Folic Acid Antagonists: Antimicrobial and Immunomodulating Mechanisms and Applications



Abstract
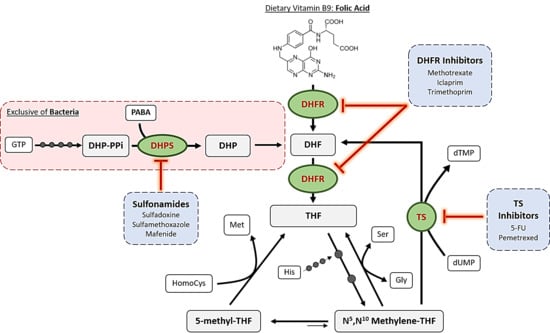
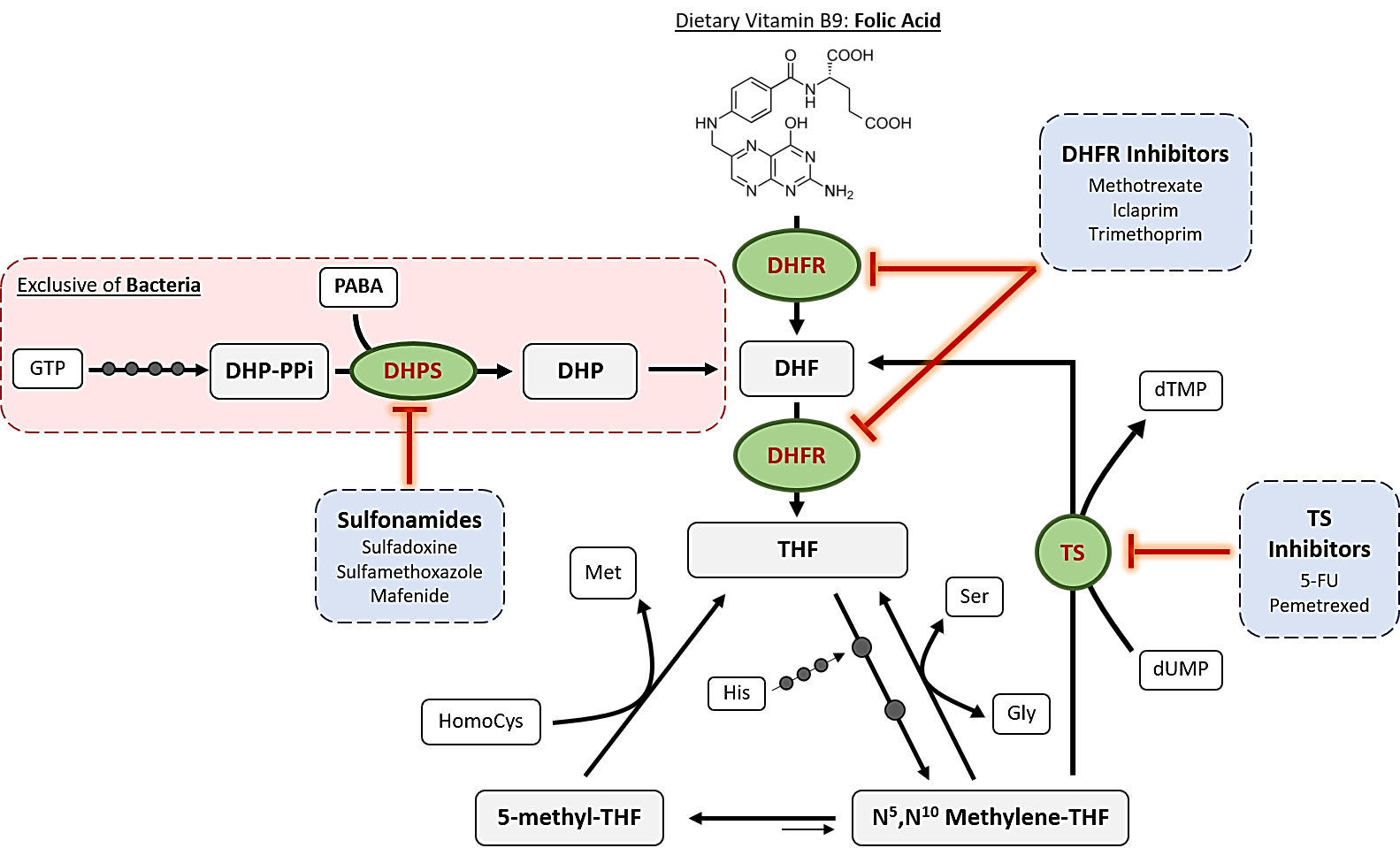
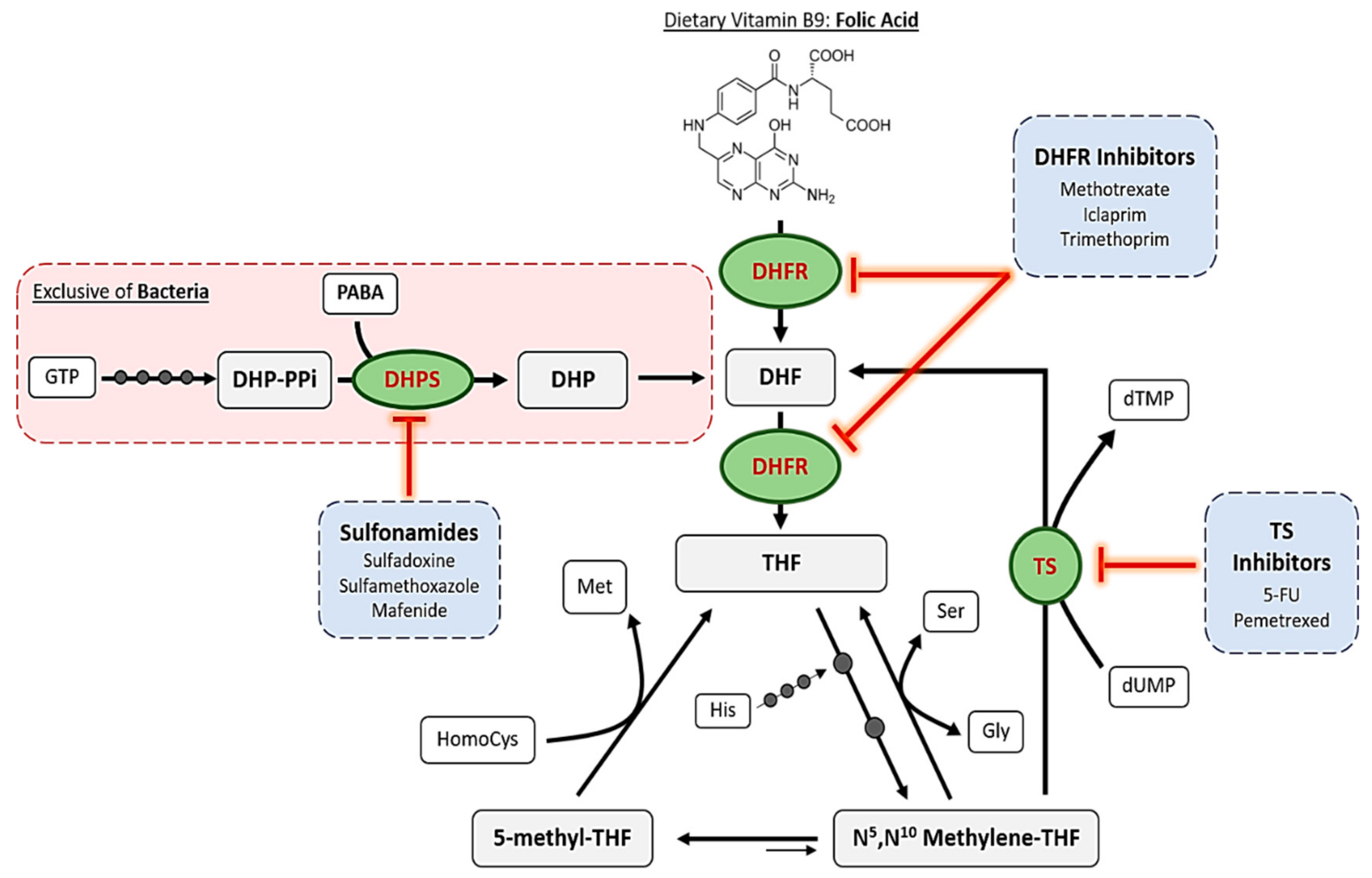
1. Introduction to Folic Acid Antagonists: Antifolates
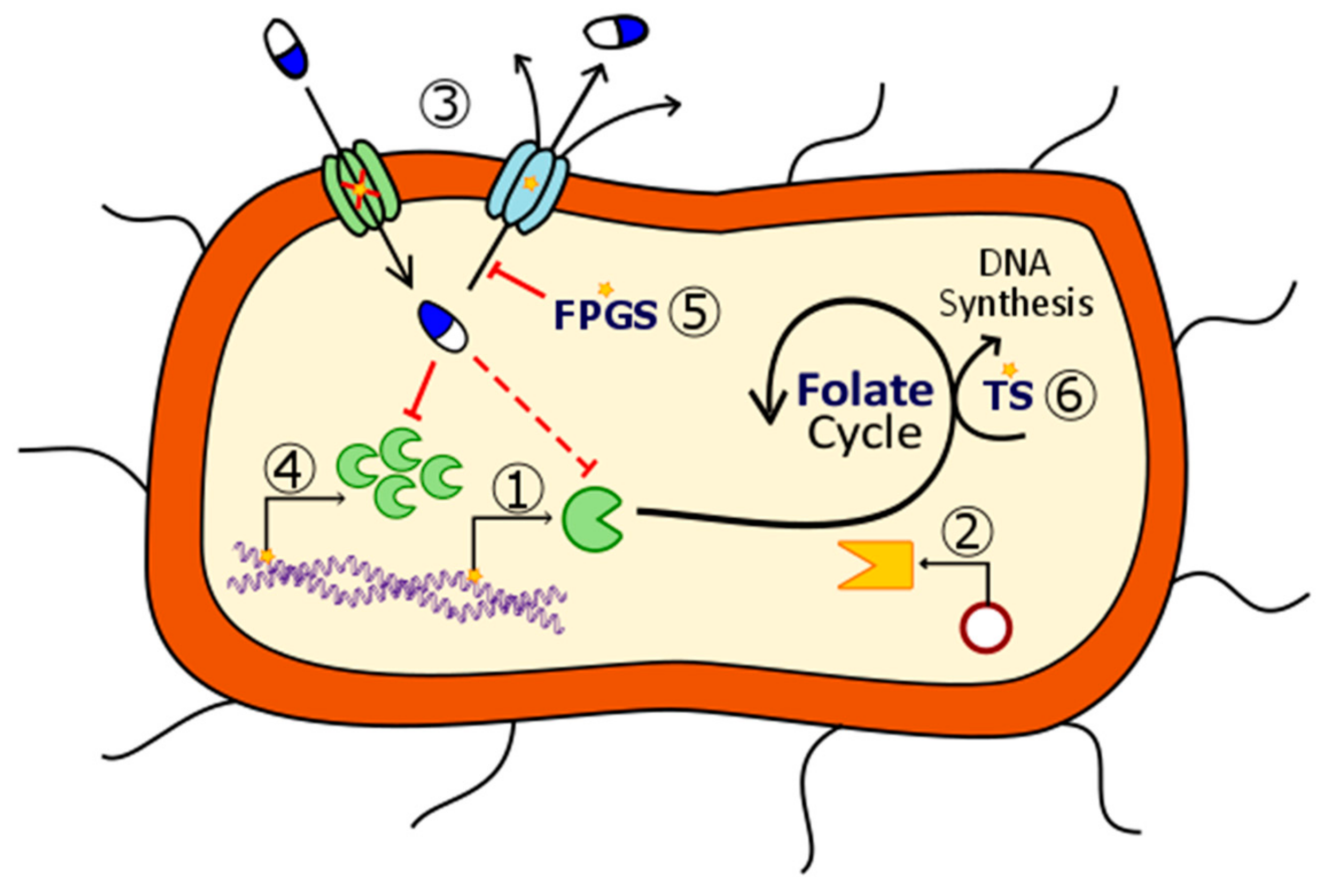
2. Mechanisms of Action of Antifolates
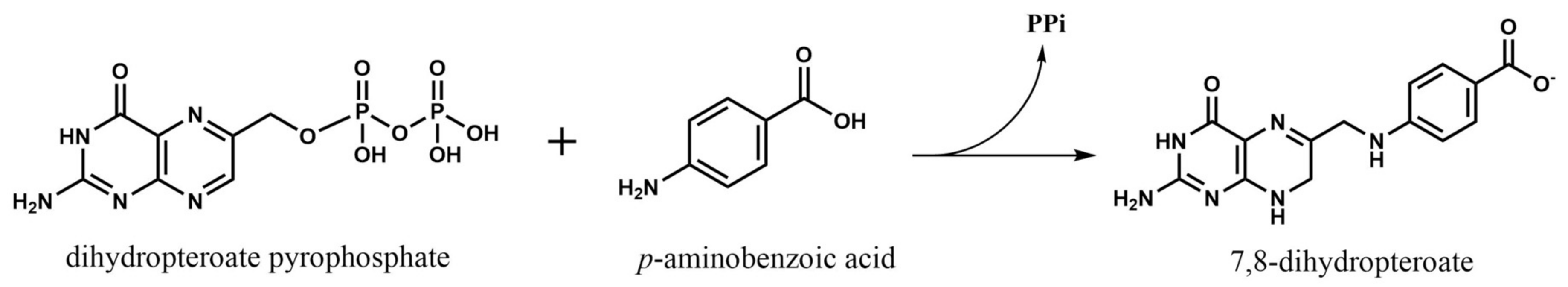
2.1. Dihydropteroate Synthase
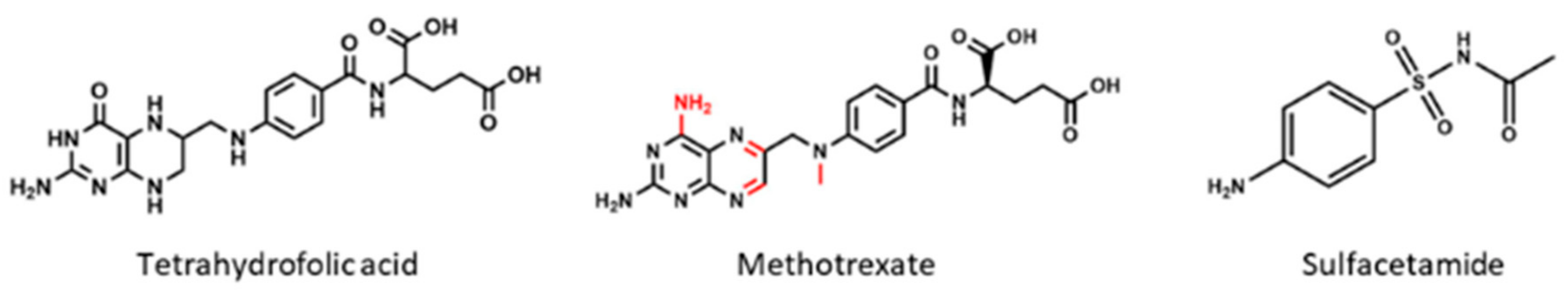
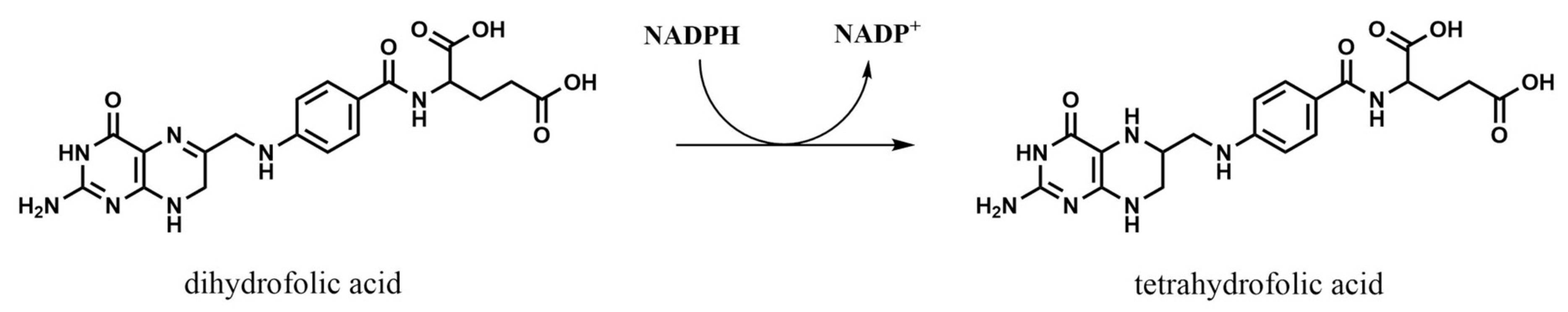
2.2. Dihydrofolate Reductase
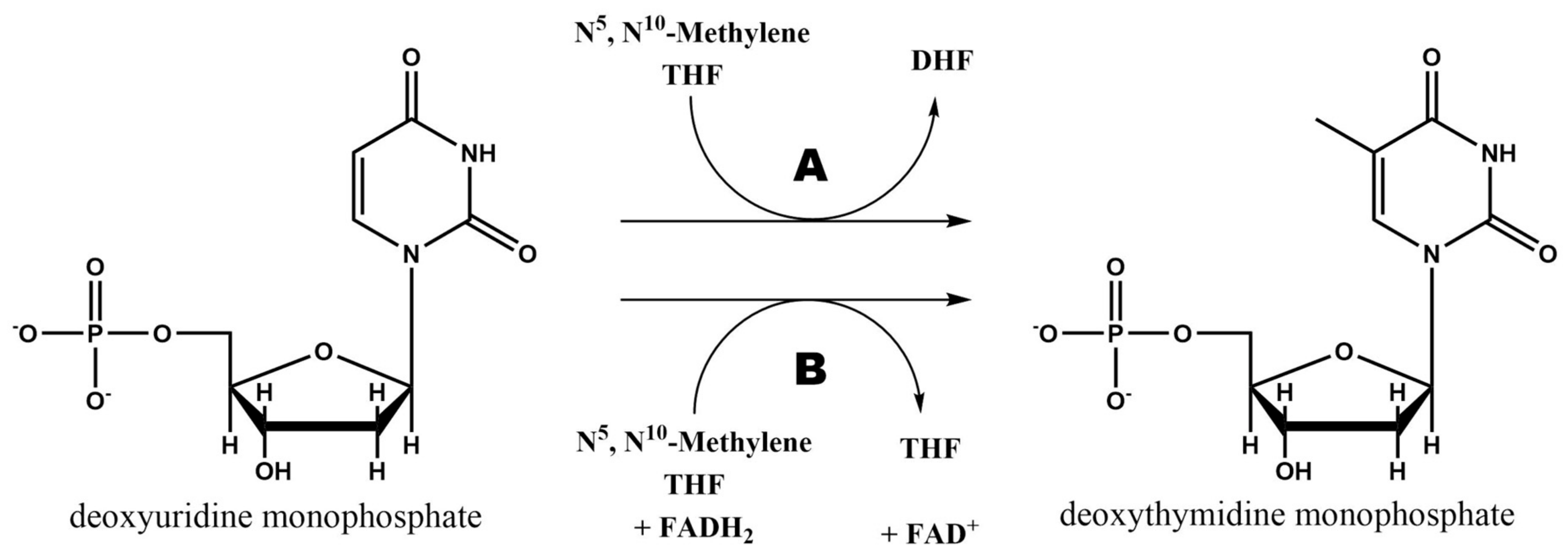
2.3. Thymidylate Synthase
2.4. Other Enzymes
2.4.1. Bifunctional DHFR-TS
2.4.2. Purine Synthetic Pathway
2.4.3. Folylpoly-γ-Glutamate Synthetase
3. Antifolates as Antibiotic Agents
3.1. Mafenide Acetate
3.2. Silver Sulfadiazine
3.3. Sulfadiazine
3.4. Sulfacetamide
3.5. Sulfisoxazole
3.6. Co-Trimoxazole: Trimethoprim and Sulfamethoxazole
3.7. Dapsone (Part I) as an Antibiotic
3.8. Antimalarial Antifolates: Proguanil and Pyrimethamine/Sulfadoxine
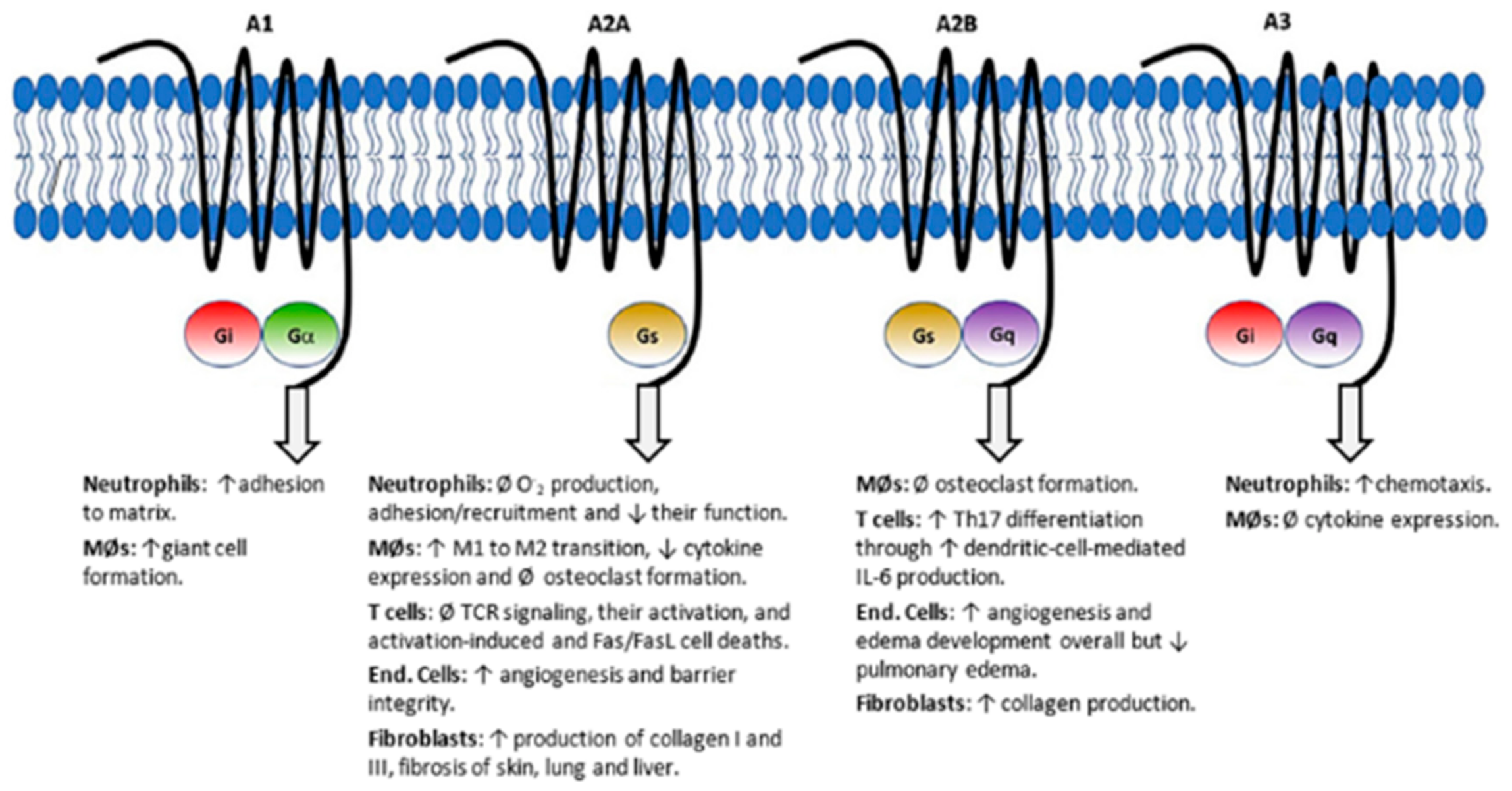
4. Antifolates as Immunomodulating Agents
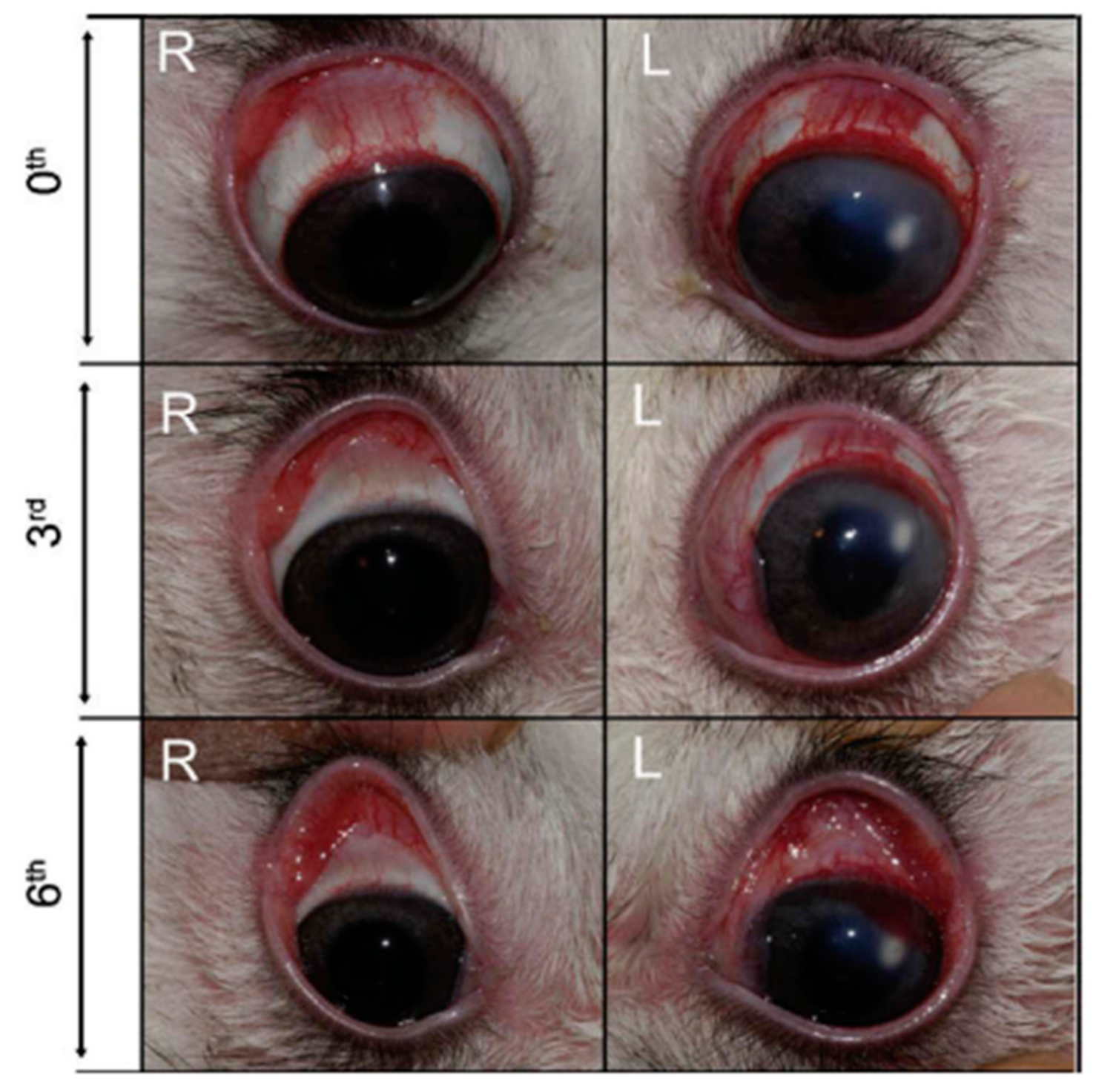
4.1. Dapsone (Part II) as an Immunomodulator
4.2. Methotrexate
4.3. Sulfasalazine
5. Development of Resistances to Antifolates
6. Current Experimental Compounds and Future Trends on Antifolates Research
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AICARFT | Aminoimidazole-4-carboxamide ribonucleotide formyltransferase |
| AIF | Apoptosis-inducing factor |
| ATIC | Aminoimidazole-4-carboxamide ribonucleotide formyltransferase/inosine monophosphate cyclohydrolase |
| DHF | Dihydrofolate |
| DHFS | Dihydrofolate synthase |
| DHFR | Dihydrofolate reductase |
| DHP-PPi | Dihydropteroate pyrophosphate |
| DHPS | Dihydropteroate synthase |
| DMARD | Disease-modifying antirheumatic drug |
| dUMP | 2′-deoxyuridine-5′-monophosphate |
| dTMP | 2′-deoxythymidine-5′-monophosphate |
| FDTS | Flavin-dependent thymidylate synthase |
| FPGS | Folylpoly-γ-glutamate synthetase |
| GARFT | Glycinamide ribonucleotide formyltransferase |
| GGH | γ-glutamyl hydrolase |
| GPCR | G-protein-coupled receptor |
| HEMA | 2-hydroxyethyl methacrylate |
| IL | Interleukin |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| PABA | p-aminobenzoic acid |
| PPi | Pyrophosphate |
| ROS | Reactive oxygen species |
| THF | Tetrahydrofolate |
| TNF-α | Tumor necrosis factor alpha |
| TRMs | Thymine-requiring mutants |
| TS | Thymidylate synthase |
References
- Zheng, Y.; Cantley, L.C. Toward a Better Understanding of Folate Metabolism in Health and Disease. J. Exp. Med. 2018. [Google Scholar] [CrossRef]
- Fowler, B. The Folate Cycle and Disease in Humans. Kidney Int. 2001, 59 (Suppl. 78), S221–S229. [Google Scholar] [CrossRef]
- Metz, J. Folic Acid Metabolism and Malaria. Food Nutr. Bull. 2007, 28 (Suppl. 4), 540–549. [Google Scholar] [CrossRef]
- Kompis, I.M.; Islam, K.; Then, R.L. DNA and RNA Synthesis: Antifolates. Chem. Rev. 2005, 105, 593–620. [Google Scholar] [CrossRef] [PubMed]
- Lucock, M. Folic Acid: Nutritional Biochemistry, Molecular Biology, and Role in Disease Processes. Mol. Genet. Metab. 2000, 71, 121–138. [Google Scholar] [CrossRef]
- Fernández-Villa, D.; Jiménez Gómez-Lavín, M.; Abradelo, C.; San Román, J.; Rojo, L. Tissue Engineering Therapies Based on Folic Acid and Other Vitamin B Derivatives. Functional Mechanisms and Current Applications in Regenerative Medicine. Int. J. Mol. Sci. 2018, 19, 4068. [Google Scholar] [CrossRef]
- Gao, Y.; Sheng, C.; Xie, R.; Sun, W.; Asztalos, E.; Moddemann, D.; Zwaigenbaum, L.; Walker, M.; Wen, S.W. New Perspective on Impact of Folic Acid Supplementation during Pregnancy on Neurodevelopment/Autism in the Offspring Children—A Systematic Review. PLoS ONE 2016, 11, e0165626. [Google Scholar] [CrossRef] [PubMed]
