Insulin Receptor Trafficking: Consequences for Insulin Sensitivity and Diabetes



Abstract
:1. Overview of Insulin Receptor (INSR) Signaling Regulation
2. Ligand-Dependent INSR Activation
3. Ligand-Dependent INSR Endocytosis: A Spatial Modulator of INSR Signaling
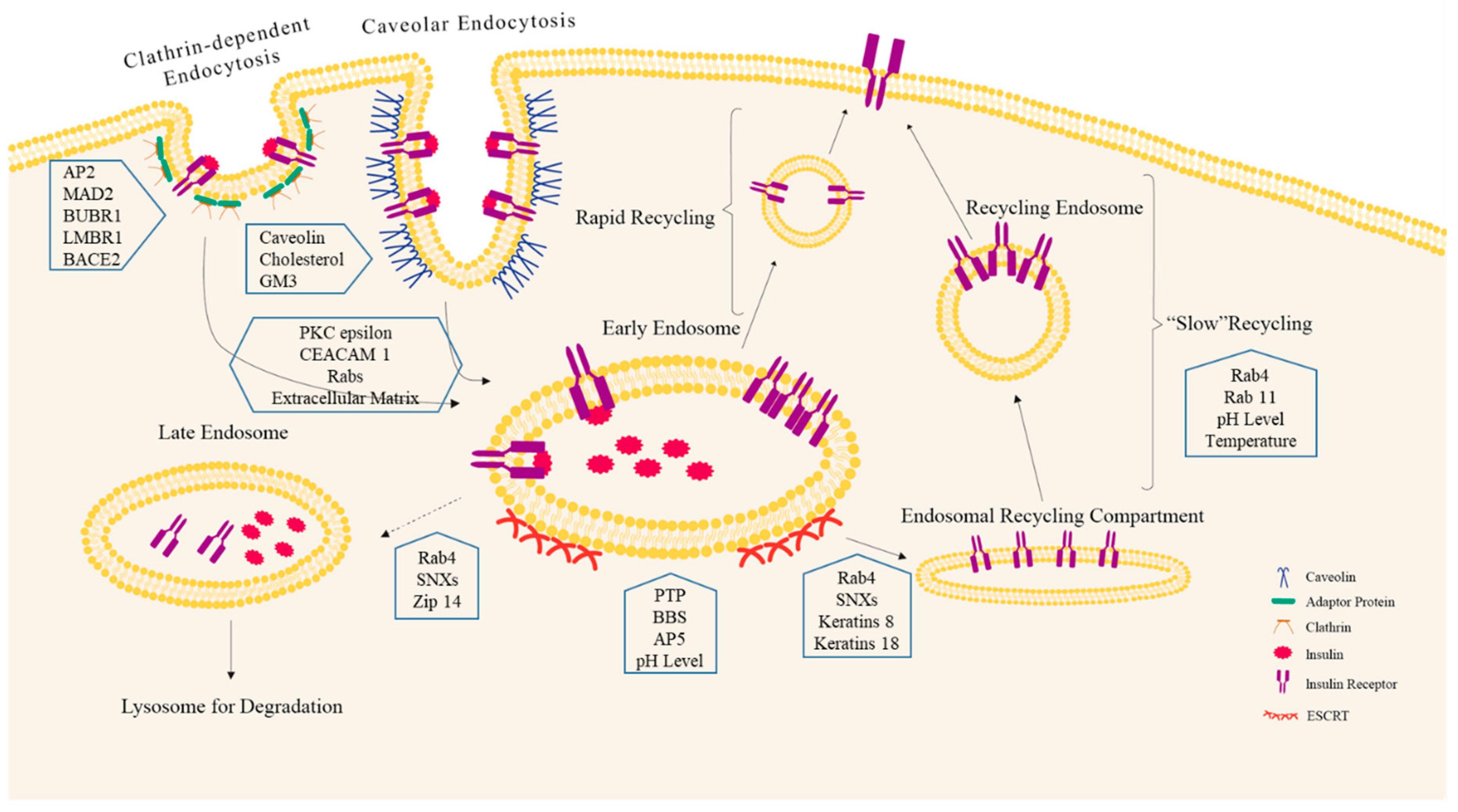
3.1. Clathrin-Mediated INSR Endocytosis
3.2. Clathrin-Independent INSR Endocytosis
3.3. Candidate Regulators of INSR Endocytosis
4. INSR Endosomal Sorting: The Fate of INSR
4.1. INSR Trafficking
4.2. Regulation of INSR Recycling
4.3. Nuclear Translocation of Internalized INSR
5. INSR Trafficking and Diabetes
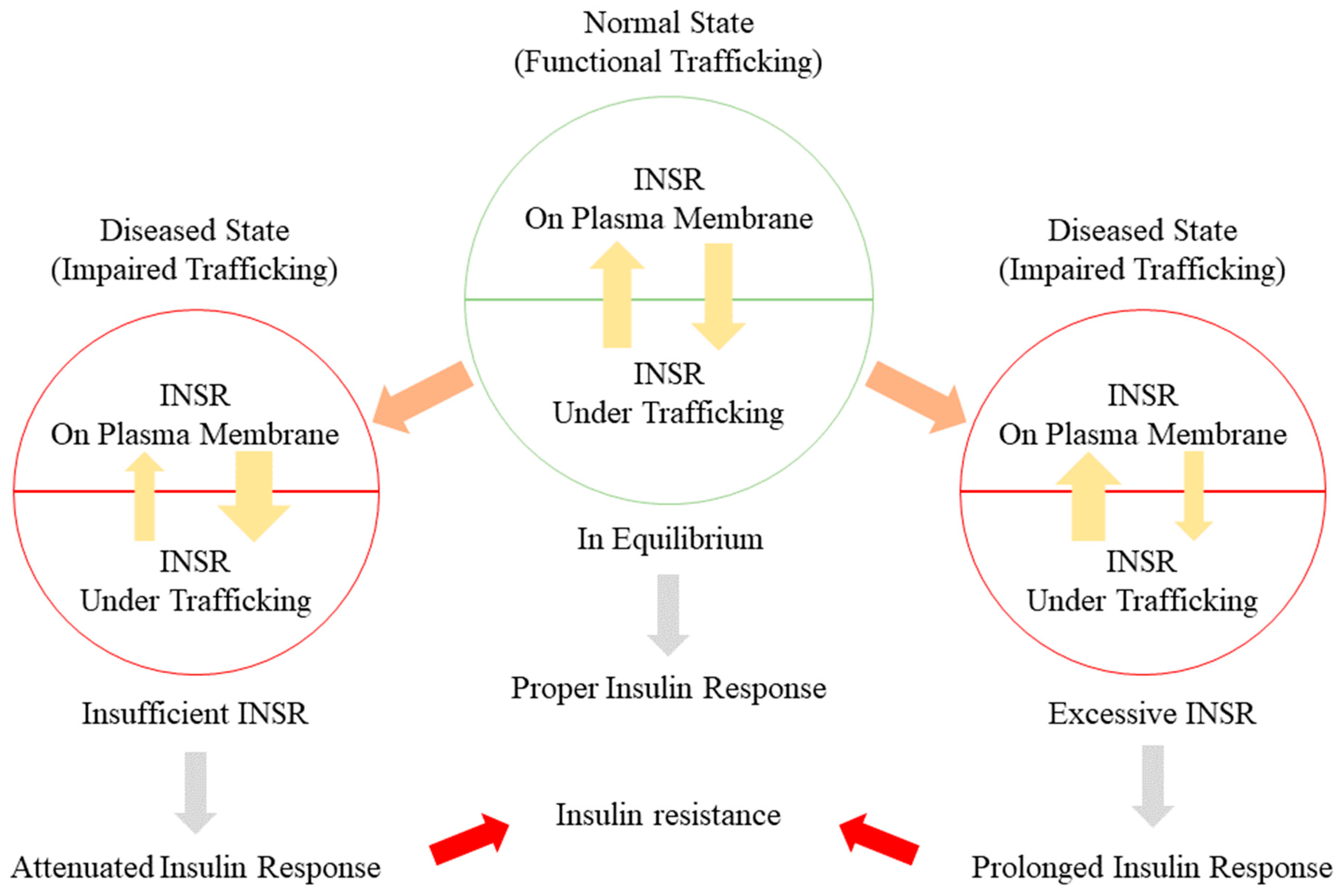
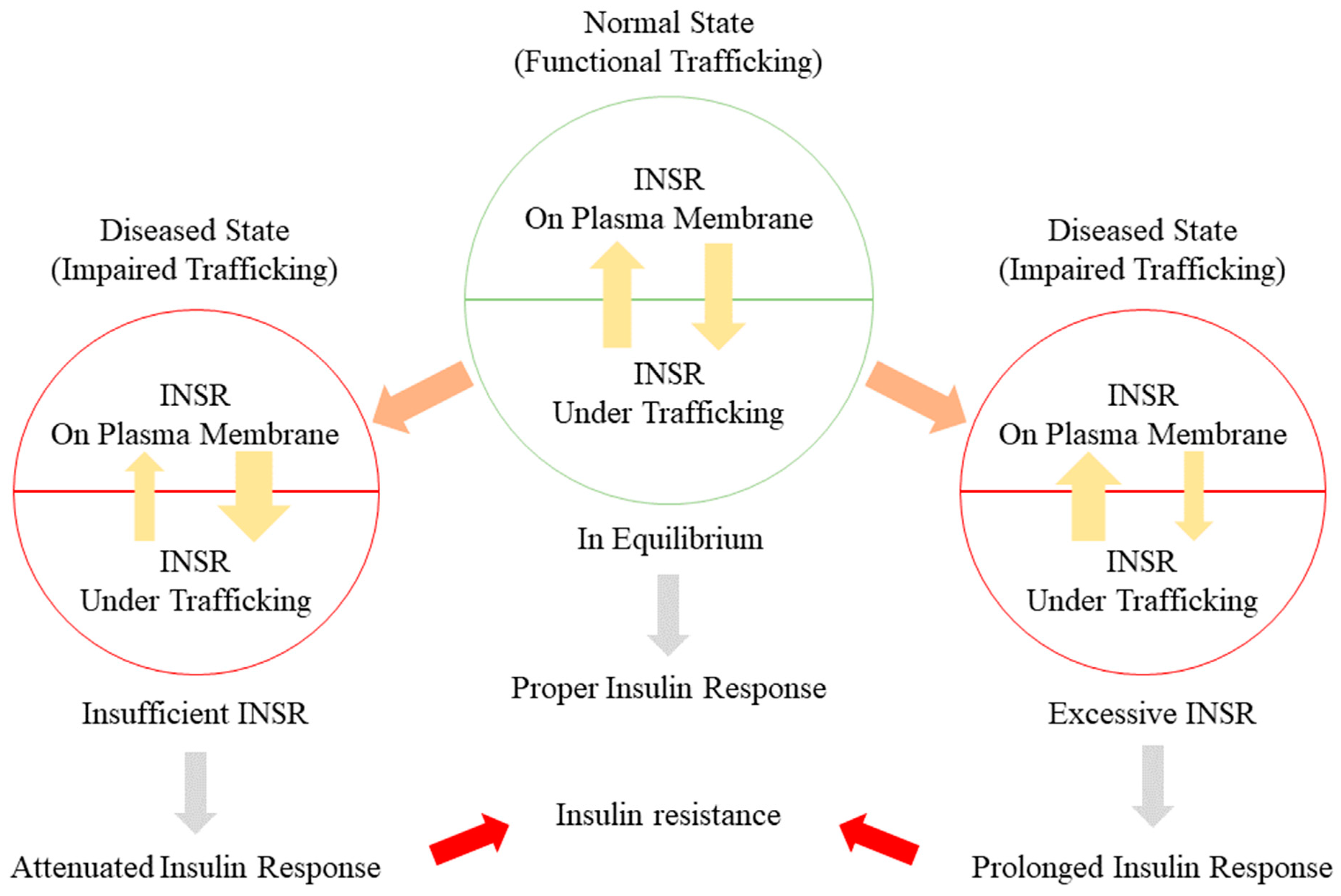
5.1. Alteration of INSR Trafficking in Insulin Resistance
5.2. Alterations of INSR in Insulin Resistance
5.3. Disturbance of the INSR Recycling Process: A Possible Initiator of T2DM and a Promising Therapeutic Target
6. Conclusions
Funding
Conflicts of Interest
References
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.W.; Lawrence, M.C. Ligand-induced activation of the insulin receptor: A multi-step process involving structural changes in both the ligand and the receptor. Bioessays 2009, 31, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Boothe, T.; Lim, G.E.; Cen, H.; Skovso, S.; Piske, M.; Li, S.N.; Nabi, I.R.; Gilon, P.; Johnson, J.D. Inter-domain tagging implicates caveolin-1 in insulin receptor trafficking and Erk signaling bias in pancreatic β-cells. Mol. Metab. 2016, 5, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Giudice, J.; Jares-Erijman, E.A.; Leskow, F.C. Insulin receptor membrane retention by a traceable chimeric mutant. Cell Commun. Signal 2013, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- De Meyts, P. The Insulin Receptor and Its Signal Transduction Network. In Endotext; Feingold, K.R., Anawalt, B., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000–2019. Available online: https://www.ncbi.nlm.nih.gov/books/NBK378978/ (accessed on 27 April 2016).
