Methionine Adenosyltransferase 1a (MAT1A) Enhances Cell Survival During Chemotherapy Treatment and is Associated with Drug Resistance in Bladder Cancer PDX Mice

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results
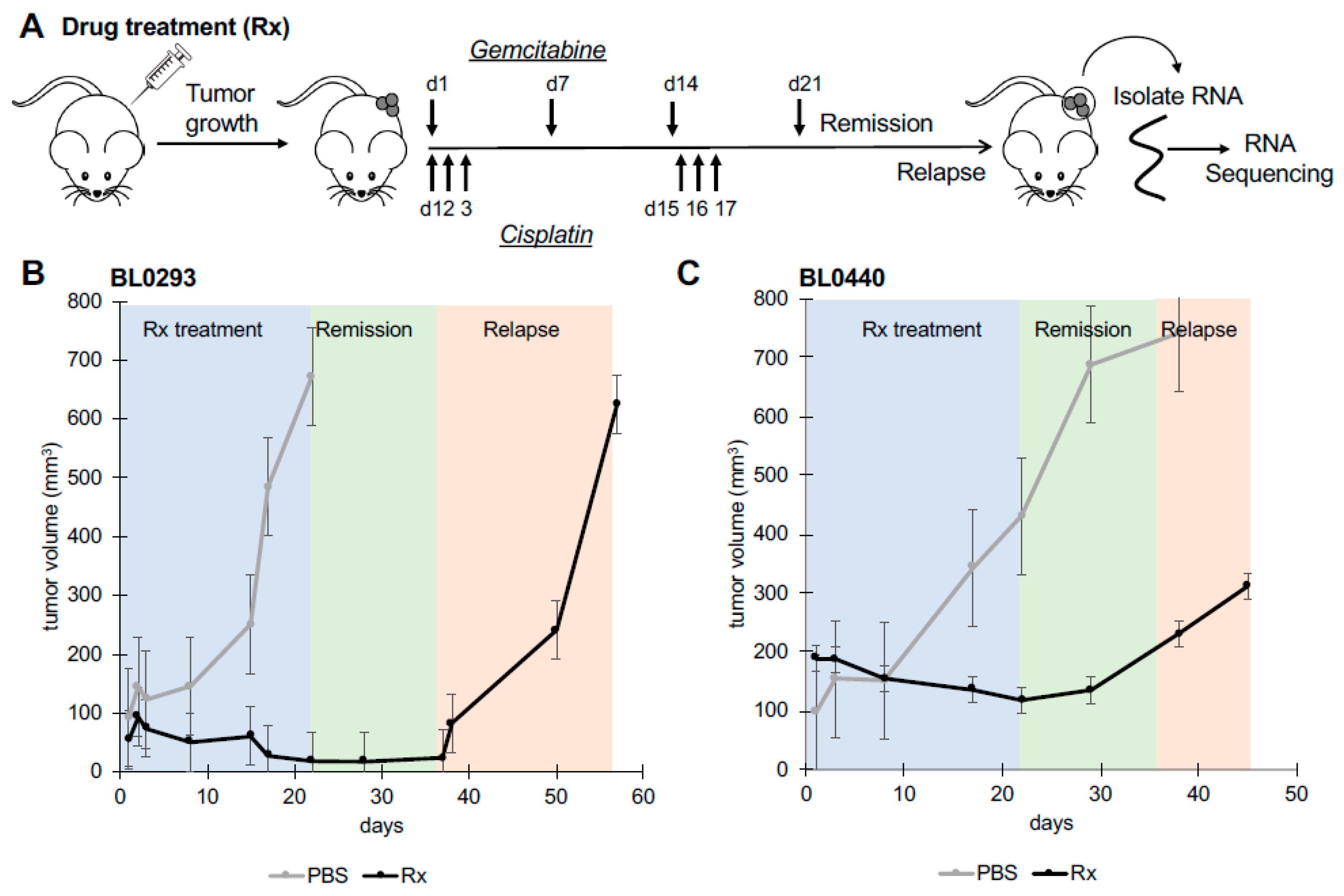