- Scocchera, E.; Wright, D.L. The Antifolates. In Antibacterials. Volume II; Fisher, J., Mobashery, S., Miller, M., Eds.; Springer: Cham, Switzerland, 2017; Volume 26, pp. 123–149. [Google Scholar] [CrossRef]
- Goulian, M.; Bleile, B.M.; Dickey, L.M.; Grafstrom, R.H.; Ingraham, H.A.; Neynaber, S.A.; Peterson, M.S.; Tseng, B.Y. Mechanism of Thymineless Death. In Purine and Pyrimidine Metabolism in Man V. Advances in Experimental Medicine and Biology; Nyhan, W.L., Thompson, L.F., Watts, R.W.E., Eds.; Springer: New York, NY, USA, 1986; Volume 195B, pp. 89–95. [Google Scholar] [CrossRef]
- Vicente-Blázquez, A.; González, M.; Álvarez, R.; del Mazo, S.; Medarde, M.; Peláez, R. Antitubulin Sulfonamides: The Successful Combination of an Established Drug Class and a Multifaceted Target. Med. Res. Rev. 2019, 39, 775–830. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. The Properties of Known Drugs. 1. Molecular Frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Bemis, G.W.; Murcko, M.A. Properties of Known Drugs. 2. Side Chains. J. Med. Chem. 1999, 42, 5095–5099. [Google Scholar] [CrossRef] [PubMed]
- Apaydın, S.; Török, M. Sulfonamide Derivatives as Multi-Target Agents for Complex Diseases. Bioorg. Med. Chem. Lett. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gulçin, İ.; Taslimi, P. Sulfonamide Inhibitors: A Patent Review 2013-Present. Expert Opin. Ther. Pat. 2018, 28, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Chauhan, P.M.S. Dihydrofolate Reductase as a Therapeutic Target for Infectious Diseases: Opportunities and Challenges. Future Med. Chem. 2012, 4, 1335–1365. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.R.; Morrison, J.F. Mechanism of Inhibition of Dihydrofolate Reductases from Bacterial and Vertebrate Sources by Various Classes of Folate Analogues. Biochim. Biophys. Acta (BBA)/Protein Struct. Mol. 1986, 869, 275–285. [Google Scholar] [CrossRef]
- Raimondi, M.V.; Randazzo, O.; La Franca, M.; Barone, G.; Vignoni, E.; Rossi, D.; Collina, S. DHFR Inhibitors: Reading the Past for Discovering Novel Anticancer Agents. Molecules 2019, 24, 1140. [Google Scholar] [CrossRef]
- Koehn, E.M.; Perissinotti, L.L.; Moghram, S.; Prabhakar, A.; Lesley, S.A.; Mathews, I.I.; Kohen, A. Folate Binding Site of Flavin-Dependent Thymidylate Synthase. Proc. Natl. Acad. Sci. USA 2012, 109, 15722–15727. [Google Scholar] [CrossRef] [PubMed]
- Kögler, M.; Vanderhoydonck, B.; De Jonghe, S.; Rozenski, J.; Van Belle, K.; Herman, J.; Louat, T.; Parchina, A.; Sibley, C.; Lescrinier, E.; et al. Synthesis and Evaluation of 5-Substituted 2′-Deoxyuridine Monophosphate Analogues as Inhibitors of Flavin-Dependent Thymidylate Synthase in Mycobacterium Tuberculosis. J. Med. Chem. 2011, 54, 4847–4862. [Google Scholar] [CrossRef]
- Modranka, J.; Li, J.; Parchina, A.; Vanmeert, M.; Dumbre, S.; Salman, M.; Myllykallio, H.; Becker, H.F.; Vanhoutte, R.; Margamuljana, L.; et al. Synthesis and Structure–Activity Relationship Studies of Benzo[b][1,4]Oxazin-3(4H)-One Analogues as Inhibitors of Mycobacterial Thymidylate Synthase X. ChemMedChem 2019, 14, 645–662. [Google Scholar] [CrossRef]
- Negrya, S.D.; Efremenkova, O.V.; Solyev, P.N.; Chekhov, V.O.; Ivanov, M.A.; Sumarukova, I.G.; Karpenko, I.L.; Kochetkov, S.N.; Alexandrova, L.A. Novel 5-Substituted Derivatives of 2′-Deoxy-6-Azauridine with Antibacterial Activity. J. Antibiot. (Tokyo) 2019, 72, 535–544. [Google Scholar] [CrossRef]
- Yuvaniyama, J.; Chitnumsub, P.; Kamchonwongpaisan, S.; Vanichtanankul, J.; Sirawaraporn, W.; Taylor, P.; Walkinshaw, M.D.; Yuthavong, Y. Insights into Antifolate Resistance from Malarial DHFR-TS Structures. Nat. Struct. Biol. 2003, 10, 357–365. [Google Scholar] [CrossRef]