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- Ceresa, B.P.; Kao, A.W.; Santeler, S.R.; Pessin, J.E. Inhibition of clathrin-mediated endocytosis selectively attenuates specific insulin receptor signal transduction pathways. Mol. Cell Biol. 1998, 18, 3862–3870. [Google Scholar] [CrossRef]
- Hari, J.; Roth, R.A. Defective internalization of insulin and its receptor in cells expressing mutated insulin receptors lacking kinase activity. J. Biol. Chem. 1987, 262, 15341–15344. [Google Scholar]
- Andersen, M.; Norgaard-Pedersen, D.; Brandt, J.; Pettersson, I.; Slaaby, R. IGF1 and IGF2 specificities to the two insulin receptor isoforms are determined by insulin receptor amino acid 718. PLoS ONE 2017, 12, e0178885. [Google Scholar] [CrossRef]
- Backer, J.M.; Kahn, C.R.; White, M.F. Tyrosine phosphorylation of the insulin receptor is not required for receptor internalization: Studies in 2,4-dinitrophenol-treated cells. Proc. Natl. Acad. Sci. USA 1989, 86, 3209–3213. [Google Scholar] [CrossRef]
- Carpentier, J.L. Insulin receptor internalization: Molecular mechanisms and physiopathological implications. Diabetologia 1994, 37, S117–S124. [Google Scholar] [CrossRef]
- Trischitta, V.; Reaven, G.M. Evidence of a defect in insulin-receptor recycling in adipocytes from older rats. Am. J. Physiol. 1988, 254, E39–E44. [Google Scholar] [CrossRef] [PubMed]
- Podlecki, D.A.; Smith, R.M.; Kao, M.; Tsai, P.; Huecksteadt, T.; Brandenburg, D.; Lasher, R.S.; Jarett, L.; Olefsky, J.M. Nuclear translocation of the insulin receptor. A possible mediator of insulin’s long term effects. J. Biol. Chem. 1987, 262, 3362–3368. [Google Scholar] [PubMed]
- Trischitta, V.; Wong, K.Y.; Brunetti, A.; Scalisi, R.; Vigneri, R.; Goldfine, I.D. Endocytosis, recycling, and degradation of the insulin receptor. Studies with monoclonal antireceptor antibodies that do not activate receptor kinase. J. Biol. Chem. 1989, 264, 5041–5046. [Google Scholar] [PubMed]
- Razani, B.; Combs, T.P.; Wang, X.B.; Frank, P.G.; Park, D.S.; Russell, R.G.; Li, M.; Tang, B.; Jelicks, L.A.; Scherer, P.E.; et al. Caveolin-1-deficient mice are lean, resistant to diet-induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. J. Biol. Chem. 2002, 277, 8635–8647. [Google Scholar] [CrossRef] [PubMed]
- Jose, M.; Biosca, J.A.; Trujillo, R.; Itarte, E. Characterization of the hepatic insulin receptor undergoing internalization through clathrin-coated vesicles and endosomes. FEBS Lett. 1993, 334, 286–288. [Google Scholar] [CrossRef] [Green Version]
- Fagerholm, S.; Ortegren, U.; Karlsson, M.; Ruishalme, I.; Stralfors, P. Rapid insulin-dependent endocytosis of the insulin receptor by caveolae in primary adipocytes. PLoS ONE 2009, 4, e5985. [Google Scholar] [CrossRef]
- Elkin, S.R.; Lakoduk, A.M.; Schmid, S.L. Endocytic pathways and endosomal trafficking: A primer. Wien. Med. Wochenschr. 2016, 166, 196–204. [Google Scholar] [CrossRef]
- Pearse, B.M.; Smith, C.J.; Owen, D.J. Clathrin coat construction in endocytosis. Curr. Opin. Struct. Biol. 2000, 10, 220–228. [Google Scholar] [CrossRef]
- Traub, L.M.; Bonifacino, J.S. Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harb. Perspect. Biol. 2013, 5, a016790. [Google Scholar] [CrossRef]
- Carpentier, J.L.; Paccaud, J.P.; Backer, J.; Gilbert, A.; Orci, L.; Kahn, C.R.; Baecker, J. Two steps of insulin receptor internalization depend on different domains of the beta-subunit. J. Cell. Biol. 1993, 122, 1243–1252. [Google Scholar] [CrossRef]
- Backer, J.M.; Kahn, C.R.; Cahill, D.A.; Ullrich, A.; White, M.F. Receptor-mediated internalization of insulin requires a 12-amino acid sequence in the juxtamembrane region of the insulin receptor beta-subunit. J. Biol. Chem. 1990, 265, 16450–16454. [Google Scholar] [PubMed]
- Backer, J.M.; Shoelson, S.E.; Weiss, M.A.; Hua, Q.X.; Cheatham, R.B.; Haring, E.; Cahill, D.C.; White, M.F. The insulin receptor juxtamembrane region contains two independent tyrosine/beta-turn internalization signals. J. Cell Biol. 1992, 118, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.; Zhang, X.; Xing, C.; Yu, H. Mitotic Checkpoint Regulators Control Insulin Signaling and Metabolic Homeostasis. Cell 2016, 166, 567–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, L.T.; Lin, C.L.; Tzen, K.Y.; Chang, S.C.; Chang, M.F. LMBD1 protein serves as a specific adaptor for insulin receptor internalization. J. Biol. Chem. 2013, 288, 32424–32432. [Google Scholar] [CrossRef] [PubMed]
- Casas, S.; Casini, P.; Piquer, S.; Altirriba, J.; Soty, M.; Cadavez, L.; Gomis, R.; Novials, A. BACE2 plays a role in the insulin receptor trafficking in pancreatic ss-cells. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E1087–E1095. [Google Scholar] [CrossRef]
- Taghibiglou, C.; Bradley, C.A.; Gaertner, T.; Li, Y.; Wang, Y.; Wang, Y.T. Mechanisms involved in cholesterol-induced neuronal insulin resistance. Neuropharmacology 2009, 57, 268–276. [Google Scholar] [CrossRef]
- Stralfors, P. Caveolins and caveolae, roles in insulin signaling and diabetes. Adv. Exp. Med. Biol. 2012, 729, 111–126. [Google Scholar] [CrossRef]
- Kimura, A.; Mora, S.; Shigematsu, S.; Pessin, J.E.; Saltiel, A.R. The insulin receptor catalyzes the tyrosine phosphorylation of caveolin-1. J. Biol. Chem. 2002, 277, 30153–30158. [Google Scholar] [CrossRef]
- Kwon, H.; Lee, J.; Jang, D.; Choi, M.; Jeong, K.; Pak, Y. Alternative translation initiation of Caveolin-2 desensitizes insulin signaling through dephosphorylation of insulin receptor by PTP1B and causes insulin resistance. Biochim. Biophys. Acta 2018. [Google Scholar] [CrossRef]
- Gustavsson, J.; Parpal, S.; Karlsson, M.; Ramsing, C.; Thorn, H.; Borg, M.; Lindroth, M.; Peterson, K.H.; Magnusson, K.E.; Stralfors, P. Localization of the insulin receptor in caveolae of adipocyte plasma membrane. FASEB J. 1999, 13, 1961–1971. [Google Scholar] [CrossRef] [Green Version]
