by regulating the activation of chaperone-mediated autophagy. J. Hepatol. 2018, 69, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Jogasuria, A.; Yin, H.; Xu, M.J.; Hu, X.; Wang, J.; Kim, C.; Wu, J.; Lee, K.; Gao, B.; et al. The detrimental role played by lipocalin-2 in alcoholic fatty liver in mice. Am. J. Pathol. 2016, 186, 2417–2428. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Fu, X.; Xie, J.; Meng, Z.; Gu, Y.; Wang, X.; Li, L.; Pan, H.; Huang, W. MiR-26a enhances autophagy to protect against ethanol-induced acute liver injury. J. Mol. Med. (Berl.) 2015, 93, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Babuta, M.; Furi, I.; Bala, S.; Bukong, T.N.; Lowe, P.; Catalano, D.; Calenda, C.; Kodys, K.; Szabo, G. Dysregulated autophagy and lysosome function are linked to exosome production by micro-RNA 155 in alcoholic liver disease. Hepatology 2019. [Google Scholar] [CrossRef]
- Atef, M.M.; Hafez, Y.M.; Alshenawy, H.A.; Emam, M.N. Ameliorative effects of autophagy inducer, simvastatin on alcohol-induced liver disease in a rat model. J. Cell Biochem. 2018, 120, 7679–7688. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Yang, Y.; Ren, L.; Shao, T.; Li, F.; Zhao, C.; Liu, L.; Zhang, H.; McClain, C.J.; Feng, W. Activation of autophagy attenuates EtOH-LPS-induced hepatic steatosis and injury through MD2 associated TLR4 signaling. Sci. Rep. 2017, 7, 9292. [Google Scholar] [CrossRef]
- Liu, L.; Xie, P.; Li, W.; Wu, Y.; An, W. Augmenter of liver regeneration protects against ethanol-induced acute liver injury by promoting autophagy. Am. J. Pathol. 2019, 189, 552–567. [Google Scholar] [CrossRef]
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Cell Types | General Functions | Functions in ALD |
|---|---|---|
| Hepatocytes | Autophagy in hepatocytes is critical to maintain homeostasis of liver functions. See other reviews for details [11,13,25,26,27,28]. | Generally, autophagy plays a protective role in ALD, whereas different status of autophagy deficiency can lead to diverse consequences [9,14,24,25,26,71,72]. |
| Macrophages | Macrophage autophagy is important to maintain a normal immune response [64]. Macrophage-specific deletion of Atg5 can aggravate CCL4-induced liver fibrosis [67]. Impaired macrophage autophagy can promote proinflammatory macrophage polarization and increase the immune response in obese mice [69]. Inhibition of macrophage autophagy can also increase toxin-induced acute liver injury from GalN/LPS co-treatment through down-regulation of IL1β [70]. | Autophagy in macrophages is critical to protect the liver from alcohol-induced damage, either by mediating effects from other protectors or by directly protecting the liver from a “second hit” in the progression of liver injury [73,74,75]. |
| Hepatic stellate cells | Activation of HSCs requires production of energy from autophagy-mediated lipid catabolism [64,65,66]. | An in vitro study using HSC-T6, an immortalized rat HSC line, shows that autophagy may contribute to alcohol-induced HSC activation [76]. |
| Endothelial cells | Autophagy in endothelial cells is critical to maintain these cells homeostasis [64]. Impaired autophagy by deletion of Atg7 in endothelial cells does not affect liver homeostasis but amplifies liver fibrosis without increasing liver injury following CCL4-treatment [68]. | Unclear. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, S.; Khambu, B.; Hong, H.; Liu, G.; Huda, N.; Yin, X.-M. Autophagy, Metabolism, and Alcohol-Related Liver Disease: Novel Modulators and Functions. Int. J. Mol. Sci. 2019, 20, 5029. https://doi.org/10.3390/ijms20205029
Yan S, Khambu B, Hong H, Liu G, Huda N, Yin X-M. Autophagy, Metabolism, and Alcohol-Related Liver Disease: Novel Modulators and Functions. International Journal of Molecular Sciences. 2019; 20(20):5029. https://doi.org/10.3390/ijms20205029
Chicago/Turabian StyleYan, Shengmin, Bilon Khambu, Honghai Hong, Gang Liu, Nazmul Huda, and Xiao-Ming Yin. 2019. "Autophagy, Metabolism, and Alcohol-Related Liver Disease: Novel Modulators and Functions" International Journal of Molecular Sciences 20, no. 20: 5029. https://doi.org/10.3390/ijms20205029
APA StyleYan, S., Khambu, B., Hong, H., Liu, G., Huda, N., & Yin, X.-M. (2019). Autophagy, Metabolism, and Alcohol-Related Liver Disease: Novel Modulators and Functions. International Journal of Molecular Sciences, 20(20), 5029. https://doi.org/10.3390/ijms20205029