- Aboge, G.O.; Jia, H.; Terkawi, M.; Goo, Y.-K.; Batbaatar, V.; Nishikawa, Y.; Sunaga, F.; Namikawa, K.; Igarashi, I.; Fujisaki, K.; et al. Precursors of Methotrexate Target Dihydrofolate Reductase-Thymidylate Synthase of Babesia Gibsoni and Inhibit Parasite Proliferation. J. Protozool. Res. 2010, 20, 70–81. [Google Scholar]
- Baggott, J.E.; Tamura, T. Folate-Dependent Purine Nucleotide Biosynthesis in Humans. Adv. Nutr. 2015, 6, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Fales, K.R.; Njoroge, F.G.; Brooks, H.B.; Thibodeaux, S.; Torrado, A.; Si, C.; Toth, J.L.; Mc Cowan, J.R.; Roth, K.D.; Thrasher, K.J.; et al. Discovery of N-(6-Fluoro-1-Oxo-1,2-Dihydroisoquinolin-7-Yl)-5-[(3R)-3-Hydroxypyrrolidin-1-Yl]Thiophene-2-Sulfonamide (LSN 3213128), a Potent and Selective Nonclassical Antifolate Aminoimidazole-4-Carboxamide Ribonucleotide Formyltransferase (AICARFT) Inhi. J. Med. Chem. 2017, 60, 9599–9616. [Google Scholar] [CrossRef] [PubMed]
- Brooks, H.B.; Meier, T.I.; Geeganage, S.; Fales, K.R.; Thrasher, K.J.; Konicek, S.A.; Spencer, C.D.; Thibodeaux, S.; Foreman, R.T.; Hui, Y.H.; et al. Characterization of a Novel AICARFT Inhibitor Which Potently Elevates ZMP and Has Anti-Tumor Activity in Murine Models. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Reyes, P.; Rathod, P.K.; Sanchez, D.J.; Mrema, J.E.K.; Rieckmann, K.H.; Heidrich, H. Enzymes of Purine and Pyrimidine Metabolism From the Human Malaria Parasite, Plasmodium Falciparum. Mol. Biochem. Parasitol. 1982, 5, 275–290. [Google Scholar] [CrossRef]
- Yamamoto, T.; Shikano, K.; Nanki, T.; Kawai, S. Folylpolyglutamate Synthase Is a Major Determinant of Intracellular Methotrexate Polyglutamates in Patients with Rheumatoid Arthritis. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Coward, J.K.; McGuire, J.J. Mechanism-Based Inhibitors of Folylpoly-γ-Glutamate Synthetase and γ-Glutamyl Hydrolase: Control of Folylpoly-γ-Glutamate Homeostasis as a Drug Target. Vitam. Horm. 2008, 79, 347–373. [Google Scholar] [CrossRef]
- Salcedo, E.; Cortese, J.F.; Plowe, C.V.; Sims, P.F.G.; Hyde, J.E. A Bifunctional Dihydrofolate Synthetase-Folylpolyglutamate Synthetase in Plasmodium Falciparum Identified by Functional Complementation in Yeast and Bacteria. Mol. Biochem. Parasitol. 2001, 112, 239–252. [Google Scholar] [CrossRef]
- Eagon, R.G.; Mcmanus, A.T. The Effect of Mafenide on Dihydropteroate Synthase. J. Antimicrob. Chemother. 1990, 25, 25–29. [Google Scholar] [CrossRef]
- Punjataewakupt, A.; Napavichayanun, S.; Aramwit, P. The Downside of Antimicrobial Agents for Wound Healing. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 39–54. [Google Scholar] [CrossRef]
- Greenhalgh, D.G. Topical Antimicrobial Agents for Burn Wounds. Clin. Plast. Surg. 2009, 36, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Heggers, J.P.; Chinkes, D.L.; Wolf, S.E.; Hawkins, H.K.; Wolfe, R.R. Topical Sulfamylon Cream Inhibits DNA and Protein Synthesis in the Skin Donor Site Wound. Surgery 2006, 139, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Glasser, J.S.; Guymon, C.H.; Mende, K.; Wolf, S.E.; Hospenthal, D.R.; Murray, C.K. Activity of Topical Antimicrobial Agents against Multidrug-Resistant Bacteria Recovered from Burn Patients. Burns 2010, 36, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.H.; Colon, N.C.; Herndon, D.N. Burns. Pediatr. Surg. 2012, 369–384. [Google Scholar] [CrossRef]
- Dai, T.; Huang, Y.Y.; Sharma, S.K.; Hashmi, J.T.; Kurup, D.B.; Hamblin, M.R. Topical Antimicrobials for Burn Wound Infections. Recent Pat. Antiinfect. Drug Discov. 2010, 5, 124–151. [Google Scholar] [CrossRef]