- Vainio, S.; Bykov, I.; Hermansson, M.; Jokitalo, E.; Somerharju, P.; Ikonen, E. Defective insulin receptor activation and altered lipid rafts in Niemann-Pick type C disease hepatocytes. Biochem. J. 2005, 391, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Wandelmer, J.; Davalos, A.; Herrera, E.; Giera, M.; Cano, S.; de la Pena, G.; Lasuncion, M.A.; Busto, R. Inhibition of cholesterol biosynthesis disrupts lipid raft/caveolae and affects insulin receptor activation in 3T3-L1 preadipocytes. Biochim. Biophys. Acta 2009, 1788, 1731–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aerts, J.M.; Boot, R.G.; van Eijk, M.; Groener, J.; Bijl, N.; Lombardo, E.; Bietrix, F.M.; Dekker, N.; Groen, A.K.; Ottenhoff, R.; et al. Glycosphingolipids and insulin resistance. Adv. Exp. Med. Biol. 2011, 721, 99–119. [Google Scholar] [CrossRef] [PubMed]
- Kabayama, K.; Sato, T.; Kitamura, F.; Uemura, S.; Kang, B.W.; Igarashi, Y.; Inokuchi, J. TNFalpha-induced insulin resistance in adipocytes as a membrane microdomain disorder: Involvement of ganglioside GM3. Glycobiology 2005, 15, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, D.J.; Diakanastasis, B.; Stockli, J.; Schmitz-Peiffer, C. Protein kinase Cepsilon modulates insulin receptor localization and trafficking in mouse embryonic fibroblasts. PLoS ONE 2013, 8, e58046. [Google Scholar] [CrossRef] [PubMed]
- Hunker, C.M.; Kruk, I.; Hall, J.; Giambini, H.; Veisaga, M.L.; Barbieri, M.A. Role of Rab5 in insulin receptor-mediated endocytosis and signaling. Arch. Biochem. Biophys. 2006, 449, 130–142. [Google Scholar] [CrossRef]
- Boura-Halfon, S.; Voliovitch, H.; Feinstein, R.; Paz, K.; Zick, Y. Extracellular matrix proteins modulate endocytosis of the insulin receptor. J. Biol. Chem. 2003, 278, 16397–16404. [Google Scholar] [CrossRef]
- Goh, L.K.; Sorkin, A. Endocytosis of receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a017459. [Google Scholar] [CrossRef]
- Puertollano, R.; Bonifacino, J.S. Interactions of GGA3 with the ubiquitin sorting machinery. Nat. Cell Biol. 2004, 6, 244–251. [Google Scholar] [CrossRef]
- Teis, D.; Saksena, S.; Emr, S.D. SnapShot: The ESCRT machinery. Cell 2009, 137, 182. [Google Scholar] [CrossRef]
- Peschard, P.; Park, M. From Tpr-Met to Met, tumorigenesis and tubes. Oncogene 2007, 26, 1276–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eden, E.R.; White, I.J.; Tsapara, A.; Futter, C.E. Membrane contacts between endosomes and ER provide sites for PTP1B-epidermal growth factor receptor interaction. Nat. Cell Biol. 2010, 12, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Rosen, O.M.; Herrera, R.; Olowe, Y.; Petruzzelli, L.M.; Cobb, M.H. Phosphorylation activates the insulin receptor tyrosine protein kinase. Proc. Natl. Acad. Sci. USA 1983, 80, 3237–3240. [Google Scholar] [CrossRef] [PubMed]
- Drake, P.G.; Posner, B.I. Insulin receptor-associated protein tyrosine phosphatase(s): Role in insulin action. Mol. Cell Biochem. 1998, 182, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.N.; Baquiran, G.; Brule, C.; Burgess, J.; Foster, B.; Bergeron, J.J.; Posner, B.I. Internalization and activation of the rat liver insulin receptor kinase in vivo. J. Biol. Chem. 1989, 264, 12931–12940. [Google Scholar] [PubMed]
- Mooney, R.A.; Green, D.A. Insulin receptor dephosphorylation in permeabilized adipocytes is inhibitable by manganese and independent of receptor kinase activity. Biochem. Biophys. Res. Commun. 1989, 162, 1200–1206. [Google Scholar] [CrossRef]
- Mooney, R.A.; Anderson, D.L. Phosphorylation of the insulin receptor in permeabilized adipocytes is coupled to a rapid dephosphorylation reaction. J. Biol. Chem. 1989, 264, 6850–6857. [Google Scholar] [PubMed]
- Backer, J.M.; Kahn, C.R.; White, M.F. Tyrosine phosphorylation of the insulin receptor during insulin-stimulated internalization in rat hepatoma cells. J. Biol. Chem. 1989, 264, 1694–1701. [Google Scholar]
- Burgess, J.W.; Wada, I.; Ling, N.; Khan, M.N.; Bergeron, J.J.; Posner, B.I. Decrease in beta-subunit phosphotyrosine correlates with internalization and activation of the endosomal insulin receptor kinase. J. Biol. Chem. 1992, 267, 10077–10086. [Google Scholar]
- Drake, P.G.; Bevan, A.P.; Burgess, J.W.; Bergeron, J.J.; Posner, B.I. A role for tyrosine phosphorylation in both activation and inhibition of the insulin receptor tyrosine kinase in vivo. Endocrinology 1996, 137, 4960–4968. [Google Scholar] [CrossRef]
- Starks, R.D.; Beyer, A.M.; Guo, D.F.; Boland, L.; Zhang, Q.; Sheffield, V.C.; Rahmouni, K. Regulation of Insulin Receptor Trafficking by Bardet Biedl Syndrome Proteins. PLoS Genet. 2015, 11, e1005311. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Smith, B.J.; Pillay, T.S. The APS adapter protein couples the insulin receptor to the phosphorylation of c-Cbl and facilitates ligand-stimulated ubiquitination of the insulin receptor. FEBS Lett. 2000, 475, 31–34. [Google Scholar] [CrossRef]
- Kishi, K.; Mawatari, K.; Sakai-Wakamatsu, K.; Yuasa, T.; Wang, M.; Ogura-Sawa, M.; Nakaya, Y.; Hatakeyama, S.; Ebina, Y. APS-mediated ubiquitination of the insulin receptor enhances its internalization, but does not induce its degradation. Endocr. J. 2007, 54, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Morcavallo, A.; Genua, M.; Palummo, A.; Kletvikova, E.; Jiracek, J.; Brzozowski, A.M.; Iozzo, R.V.; Belfiore, A.; Morrione, A. Insulin and insulin-like growth factor II differentially regulate endocytic sorting and stability of insulin receptor isoform A. J. Biol. Chem. 2012, 287, 11422–11436. [Google Scholar] [CrossRef] [PubMed]
- Del Prato, S.; Tiengo, A. The importance of first-phase insulin secretion: Implications for the therapy of type 2 diabetes mellitus. Diabetes Metab. Res. Rev. 2001, 17, 164–174. [Google Scholar] [CrossRef]
- Fehlmann, M.; Carpentier, J.L.; Van Obberghen, E.; Freychet, P.; Thamm, P.; Saunders, D.; Brandenburg, D.; Orci, L. Internalized insulin receptors are recycled to the cell surface in rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 5921–5925. [Google Scholar] [CrossRef] [PubMed]