- European Chemicals Agency. Mafenide—Annex III inventory—ECHA. Available online: https://echa.europa.eu/information-on-chemicals/annex-iii-inventory/-/dislist/details/AIII-100.004.843 (accessed on 13 August 2019).
- Hashmi, D.L.; Haith, L. The Current State of Topical Burn Treatments: A Review. Curr. Trauma Rep. 2019, 160–168. [Google Scholar] [CrossRef]
- Nalbandi, B.; Amiri, S. Antibacterial Activity of PVA-Based Nanofibers Loaded with Silver Sulfadiazine/Cyclodextrin Nanocapsules. Int. J. Polym. Mater. Polym. Biomater. 2019, 68, 647–659. [Google Scholar] [CrossRef]
- Patel, K.K.; Surekha, D.B.; Tripathi, M.; Anjum, M.; Muthu, M.S.; Tilak, R.; Agrawal, A.K.; Singh, S. Antibiofilm Potential of Silver Sulfadiazine-Loaded Nanoparticle Formulations: A Study on the Effect of DNase-I on Microbial Biofilm and Wound Healing Activity. Mol Pharm. 2019. [Google Scholar] [CrossRef]
- Vigani, B.; Rossi, S.; Sandri, G.; Bonferoni, M.C.; Caramella, C.M.; Ferrari, F. Hyaluronic Acid and Chitosan-Based Nanosystems: A New Dressing Generation for Wound Care. Expert Opin. Drug Deliv. 2019, 16, 1–26. [Google Scholar] [CrossRef]
- Scholar, E. Sulfadiazine. xPharm Compr. Pharmacol. Ref. 2007, 1–5. [Google Scholar] [CrossRef]
- Kurowska, A.; Ghate, V.; Kodoth, A.; Shah, A.; Shah, A.; Vishalakshi, B.; Prakash, B.; Lewis, S.A. Non-Propellant Foams of Green Nano-Silver and Sulfadiazine: Development and In Vivo Evaluation for Burn Wounds. Pharm. Res. 2019, 36. [Google Scholar] [CrossRef] [PubMed]
- Catalano-Pons, C.; Bargy, S.; Schlecht, D.; Tabone, M.D.; Deschênes, G.; Bensman, A.; Ulinski, T. Sulfadiazine-Induced Nephrolithiasis in Children. Pediatr. Nephrol. 2004, 19, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Moya, F.; Thorndike, V. Passage of Drugs across the Placenta. Am. J. Obstet. Gynecol. 1962, 84, 1778–1798. [Google Scholar] [CrossRef]
- Mofenson, L.M.; Brady, M.T.; Danner, S.P.; Dominguez, K.L.; Hazra, R.; Handelsman, E.; Havens, P.; Nesheim, S.; Read, J.S.; Serchuck, L.; et al. Guidelines for the Prevention and Treatment of Opportunistic Infections among HIV-Exposed and HIV-Infected Children. Morb. Mortal. Wkly. Rep. 2009, 58, 1–166. [Google Scholar]
- Krátký, M.; Dzurková, M.; Janoušek, J.; Konečná, K.; Trejtnar, F.; Stolaříková, J.; VinŠová, J. Sulfadiazine Salicylaldehyde-Based Schiff Bases: Synthesis, Antimicrobial Activity and Cytotoxicity. Molecules 2017, 22, 1573. [Google Scholar] [CrossRef] [PubMed]
- Channar, P.A.; Saeed, A.; Albericio, F.; Larik, F.A.; Abbas, Q.; Hassan, M.; Raza, H.; Seo, S.Y. Sulfonamide-Linked Ciprofloxacin, Sulfadiazine and Amantadine Derivatives as a Novel Class of Inhibitors of Jack Bean Urease; Synthesis, Kinetic Mechanism and Molecular Docking. Molecules 2017, 22, 1352. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, A.; Bershad, S. Topical Acne Drugs: Review of Clinical Properties, Systemic Exposure, and Safety. Am. J. Clin. Dermatol. 2003, 4, 473–492. [Google Scholar] [CrossRef]
- Borda, L.J.; Perper, M.; Keri, J.E. Treatment of Seborrheic Dermatitis: A Comprehensive Review. J. Dermatol. Treat. 2019, 30, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Friedlaender, M.H. A Review of the Causes and Treatment of Bacterial and Allergic Conjunctivitis. Clin. Ther. 1995, 17, 800–810. [Google Scholar] [CrossRef]
- Lee, K. Dermatologic Applications of Sodium Sulfacetamide. J. Am. Acad. Dermatol. 2018, 79, AB102. [Google Scholar] [CrossRef]
- Pelle, M.T.; Crawford, G.H.; James, W.D. Rosacea: II. Therapy. J. Am. Acad. Dermatol. 2004, 51, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Sensoy, D.; Cevher, E.; Sarici, A.; Yilmaz, M.; Özdamar, A.; Bergişadi, N. Bioadhesive Sulfacetamide Sodium Microspheres: Evaluation of Their Effectiveness in the Treatment of Bacterial Keratitis Caused by Staphylococcus Aureus and Pseudomonas Aeruginosa in a Rabbit Model. Eur. J. Pharm. Biopharm. 2009, 72, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.D.; Ginsburg, C.M.; Clahsen, J.C.; Jackson, L.H. Treatment of Acute Otitis Media of Infancy With Cefaclor. Arch. Pediatr. Adolesc. Med. 1978, 132, 992. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.E.; Nelson, J.D.; Clahsen, J.; Jackson, L.H. Otitis Media of Infancy and Early Childhood. Am. J. Dis. Child. 1976, 130, 965. [Google Scholar] [CrossRef] [PubMed]
- Krause, P.J.; Owens, N.J.; Nightingale, C.H.; Klimek, J.J.; Lehmann, W.B.; Quintiliani, R.; The, S.; Diseases, I.; Jun, N. Penetration of Amoxicillin, Cefaclor, Erythromycin-Sulfisoxazole, and Trimethoprim-Sulfamethoxazole into the Middle Ear Fluid of Patients with Chronic Serous Otitis Media. J. Infect. Dis. 1982, 145, 815–821. [Google Scholar] [CrossRef]
- Perrin, J.M.; Charney, E.; MacWhinney, J.B., Jr.; McInerny, T.K.; Miller, R.L.; Nazarian, L.F. Sulfisoxazole as Chemoprophylaxis for Recurrent Otitis Media. N. Engl. J. Med. 1974, 291, 664–667. [Google Scholar] [CrossRef]
- Koch-weser, J.; Sidel, V.W.; Dexter, M.; Parish, C.; Finer, D.C.; Kanarek, P. Adverse Reactions to Sulfisoxazole, Sulfamethoxazole, and Nitrofurantoin. Arch. Intern. Med. 1971, 128, 399. [Google Scholar] [CrossRef]
- Kaplan, S.A.; Weinfeld, R.E.; Abruzzo, C.W.; Lewis, M. Pharmacokinetic Profile of Sulfisoxazole Following Intravenous, Intramuscular, and Oral Administration to Man. J. Pharm. Sci. 1972, 61, 773–778. [Google Scholar] [CrossRef]
- Aytac, Z.; Sen, H.S.; Durgun, E.; Uyar, T. Sulfisoxazole/Cyclodextrin Inclusion Complex Incorporated in Electrospun Hydroxypropyl Cellulose Nanofibers as Drug Delivery System. Colloids Surf. B Biointerfaces 2015, 128, 331–338. [Google Scholar] [CrossRef]
- Yildiz, Z.I.; Celebioglu, A.; Uyar, T. Polymer-Free Electrospun Nanofibers from Sulfobutyl Ether7-Beta-Cyclodextrin (SBE7-β-CD) Inclusion Complex with Sulfisoxazole: Fast-Dissolving and Enhanced Water-Solubility of Sulfisoxazole. Int. J. Pharm. 2017, 531, 550–558. [Google Scholar] [CrossRef]
- Syed, H.; Safa, R.; Chidlow, G.; Osborne, N.N. Sulfisoxazole, an Endothelin Receptor Antagonist, Protects Retinal Neurones from Insults of Ischemia/Reperfusion or Lipopolysaccharide. Neurochem. Int. 2006, 48, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Uchino, T.; Sanyal, S.N.; Yamabe, M.; Kaku, T.; Takebayashi, S.; Shimaoka, T.; Shimada, T.; Noguchi, T.; Ono, K. Rescue of Pulmonary Hypertension with an Oral Sulfonamide Antibiotic Sulfisoxazole by Endothelin Receptor Antagonistic Actions. Hypertens. Res. 2008, 31, 1781–1790. [Google Scholar] [CrossRef] [PubMed]
- Im, E.-J.; Lee, C.-H.; Moon, P.-G.; Rangaswamy, G.G.; Lee, B.; Lee, J.M.; Lee, J.-C.; Jee, J.-G.; Bae, J.-S.; Kwon, T.-K.; et al. Sulfisoxazole Inhibits the Secretion of Small Extracellular Vesicles by Targeting the Endothelin Receptor A. Nat. Commun. 2019, 10, 1387. [Google Scholar] [CrossRef]
- Palomino, J.C.; Martin, A. The Potential Role of Trimethoprim-Sulfamethoxazole in the Treatment of Drug-Resistant Tuberculosis. Future Microbiol. 2016, 11, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Gleckman, R.; Blagg, N.; Joubert, D.W. Trimethoprim: Mechanisms of Action, Antimicrobial Activity, Bacterial Resistance, Pharmacokinetics, Adverse Reactions, and Therapeutic Indications. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1981, 1, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Bowen, A.C.; Carapetis, J.R.; Currie, B.J.; Fowler, V.; Chambers, H.F.; Tong, S.Y.C. Sulfamethoxazole-Trimethoprim (Cotrimoxazole) for Skin and Soft Tissue Infections Including Impetigo, Cellulitis, and Abscess. Open Forum Infect. Dis. 2017, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.M.