- David, R. Endocytosis: Sorting the recycling. Nat. Rev. Mol. Cell Biol. 2011, 12, 3. [Google Scholar] [CrossRef]
- Li, G.; Marlin, M.C. Rab family of GTPases. Methods Mol. Biol. 2015, 1298, 1–15. [Google Scholar] [CrossRef]
- Bergqvist, N.; Nyman, E.; Cedersund, G.; Stenkula, K.G. A systems biology analysis connects insulin receptor signaling with glucose transporter translocation in rat adipocytes. J. Biol. Chem. 2017, 292, 11206–11217. [Google Scholar] [CrossRef] [Green Version]
- Worby, C.A.; Dixon, J.E. Sorting out the cellular functions of sorting nexins. Nat. Rev. Mol. Cell Biol. 2002, 3, 919–931. [Google Scholar] [CrossRef]
- Haft, C.R.; de la Luz Sierra, M.; Barr, V.A.; Haft, D.H.; Taylor, S.I. Identification of a family of sorting nexin molecules and characterization of their association with receptors. Mol. Cell Biol. 1998, 18, 7278–7287. [Google Scholar] [CrossRef] [PubMed]
- MaCaulay, S.L.; Stoichevska, V.; Grusovin, J.; Gough, K.H.; Castelli, L.A.; Ward, C.W. Insulin stimulates movement of sorting nexin 9 between cellular compartments: A putative role mediating cell surface receptor expression and insulin action. Biochem. J. 2003, 376, 123–134. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, K.M.; Ayllon, V.; O’Keeffe, J.; Wang, Y.; Cox, O.T.; Loughran, G.; Forgac, M.; O’Connor, R. Heme-binding protein HRG-1 is induced by insulin-like growth factor I and associates with the vacuolar H+-ATPase to control endosomal pH and receptor trafficking. J. Biol. Chem. 2010, 285, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Van Putten, J.P.; Wieringa, T.; Krans, H.M. Low pH and ketoacids induce insulin receptor binding and postbinding alterations in cultured 3T3 adipocytes. Diabetes 1985, 34, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Roux, A.; Loranger, A.; Lavoie, J.N.; Marceau, N. Keratin 8/18 regulation of insulin receptor signaling and trafficking in hepatocytes through a concerted phosphoinositide-dependent Akt and Rab5 modulation. FASEB J. 2017, 31, 3555–3573. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, T.B.; Troche, C.; Kim, M.H.; Cousins, R.J. Hepatic ZIP14-mediated Zinc Transport Contributes to Endosomal Insulin Receptor Trafficking and Glucose Metabolism. J. Biol. Chem. 2016, 291, 23939–23951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polidori, D.C.; Bergman, R.N.; Chung, S.T.; Sumner, A.E. Hepatic and Extrahepatic Insulin Clearance Are Differentially Regulated: Results From a Novel Model-Based Analysis of Intravenous Glucose Tolerance Data. Diabetes 2016, 65, 1556–1564. [Google Scholar] [CrossRef] [Green Version]
- Frank, H.J.; Donohoe, M.T.; Morris, W.L. Effect of glyburide on in vivo recycling of the hepatic insulin receptor. Am. J. Med. 1985, 79, 53–58. [Google Scholar] [CrossRef]
- Chvatchko, Y.; Van Obberghen, E.; Fehlmann, M. Internalization and recycling of insulin receptors in hepatoma cells. Absence of regulation by receptor occupancy. Biochem. J. 1984, 222, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Carpentier, J.L.; Gazzano, H.; Van Obberghen, E.; Fehlmann, M.; Freychet, P.; Orci, L. Internalization and recycling of 125I-photoreactive insulin-receptor complexes in hepatocytes in primary culture. Mol. Cell Endocrinol. 1986, 47, 243–255. [Google Scholar] [CrossRef]
- Soubigou, P.; Pringault, E.; Plas, C. Cell-surface insulin receptor cycling and its implication in the glycogenic response in cultured foetal hepatocytes. Biochem. J. 1986, 239, 609–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, J.; Hammond, V.A.; Taylor, R.; Alberti, K.G. Effects of monensin on insulin interactions with isolated hepatocytes. Evidence for inhibition of receptor recycling and insulin degradation. Biochem. J. 1986, 234, 463–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, S.; Olefsky, J.M. Separate intracellular pathways for insulin receptor recycling and insulin degradation in isolated rat adipocytes. J. Cell Physiol. 1983, 117, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S. Kinetics of insulin receptor internalization and recycling in adipocytes. Shunting of receptors to a degradative pathway by inhibitors of recycling. J. Biol. Chem. 1985, 260, 4136–4144. [Google Scholar] [PubMed]
- Arsenis, G.; Hayes, G.R.; Livingston, J.N. Insulin receptor cycling and insulin action in the rat adipocyte. J. Biol. Chem. 1985, 260, 2202–2207. [Google Scholar] [PubMed]
- Sato, H.; Terasaki, T.; Mizuguchi, H.; Okumura, K.; Tsuji, A. Receptor-recycling model of clearance and distribution of insulin in the perfused mouse liver. Diabetologia 1991, 34, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, A.; Krolenko, S.; Kudrjavtceva, N.; Lazebnik, J.; Teslenko, L.; Soderquist, A.M.; Nikolsky, N. Recycling of epidermal growth factor-receptor complexes in A431 cells: Identification of dual pathways. J. Cell Biol. 1991, 112, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanyaloglu, A.C.; von Zastrow, M. A novel sorting sequence in the beta2-adrenergic receptor switches recycling from default to the Hrs-dependent mechanism. J. Biol. Chem. 2007, 282, 3095–3104. [Google Scholar] [CrossRef] [PubMed]
- Desbuquois, B.; Lopez, S.; Janicot, M.; Burlet, H.; de Galle, B.; Fouque, F. Role of acidic subcellular compartments in the degradation of internalized insulin and in the recycling of the internalized insulin receptor in liver cells: In vivo and in vitro studies. Diabete Metab. 1992, 18, 104–112. [Google Scholar] [PubMed]
- Cromlish, W.A.; Tang, M.; Kyskan, R.; Tran, L.; Kennedy, B.P. PTP1B-dependent insulin receptor phosphorylation/residency in the endocytic recycling compartment of CHO-IR cells. Biochem. Pharmacol. 2006, 72, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Kesten, D.; Horovitz-Fried, M.; Brutman-Barazani, T.; Sampson, S.R. Insulin-induced translocation of IR to the nucleus in insulin responsive cells requires a nuclear translocation sequence. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 551–559. [Google Scholar] [CrossRef]