-W.; Juurlink, D.N. Considerations When Prescribing Trimethoprim—Sulfamethoxazole. Can. Med. Assoc. J. 2011, 183, 1851–1858. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.V.; Thota, P.; Pellegrino, D.; Pasupuleti, V.; Benites-Zapata, V.A.; Deshpande, A.; Penalva de Oliveira, A.C.; Vidal, J.E. A Systematic Review and Meta-Analysis of the Relative Efficacy and Safety of Treatment Regimens for HIV-Associated Cerebral Toxoplasmosis: Is Trimethoprim-Sulfamethoxazole a Real Option? HIV Med. 2017, 18, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Crider, K.S.; Cleves, M.A.; Reefhuis, J.; Berry, R.J.; Hobbs, C.A.; Hu, D.J. Antibacterial Medication Use During Pregnancy and Risk of Birth Defects. Arch. Pediatr. Adolesc. Med. 2009, 163, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Matok, I.; Gorodischer, R.; Koren, G.; Landau, D.; Wiznitzer, A.; Levy, A. Exposure to Folic Acid Antagonists during the First Trimester of Pregnancy and the Risk of Major Malformations. Br. J. Clin. Pharmacol. 2009, 68, 956–962. [Google Scholar] [CrossRef]
- Michałek, K.; Lechowicz, M.; Pastuszczak, M.; Wojas-Pelc, A. The Use of Trimethoprim and Sulfamethoxazole (TMP-SMX) in Dermatology. Folia Med. Cracov. 2015, 55, 35–41. [Google Scholar] [PubMed]
- Aronson, J.K. Dapsone and Analogues. In Meyler’s Side Effects of Drugs, 16th ed.; Aronson, J.K., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2016; pp. 824–830. [Google Scholar] [CrossRef]
- Sundeep Chaitanya, V.; Das, M.; Bhat, P.; Ebenezer, M. Computational Modelling of Dapsone Interaction with Dihydropteroate Synthase in Mycobacterium Leprae; Insights into Molecular Basis of Dapsone Resistance in Leprosy. J. Cell. Biochem. 2015, 116, 2293–2303. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.S.; Paidesetty, S.K.; Dehury, B.; Sahoo, J.; Vedithi, S.C.; Mahapatra, N.; Hussain, T.; Padhy, R.N. Molecular Docking and Simulation Study for Synthesis of Alternative Dapsone Derivative as a Newer Antileprosy Drug in Multidrug Therapy. J. Cell. Biochem. 2018, 119, 9838–9852. [Google Scholar] [CrossRef] [PubMed]
- Kneiber, D.; Kowalski, E.H.; Amber, K.T. Dapsone, Two Birds with One Stone: A Response to “Dapsone Advantages over Trimethoprim-Sulfamethoxazole for Pneumocystis Pneumonia Prophylaxis in Immunobullous Patients”. J. Am. Acad. Dermatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.I.; Stiller, M.J. Dapsone and Sulfones in Dermatology: Overview and Update. J. Am. Acad. Dermatol. 2001, 45, 420–434. [Google Scholar] [CrossRef]
- Geyfman, M.; Debabov, D.; Poloso, N.; Alvandi, N. Mechanistic Insight into the Activity of a Sulfone Compound Dapsone on Propionibacterium (Newly Reclassified as Cutibacterium) Acnes-Mediated Cytokine Production. Exp. Dermatol. 2019, 28, 190–197. [Google Scholar] [CrossRef]
- Chan, H.-L.; Lee, K.-O. Tonsillar Membrane in the DDS (Dapsone) Syndrome. Int. J. Dermatol. 1991, 30, 216–217. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.E. An Historical and Clinical Review of the Interaction of Leprosy and Pregnancy: A Cycle to Be Broken. Soc. Sci. Med. 1993, 37, 457–472. [Google Scholar] [CrossRef]
- Chaves, L.L.; Silveri, A.; Vieira, A.C.C.; Ferreira, D.; Cristiano, M.C.; Paolino, D.; Di Marzio, L.; Lima, S.C.; Reis, S.; Sarmento, B.; et al. PH-Responsive Chitosan Based Hydrogels Affect the Release of Dapsone: Design, Set-up, and Physicochemical Characterization. Int. J. Biol. Macromol. 2019, 133, 1268–1279. [Google Scholar] [CrossRef]
- World Health Organization. Overview of Malaria Treatment. Available online: https://www.who.int/malaria/areas/treatment/overview/en/ (accessed on 2 September 2019).
- Nothdurft, H.D.; Kain, K.C. Malaria Prevention. Travel Trop. Med. Man. 2017, 71–90. [Google Scholar] [CrossRef]
- Nzila, A.; Ward, S.A.; Marsh, K.; Sims, P.F.G.; Hyde, J.E. Comparative Folate Metabolism in Humans and Malaria Parasites (Part I): Pointers for Malaria Treatment from Cancer Chemotherapy. Trends Parasitol. 2005, 21, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Wozel, G.; Blasum, C. Dapsone in Dermatology and Beyond. Arch. Dermatol. Res. 2014, 103–124. [Google Scholar] [CrossRef] [PubMed]
- Rojo, L.; Fernandez-Gutierrez, M.; Deb, S.; Stevens, M.M.; San Roman, J. Designing Dapsone Polymer Conjugates for Controlled Drug Delivery. Acta Biomater. 2015, 27, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.M.; Pratt, A.G.; Isaacs, J.D. Mechanism of Action of Methotrexate in Rheumatoid Arthritis, and the Search for Biomarkers. Nat. Rev. Rheumatol. 2016, 12, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Friedman, B.; Cronstein, B. Methotrexate Mechanism in Treatment of Rheumatoid Arthritis. Jt. Bone Spine 2019, 86, 301–307. [Google Scholar] [CrossRef]
- Municio, C.; Palacios, B.S.; Estrada-Capetillo, L.; Benguria, A.; Dopazo, A.; García-Lorenzo, E.; Fernández-Arroyo, S.; Joven, J.; Miranda-Carús, M.E.; González-Álvaro, I.; et al. Methotrexate Selectively Targets Human Proinflammatory Macrophages through a Thymidylate Synthase/P53 Axis. Ann. Rheum. Dis. 2016, 75, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, A.; Ahmad, S.; Kaur, P.; Bhatnagar, A.; Dhawan, V.; Dhir, V. Methotrexate Preferentially Affects Tc1 and Tc17 Subset of CD8 T Lymphocytes. Clin. Rheumatol. 2019, 38, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Jiménez Gómez-Lavín, M.; Abradelo, C.; San Roman, J.; Rojo, L. Bibliographic Review on the State of the Art of Strontium and Zinc Based Regenerative Therapies. Recent Developments and Clinical Applications. J. Mater. Chem. B 2019. [Google Scholar] [CrossRef]
- Fernández-Villa, D.; Aranaz, I.; Acosta, N.; Vázquez-Lasa, B.; San Román, J.; Rojo, L. Methotrexate Derivatives Bearing Divalent Cations. Antibacterial Activity and Encapsulation into Chitosan Microparticles. In Proceedings of the 30th Annual Conference of the European Society for Biomaterials, Dresden, Germany, 9 September 2019; pp. 2–9. [Google Scholar]
- Plosker, G.L.; Croom, K.F. Sulfasalazine. Drugs 2005, 65, 1825–1849. [Google Scholar] [CrossRef]
- Linares, V.; Alonso, V.; Domingo, J.L. Oxidative Stress as a Mechanism Underlying Sulfasalazine-Induced Toxicity. Expert Opin. Drug Saf. 2011, 10, 253–263. [Google Scholar] [CrossRef]
- Liptay, S.; Fulda, S.; Schanbacher, M.; Bourteele, S.; Ferri, K.F.; Kroemer, G.; Adler, G.; Debatin, K.M.; Schmid, R.M. Molecular Mechanisms of Sulfasalazine-Induced T-Cell Apoptosis. Br. J. Pharmacol. 2002, 137, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Morabito, L.; Montesinos, M.C.; Schreibman, D.M.; Balter, L.; Thompson, L.F.; Resta, R.; Carlin, G.; Huie, M.A.; Cronstein, B.N. Methotrexate and Sulfasalazine Promote Adenosine Release by a Mechanism That Requires Ecto-5′-Nucleotidase-Mediated Conversion of Adenine Nucleotides. J. Clin. Investig. 1998, 101, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Pruzanski, W.; Stefanski, E.; Vadas, P.; Ramamurthy, N.S. Inhibition of Extracellular Release of Proinflammatory Secretory Phospholipase A2 (SPLA2) by Sulfasalazine: A Novel Mechanism of Anti- Inflammatory Activity. Biochem. Pharmacol. 1997, 53, 1901–1907. [Google Scholar] [CrossRef]
- Greenfield, S.M.; Punchard, N.A.; Thompson, R.P.H. Inhibition of Red Cell Membrane Lipid Peroxidation by Sulphasalazine and 5-Aminosallcylic Acid. Gut 1991, 32, 1156–1159. [Google Scholar] [CrossRef] [PubMed]
- Brownfoot, F.C.; Hannan, N.J.; Cannon, P.; Nguyen, V.; Hastie, R.; Parry, L.J.; Senadheera, S.; Tuohey, L.; Tong, S.; Kaitu’u-Lino, T.J. Sulfasalazine Reduces Placental Secretion of Antiangiogenic Factors, up-Regulates the Secretion of Placental Growth Factor and Rescues Endothelial Dysfunction. EBioMedicine 2019, 41, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Dowd, F.J.; Johnson, B.S.; Mariotti, A.J.; Kumar, P. Pharmacology of Specific Drug Groups: Antibiotic Therapy. Pharmacol. Ther. Dent. 2017, 457–487. [Google Scholar] [CrossRef]
- Then, R.L. Mechanisms of Resistance to Trimethoprim, the Sulfonamides, and Trimethoprim-Sulfamethoxazole. Rev. Infect. Dis. 1982, 4, 261–269. [Google Scholar] [CrossRef]
- Pato, M.L.; Brown, G.M. Mechanisms of Resistance of Escherichia Coli to Sulfonamides. Arch. Biochem. Biophys. 1963, 103, 443–448. [Google Scholar] [CrossRef]
- Dallas, W.S.; Gowen, J.E.; Ray, P.H.; Cox, M.J.; Dev, I.K. Cloning, Sequencing, and Enhanced Expression of the Dihydropteroate Synthase Gene of Escherichia Coli MC4100. J. Bacteriol. 1992, 174, 5961–5970. [Google Scholar] [CrossRef][Green Version]
- Sköld, O. Sulfonamide Resistance: Mechanisms and Trends. Drug Resist. Updates 2000, 3, 155–160. [Google Scholar] [CrossRef]
- Hampele, I.C.; D’Arcy, A.; Dale, G.E.; Kostrewa, D.; Nielsen, J.; Oefner, C.; Page, M.G.P.; Schönfeld, H.J.; Stüber, D.; Then, R.L. Structure and Function of the Dihydropteroate Synthase from Staphylococcus Aureus. J. Mol. Biol. 1997, 268, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Radstrom, P.; Fermer, C.; Kristiansen, B.E.; Jenkins, A.; Skold, O.; Swedberg, G. Transformational Exchanges in the Dihydropteroate Synthase Gene of Neisseria Meningitidis: A Novel Mechanism for Acquisition of Sulfonamide Resistance. J. Bacteriol. 1992, 174, 6386–6393. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alekshun, M.N.; Levy, S.B. Molecular Mechanisms of Antibacterial Multidrug Resistance. Cell 2007, 128, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Reviews, S.; Trimethoprim, S.; Burchall, J.J.; Elwell, L.P.; Fling, M.E. Molecular Mechanisms of Resistance to Trimethoprim. Available online: http://www.Jstor.Org/Stable/4452715 (accessed on 29 August 2019).