- Sehat, B.; Tofigh, A.; Lin, Y.; Trocme, E.; Liljedahl, U.; Lagergren, J.; Larsson, O. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Sci. Signal. 2010, 3, ra10. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Lin, Y.; Badin, M.; Vasilcanu, D.; Stromberg, T.; Jernberg-Wiklund, H.; Sehat, B.; Larsson, O. Over-accumulation of nuclear IGF-1 receptor in tumor cells requires elevated expression of the receptor and the SUMO-conjugating enzyme Ubc9. Biochem. Biophys. Res. Commun. 2011, 404, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, J.; Fang, Q.; Liu, M.; Liu, X.; Jia, W.; Dong, L.Q.; Liu, F. Autophagy-mediated insulin receptor down-regulation contributes to endoplasmic reticulum stress-induced insulin resistance. Mol. Pharmacol. 2009, 76, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Grunberger, G.; Geiger, D.; Carpentier, J.L.; Robert, A.; Gorden, P. Receptor-mediated endocytosis of insulin: Inhibition of [125I]iodoinsulin internalization in insulin resistant diabetic states of man. Acta Endocrinol. (Copenh) 1989, 121, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Hamer, I.; Foti, M.; Emkey, R.; Cordier-Bussat, M.; Philippe, J.; De Meyts, P.; Maeder, C.; Kahn, C.R.; Carpentier, J.L. An arginine to cysteine(252) mutation in insulin receptors from a patient with severe insulin resistance inhibits receptor internalisation but preserves signaling events. Diabetologia 2002, 45, 657–667. [Google Scholar] [CrossRef]
- Huang, Z.; Liu, J.; Ng, K.; Wan, X.; Xu, L.; He, X.; Liao, Z.; Li, Y. Glimepiride treatment in a patient with type A insulin resistance syndrome due to a novel heterozygous missense mutation in the insulin receptor gene. J. Diabetes Investig. 2018. [Google Scholar] [CrossRef]
- Paprocki, E.; Barral, R.L.; Vanden Brink, H.; Lujan, M.; Burgert, T.S. GnRH Agonist Improves Hyperandrogenism in an Adolescent Girl With an Insulin Receptor Gene Mutation. J. Endocr. Soc. 2019, 3, 1196–1200. [Google Scholar] [CrossRef]
- Iwanishi, M.; Kusakabe, T.; Azuma, C.; Tezuka, Y.; Yamamoto, Y.; Ito-Kobayashi, J.; Washiyama, M.; Morimoto, M.; Ebihara, K. Clinical characteristics in two patients with partial lipodystrophy and Type A insulin resistance syndrome due to a novel heterozygous missense mutation in the insulin receptor gene. Diabetes Res. Clin. Pract. 2019, 152, 79–87. [Google Scholar] [CrossRef]
- Krishnamurthy, M.; Pingul, M.M. A novel insulin receptor mutation in an adolescent with acanthosis nigricans and hyperandrogenism. J. Pediatr. Endocrinol. Metab. 2016, 29, 1201–1205. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.; Smith, G.; Isaac, I.; Hutchinson, D.; Semple, R.K. Novel mutation in insulin receptor gene identified after muscle biopsy in a Niuean woman with severe insulin resistance. Diabet. Med. 2015, 32, e24–e28. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Garcia, A.; Martinez, R.; Urrutia, I.; Garin, I.; Castano, L. Identification of a novel insulin receptor gene heterozygous mutation in a patient with type A insulin resistance syndrome. J. Pediatr. Endocrinol. Metab. 2014, 27, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S.; Zou, M.; Baitei, E.Y.; Parhar, R.S.; Al-Kahtani, N.; Raef, H.; Almahfouz, A.; Amartey, J.K.; Al-Rijjal, R.; Hammami, R.; et al. Molecular characterization of a novel p.R118C mutation in the insulin receptor gene from patients with severe insulin resistance. Clin. Endocrinol. (Oxf) 2012, 76, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Li, Y.; Tang, T.; Xu, W.; Liao, Z.; Yao, B.; Hu, G.; Weng, J. Hyperinsulinaemic hypoglycaemia associated with a heterozygous missense mutation of R1174W in the insulin receptor (IR) gene. Clin. Endocrinol. (Oxf) 2009, 71, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Innaurato, S.; Brierley, G.V.; Grasso, V.; Massimi, A.; Gaudino, R.; Sileno, S.; Bernardini, S.; Semple, R.; Barbetti, F. Severe insulin resistance in disguise: A familial case of reactive hypoglycemia associated with a novel heterozygous INSR mutation. Pediatr. Diabetes 2018, 19, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Carmody, D.; Ladsaria, S.S.; Buikema, R.K.; Semple, R.K.; Greeley, S.A. Successful rhIGF1 treatment for over 5 years in a patient with severe insulin resistance due to homozygous insulin receptor mutation. Diabet. Med. 2016, 33, e8–e12. [Google Scholar] [CrossRef] [PubMed]
- Tuhan, H.; Ceylaner, S.; Nalbantoglu, O.; Acar, S.; Abaci, A.; Bober, E.; Demir, K. A Mutation in INSR in a Child Presenting with Severe Acanthosis Nigricans. J. Clin. Res. Pediatr. Endocrinol. 2017, 9, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, I.; Yamada, Y.; Kadowaki, H.; Horikoshi, M.; Kadowaki, T.; Narita, T.; Tsuchida, S.; Noguchi, A.; Koizumi, A.; Takahashi, T. Phenotypical variety of insulin resistance in a family with a novel mutation of the insulin receptor gene. Endocr. J. 2010, 57, 509–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashiramoto, M.; Osawa, H.; Ando, M.; Murakami, A.; Nishimiya, T.; Nakano, M.; Nishida, W.; Onuma, H.; Makino, H. A nonsense mutation in the Arg345 of the insulin receptor gene in a Japanese type A insulin-resistant patient. Endocr. J. 2005, 52, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Kirel, B.; Bozdag, O.; Kosger, P.; Aydogdu, S.D.; Alincak, E.; Tekin, N. A case of Donohue syndrome “Leprechaunism” with a novel mutation in the insulin receptor gene. Turk. Pediatri. Ars. 2017, 52, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Hovnik, T.; Bratanic, N.; Podkrajsek, K.T.; Kovac, J.; Paro, D.; Podnar, T.; Bratina, N.; Battelino, T. Severe progressive obstructive cardiomyopathy and renal tubular dysfunction in Donohue syndrome with decreased insulin receptor autophosphorylation due to a novel INSR mutation. Eur. J. Pediatr. 2013, 172, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Kadowaki, H.; Momomura, K.; Fukushima, Y.; Orban, T.; Okai, T.; Taketani, Y.; Akanuma, Y.; Yazaki, Y.; Kadowaki, T. A homozygous kinase-defective mutation in the insulin receptor gene in a patient with leprechaunism. Diabetologia 1997, 40, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.; Li, X.; Hou, Q.; Wang, H.; Lou, G.; Li, T.; Wang, L.; Liu, H.; Li, X.; Liao, S. Novel heterozygous mutations of the INSR gene in a familial case of Donohue syndrome. Clin. Chim. Acta 2017, 473, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Azzabi, O.; Jilani, H.; Rejeb, I.; Siala, N.; Elaribi, Y.; Hizem, S.; Selmi, I.; Halioui, S.; Lascols, O.; Jemaa, L.B.; et al. Arg924X homozygous mutation in insulin receptor gene in a Tunisian patient with Donohue syndrome. J. Pediatr. Endocrinol. Metab. 2016, 29, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Nobile, S.; Semple, R.K.; Carnielli, V.P. A novel mutation of the insulin receptor gene in a preterm infant with Donohue syndrome and heart failure. J. Pediatr. Endocrinol. Metab. 2012, 25, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Kang, M.; Kim, J.H.; Cho, J.; Kim, G.H.; Yoo, H.W. Identification and Functional Characterization of Two Novel Nonsense Mutations in the beta-Subunit of INSR That Cause Severe Insulin Resistance Syndrome. Horm. Res. Paediatr. 2015, 84, 73–78. [Google Scholar] [CrossRef]
- Siala-Sahnoun, O.; Dhieb, D.; Ben Thabet, A.; Hmida, N.; Belguith, N.; Fakhfakh, F. First molecular diagnosis of Donohue syndrome in Africa: Novel unusual insertion/deletion mutation in the INSR gene. Mol. Biol. Rep. 2016, 43, 165–173. [Google Scholar] [CrossRef]
- Ben Abdelaziz, R.; Ben Chehida, A.; Azzouz, H.; Boudabbous, H.; Lascols, O.; Ben Turkia, H.; Tebib, N. A novel homozygous missense mutation in the insulin receptor gene results in an atypical presentation of Rabson-Mendenhall syndrome. Eur. J. Med. Genet. 2016, 59, 16–19. [Google Scholar] [CrossRef]
- Abe, Y.; Sato, T.; Takagi, M.; Watanabe, T.; Nagayama, Y.; Hasegawa, T.; Abe, T. A case of Rabson-Mendenhall syndrome with a novel mutation in the tyrosine kinase domain of the insulin receptor gene complicated by medullary sponge kidney. J. Pediatr. Endocrinol. Metab. 2012, 25, 587–590. [Google Scholar] [CrossRef]
- Tuthill, A.; Semple, R.K.; Day, R.; Soos, M.A.; Sweeney, E.; Seymour, P.J.; Didi, M.; O’Rahilly, S. Functional characterization of a novel insulin receptor mutation contributing to Rabson-Mendenhall syndrome. Clin. Endocrinol. (Oxf) 2007, 66, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, H.; Wu, B.; Dong, X.; Liu, B.; Chen, H.; Lu, Y.; Zhou, W.; Yang, L. One Novel 2.43Kb Deletion and One Single Nucleotide Mutation of the INSR Gene in a Chinese Neonate with Rabson-Mendenhall Syndrome. J. Clin. Res. Pediatr. Endocrinol. 2018, 10, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Bastaki, F.; Nair, P.; Mohamed, M.; Khadora, M.M.; Saif, F.; Tawfiq, N.; Al-Ali, M.T.; Hamzeh, A.R. Identification of a Novel Homozygous INSR Variant in a Patient with Rabson-Mendenhall Syndrome from the United Arab Emirates. Horm. Res. Paediatr. 2017, 87, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Raffan, E.; Soos, M.A.; Rocha, N.; Tuthill, A.; Thomsen, A.R.; Hyden, C.S.; Gregory, J.W.; Hindmarsh, P.; Dattani, M.; Cochran, E.; et al. Founder effect in the Horn of Africa for an insulin receptor mutation that may impair receptor recycling. Diabetologia 2011, 54, 1057–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hojlund, K.; Hansen, T.; Lajer, M.; Henriksen, J.E.; Levin, K.; Lindholm, J.; Pedersen, O.; Beck-Nielsen, H. A novel syndrome of autosomal-dominant hyperinsulinemic hypoglycemia linked to a mutation in the human insulin receptor gene. Diabetes 2004, 53, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Kuroda, Y.; Fukui, K.; Iwamoto, R.; Kozawa, J.; Watanabe, T.; Yamada, Y.; Imagawa, A.; Iwahashi, H.; Shimomura, I. Hyperinsulinemia and Insulin Receptor Gene Mutation in Nonobese Healthy Subjects in Japan. J. Endocr. Soc. 2017, 1, 1351–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorwerk, P.; Christoffersen, C.T.; Muller, J.; Vestergaard, H.; Pedersen, O.; De Meyts, P. Alternative splicing of exon 17 and a missense mutation in exon 20 of the insulin receptor gene in two brothers with a novel syndrome of insulin resistance (congenital fiber-type disproportion myopathy). Horm. Res. 1999, 52, 211–220. [Google Scholar] [CrossRef]
- Morgan, R.; Bishop, A.; Owens, D.R.; Luzio, S.D.; Peters, J.R.; Rees, A. Allelic variants at insulin-receptor and insulin gene loci and susceptibility to NIDDM in Welsh population. Diabetes 1990, 39, 1479–1484. [Google Scholar] [CrossRef]
- Zhu, A.N.; Yang, X.X.; Sun, M.Y.; Zhang, Z.X.; Li, M. Associations between INSR and MTOR polymorphisms in type 2 diabetes mellitus and diabetic nephropathy in a Northeast Chinese Han population. Genet. Mol. Res. 2015, 14, 1808–1818. [Google Scholar] [CrossRef]
- Bodhini, D.; Sandhiya, M.; Ghosh, S.; Majumder, P.P.; Rao, M.R.; Mohan, V.; Radha, V. Association of His1085His INSR gene polymorphism with type 2 diabetes in South Indians. Diabetes Technol. Ther. 2012, 14, 696–700. [Google Scholar] [CrossRef]
- Ardon, O.; Procter, M.; Tvrdik, T.; Longo, N.; Mao, R. Sequencing analysis of insulin receptor defects and detection of two novel mutations in INSR gene. Mol. Genet. Metab. Rep. 2014, 1, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Krook, A.; O’Rahilly, S. Homozygous mutation in the insulin receptor. Clin. Endocrinol. (Oxf) 1996, 45, 237–238. [Google Scholar] [CrossRef] [PubMed]
- Ben Harouch, S.; Klar, A.; Falik Zaccai, T.C. INSR-Related Severe Syndromic Insulin Resistance. In GeneReviews((R); Adam, M.P., Ardinger, H.H., Eds.; University of Washington: Seattle, WA, USA, 1993–2019; Available online: https://europepmc.org/abstract/med/29369573 (accessed on 25 January 2018).