- Swedberg, G.; Skold, O. Characterization of Different Plasmid-Borne Dihydropteroate Synthases Mediating Bacterial Resistance to Sulfonamides. J. Bacteriol. 1980, 142, 1–7. [Google Scholar] [PubMed]
- Wise, E.M.; Abou-Donia, M.M. Sulfonamide Resistance Mechanism in Escherichia Col: R Plasmids Can Determine Sulfonamide-Resistant Dihydropteroate Synthases (Citrobacter/Klebsiella Pneumoniae/Multiple Drug Resistance). Biochemistry 1975, 72, 2621–2625. [Google Scholar]
- Zhao, R.; Goldman, I.D. Resistance to Antifolates. Oncogene 2003, 22, 7431–7457. [Google Scholar] [CrossRef] [PubMed]
- Swedberg, G.; Castensson, S.; Skold, O. Characterization of Mutationally Altered Dihydropteroate Synthase and Its Ability to Form a Sulfonamide-Containing Dihydrofolate Analog. J. Bacteriol. 1979, 137, 129–136. [Google Scholar]
- Estrada, A.; Wright, D.L.; Anderson, A.C. Antibacterial Antifolates: From Development through Resistance to the next Generation. Cold Spring Harb. Perspect. Med. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Zhao, Y.; Shadrick, W.R.; Wallace, M.J.; Wu, Y.; Griffith, E.C.; Qi, J.; Yun, M.-K.; White, S.W.; Lee, R.E. Pterin–sulfa Conjugates as Dihydropteroate Synthase Inhibitors and Antibacterial Agents. Bioorg. Med. Chem. Lett. 2016, 26, 3950–3954. [Google Scholar] [CrossRef]
- Qi, J.; Virga, K.G.; Das, S.; Zhao, Y.; Yun, M.-K.; White, S.W.; Lee, R.E. Synthesis of Bi-Substrate State Mimics of Dihydropteroate Synthase as Potential Inhibitors and Molecular Probes. Bioorg. Med. Chem. 2011, 19, 1298–1305. [Google Scholar] [CrossRef][Green Version]
- Dennis, M.L.; Lee, M.D.; Harjani, J.R.; Ahmed, M.; DeBono, A.J.; Pitcher, N.P.; Wang, Z.-C.; Chhabra, S.; Barlow, N.; Rahmani, R.; et al. 8-Mercaptoguanine Derivatives as Inhibitors of Dihydropteroate Synthase. Chem. A Eur. J. 2018, 24, 1922–1930. [Google Scholar] [CrossRef] [PubMed]
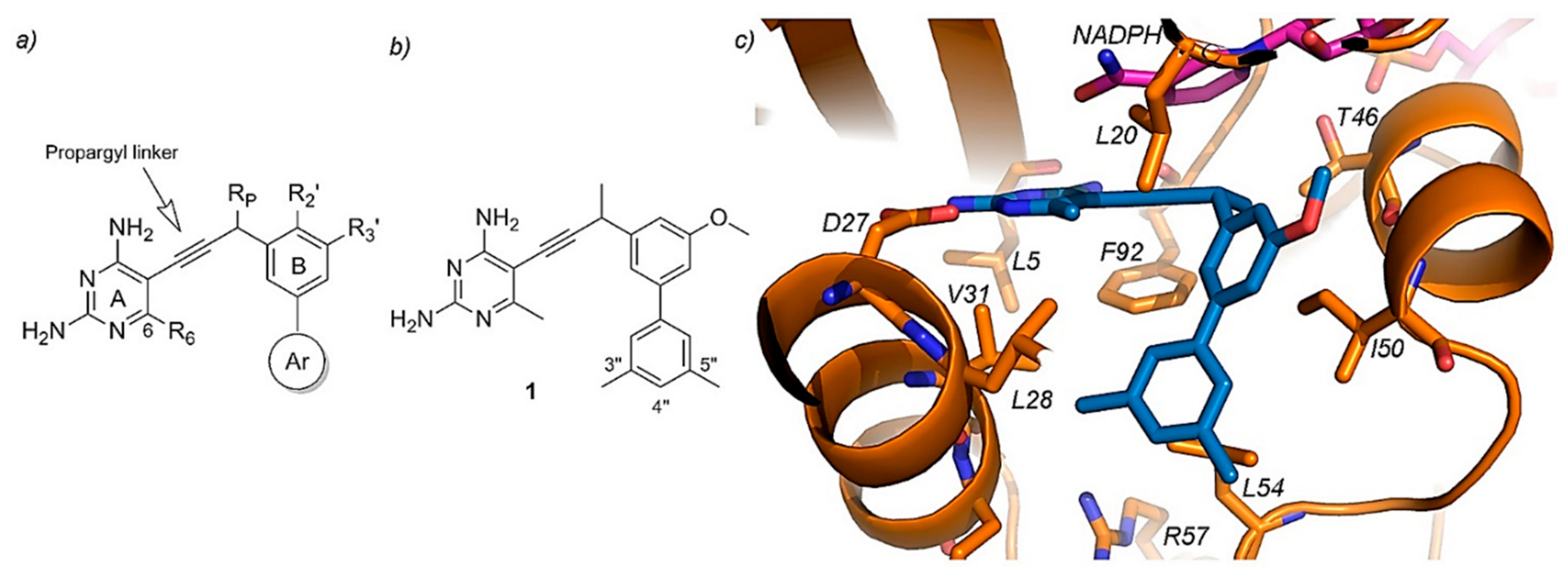
- Lombardo, M.N.; G-Dayanandan, N.; Keshipeddy, S.; Zhou, W.; Si, D.; Reeve, S.M.; Alverson, J.; Barney, P.; Walker, L.; Hoody, J.; et al. Structure-Guided In Vitro to In Vivo Pharmacokinetic Optimization of Propargyl-Linked Antifolates. Drug Metab. Dispos. 2019, 47, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, K.; Frey, K.M.; Scocchera, E.W.; Martin, B.D.; Swain, P.W.; Alverson, J.B.; Priestley, N.D.; Anderson, A.C.; Wright, D.L. Toward New Therapeutics for Skin and Soft Tissue Infections: Propargyl-Linked Antifolates Are Potent Inhibitors of MRSA and Streptococcus Pyogenes. PLoS ONE 2012, 7, e29434. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.B.; Dryden, M. Iclaprim, a Dihydrofolate Reductase Inhibitor Antibiotic in Phase III of Clinical Development: A Review of Its Pharmacology, Microbiology and Clinical Efficacy and Safety. Future Microbiol. 2018, 13, 957–969. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Mode of Action | Indication | Administration | Toxicity | References | |
|---|---|---|---|---|---|---|
Mafenide | Unknown | Severe burns wounds | Topical: creams or powders for solution | Metabolic acidosis, allergies | [31,32,33,34,35,36,37,38] | |
Silver Sulfadiazine | DHPS inhibitor + silver biocide effect | Burns and ulcers | Topical: creams or hydrogels | Allergies, sensitivity to silver or propylene glycol. Rare: hemolysis, argyria or pseudo-eschar formation | [32,33,39,40,41,42] | |
Sulfadiazine | DHPS inhibitor | Urinary tract infections, otitis media, encephalitis, meningitis. Prophylaxis for rheumatic fevers. Parasitic infections: malaria and toxoplasmosis | Oral: tablets | Allergies and gastrointestinal upset. Rare: encephalopathies, renal failure, nephrolithiasis | [43,44,45,47,48,49] | |
Sulfacetamide | DHPS inhibitor | Skin, ocular and urinary tract infections | Ophthalmic: solution/drops Topical: ointment or lotions Vaginal: creams | Rare: mild cutaneous reactions. Category C drug in pregnancy | [50,51,52,53,54,55] | |
Sulfisoxazole | DHPS inhibitor | Severe, repeated, or long-lasting urinary tract infections, meningococcal meningitis, acute otitis media, ocular infections, etc. | Oral: tablets or suspensions with or without erythromycin | Rare. Allergies, gastrointestinal disturbances | [56,57,58,59,60,61,62,63,64,65,66] | |
| Co-trimoxazole | Sulfamethoxazole | DHPS inhibitor | Bacterial bronchitis, prostatitis and urinary tract infections. | Oral: tablets of both compounds (co-trimoxazole) | Allergies, nausea, vomiting, diarrhea and hematologic alterations. Category D drug in pregnancy | [8,67,68,69,70,71,72,73,74] |
Trimethoprim | DHFR inhibitor | |||||
Dapsone | DHPS inhibitor | Leprosy. Prophylaxis for co-trimoxazole-resistant P. jirovecii infections. With pyrimethamine: malaria | Oral: tablets or suspensions Topical: gels | Hemolysis (usually mild), dapsone syndrome, gastrointestinal upset. Category C drug in pregnancy | [75,76,77,78,79,80,81,82,83] | |
| Antimalarial Agents | Proguanil | DHFR inhibitor | Malaria treatment and prophylaxis | Oral: tablets | Hypersensitivity reactions, bone marrow suppression | [3,84,85,86] |
Pyrimethamine | DHFR inhibitor | |||||
Sulfadoxine | DHPS inhibitor | |||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Villa, D.; Aguilar, M.R.; Rojo, L. Folic Acid Antagonists: Antimicrobial and Immunomodulating Mechanisms and Applications. Int. J. Mol. Sci. 2019, 20, 4996. https://doi.org/10.3390/ijms20204996
Fernández-Villa D, Aguilar MR, Rojo L. Folic Acid Antagonists: Antimicrobial and Immunomodulating Mechanisms and Applications. International Journal of Molecular Sciences. 2019; 20(20):4996. https://doi.org/10.3390/ijms20204996
Chicago/Turabian StyleFernández-Villa, Daniel, Maria Rosa Aguilar, and Luis Rojo. 2019. "Folic Acid Antagonists: Antimicrobial and Immunomodulating Mechanisms and Applications" International Journal of Molecular Sciences 20, no. 20: 4996. https://doi.org/10.3390/ijms20204996
APA StyleFernández-Villa, D., Aguilar, M. R., & Rojo, L. (2019). Folic Acid Antagonists: Antimicrobial and Immunomodulating Mechanisms and Applications. International Journal of Molecular Sciences, 20(20), 4996. https://doi.org/10.3390/ijms20204996