- Krischer, J.; Gilbert, A.; Gorden, P.; Carpentier, J.L. Endocytosis is inhibited in hepatocytes from diabetic rats. Diabetes 1993, 42, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.W.; Hnasko, R.; Schubert, W.; Lisanti, M.P. Role of caveolae and caveolins in health and disease. Physiol. Rev. 2004, 84, 1341–1379. [Google Scholar] [CrossRef]
- Ikonen, E.; Vainio, S. Lipid microdomains and insulin resistance: Is there a connection? Sci. STKE 2005, 2005, pe3. [Google Scholar] [CrossRef]
- Veillon, L.; Go, S.; Matsuyama, W.; Suzuki, A.; Nagasaki, M.; Yatomi, Y.; Inokuchi, J. Identification of Ganglioside GM3 Molecular Species in Human Serum Associated with Risk Factors of Metabolic Syndrome. PLoS ONE 2015, 10, e0129645. [Google Scholar] [CrossRef]
- Lipina, C.; Hundal, H.S. Ganglioside GM3 as a gatekeeper of obesity-associated insulin resistance: Evidence and mechanisms. FEBS Lett. 2015, 589, 3221–3227. [Google Scholar] [CrossRef] [Green Version]
- Smahelova, A.; Hyspler, R.; Haas, T. Relation of cholesterol metabolism and non-cholesterol sterols to insulin resistance. Physiol. Res. 2007, 56, 749–755. [Google Scholar]
- Cohen, A.W.; Razani, B.; Wang, X.B.; Combs, T.P.; Williams, T.M.; Scherer, P.E.; Lisanti, M.P. Caveolin-1-deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am. J. Physiol. Cell Physiol. 2003, 285, C222–C235. [Google Scholar] [CrossRef] [Green Version]
- Palacios-Ortega, S.; Varela-Guruceaga, M.; Martinez, J.A.; de Miguel, C.; Milagro, F.I. Effects of high glucose on caveolin-1 and insulin signaling in 3T3-L1 adipocytes. Adipocyte 2016, 5, 65–80. [Google Scholar] [CrossRef]
- Ormazabal, P.; Romero, C.; Gabler, F.; Quest, A.F.; Vega, M. Decreased phosphorylation of Y14 caveolin-1 in endometrial tissue of polycystic ovary syndrome patients may be related with an insulin resistant state in this tissue. Horm. Metab. Res. 2013, 45, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Capozza, F.; Combs, T.P.; Cohen, A.W.; Cho, Y.R.; Park, S.Y.; Schubert, W.; Williams, T.M.; Brasaemle, D.L.; Jelicks, L.A.; Scherer, P.E.; et al. Caveolin-3 knockout mice show increased adiposity and whole body insulin resistance, with ligand-induced insulin receptor instability in skeletal muscle. Am. J. Physiol. Cell Physiol. 2005, 288, C1317–C1331. [Google Scholar] [CrossRef] [PubMed]
- Bertacca, A.; Ciccarone, A.; Cecchetti, P.; Vianello, B.; Laurenza, I.; Del Prato, S.; Benzi, L. High insulin levels impair intracellular receptor trafficking in human cultured myoblasts. Diabetes Res. Clin. Pract. 2007, 78, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, J.L.; Fehlmann, M.; Van Obberghen, E.; Gorden, P.; Orci, L. Insulin receptor internalization and recycling: Mechanism and significance. Biochimie 1985, 67, 1143–1145. [Google Scholar] [CrossRef]
- Livingston, J.N.; Saran, B.R.; Rose, C.D.; Anderson, C.L. Rapid effects of insulin on the cycling of the insulin receptor in a human monocyte cell line (U-937). Diabetes 1985, 34, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.I. Lilly Lecture: Molecular mechanisms of insulin resistance. Lessons from patients with mutations in the insulin-receptor gene. Diabetes 1992, 41, 1473–1490. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, H.; Kadowaki, T.; Cama, A.; Marcus-Samuels, B.; Rovira, A.; Bevins, C.L.; Taylor, S.I. Mutagenesis of lysine 460 in the human insulin receptor. Effects upon receptor recycling and cooperative interactions among binding sites. J. Biol. Chem. 1990, 265, 21285–21296. [Google Scholar]
- Trischitta, V.; Brunetti, A.; Chiavetta, A.; Benzi, L.; Papa, V.; Vigneri, R. Defects in insulin-receptor internalization and processing in monocytes of obese subjects and obese NIDDM patients. Diabetes 1989, 38, 1579–1584. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Kahn, C.R. Insulin signaling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef]
- Sesti, G.; D’Alfonso, R.; Vargas Punti, M.D.; Tullio, A.N.; Liu, Y.Y.; Federici, M.; Borboni, P.; Marini, M.A.; Lauro, R.; Fusco, A. Delayed intracellular dissociation of the insulin-receptor complex impairs receptor recycling and insulin processing in cultured Epstein-Barr virus-transformed lymphocytes from insulin-resistant subjects. Diabetologia 1996, 39, 289–295. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Condition | Study | Mutation | References |
|---|---|---|---|
| Type A Insulin Resistance Syndrome | Case Study | Missense Mutation | [88,89,90,91,92,93,94,95,96,97] |
| Case Study | Splice Site Mutation | [98,99] | |
| Case Study | Nonsense Mutation | [100,101] | |
| Donohue Syndrome (Leprechaunism) | Case Study | Missense Mutation | [102,103,104,105] |
| Case Study | Nonsense Mutation | [106,107,108] | |
| Regional Study | Frameshift Mutation | [109] | |
| Rabson–Mendenhall Syndrome | Case Study | Missense Mutation | [110,111,112,113,114] |
| Regional Study | Missense Mutation | [115] | |
| Familial Hyperinsulinemic Hypoglycemia-5 | Case Study | Missense Mutation | [116] |
| Asymptomatic Hyperinsulinemia | Cohort Study | Nonsense Mutation | [117] |
| Congenital Muscle Fiber-Type Disproportion Myopathy | Case Study | Missense Mutation | [118] |
| Non-Insulin Dependent Diabetes Mellitus | Cohort Study | Polymorphism | [119,120,121] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Huang, L.; Qi, X.; Chen, C. Insulin Receptor Trafficking: Consequences for Insulin Sensitivity and Diabetes. Int. J. Mol. Sci. 2019, 20, 5007. https://doi.org/10.3390/ijms20205007
Chen Y, Huang L, Qi X, Chen C. Insulin Receptor Trafficking: Consequences for Insulin Sensitivity and Diabetes. International Journal of Molecular Sciences. 2019; 20(20):5007. https://doi.org/10.3390/ijms20205007
Chicago/Turabian StyleChen, Yang, Lili Huang, Xinzhou Qi, and Chen Chen. 2019. "Insulin Receptor Trafficking: Consequences for Insulin Sensitivity and Diabetes" International Journal of Molecular Sciences 20, no. 20: 5007. https://doi.org/10.3390/ijms20205007