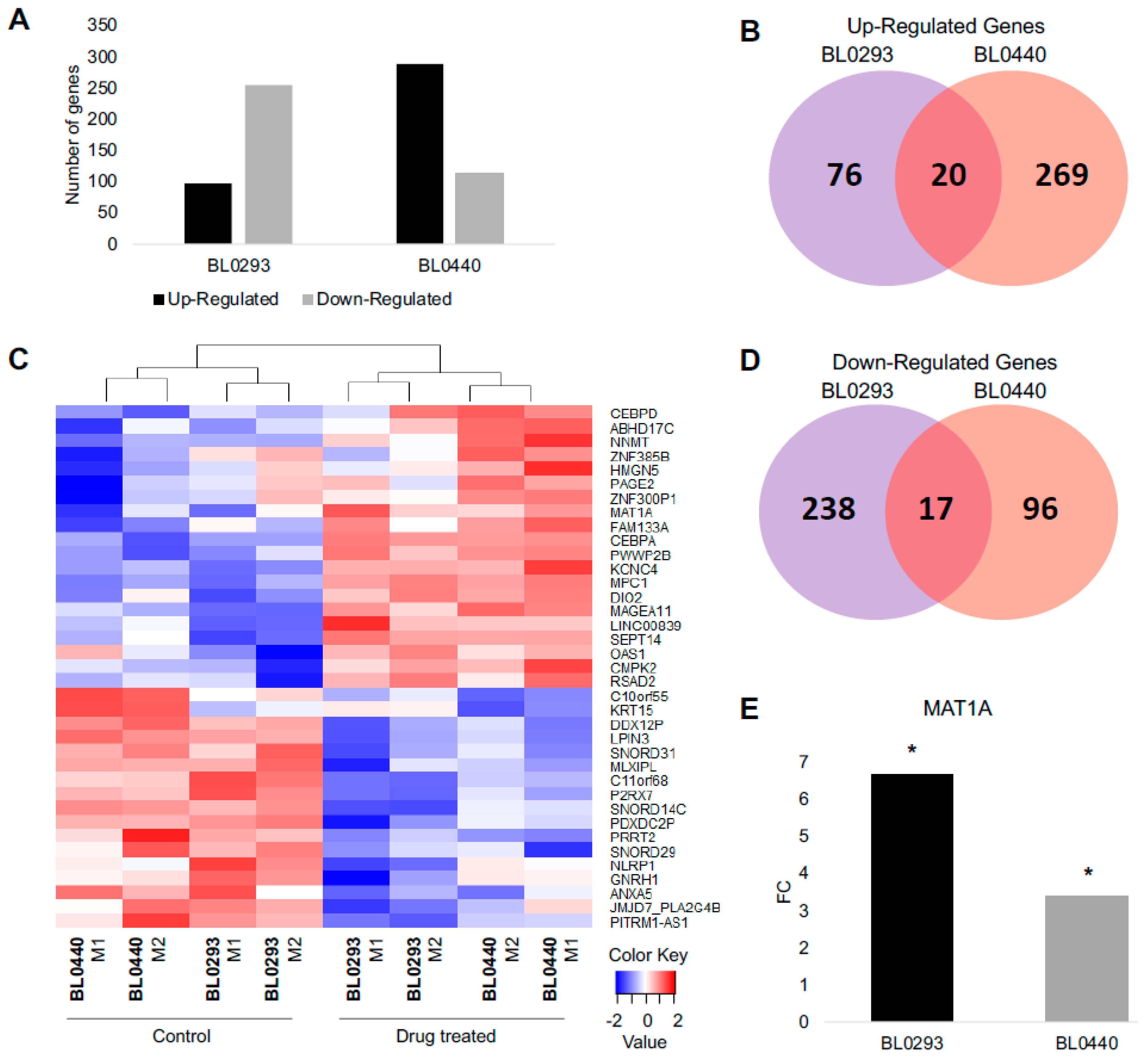
2.1. Significant Changes in the Transcriptome of Bladder Cancer Patient-Derived Xenografts are Associated with Drug Relapse Following Treatment.
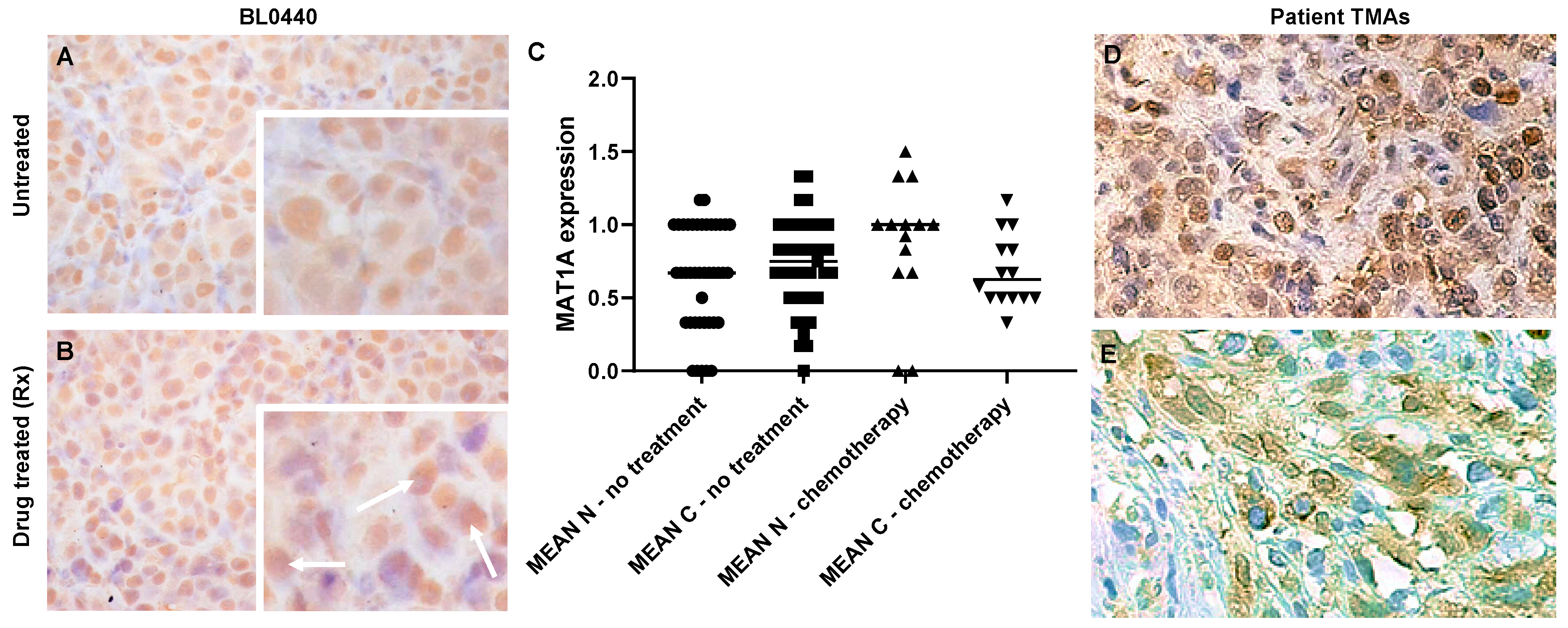
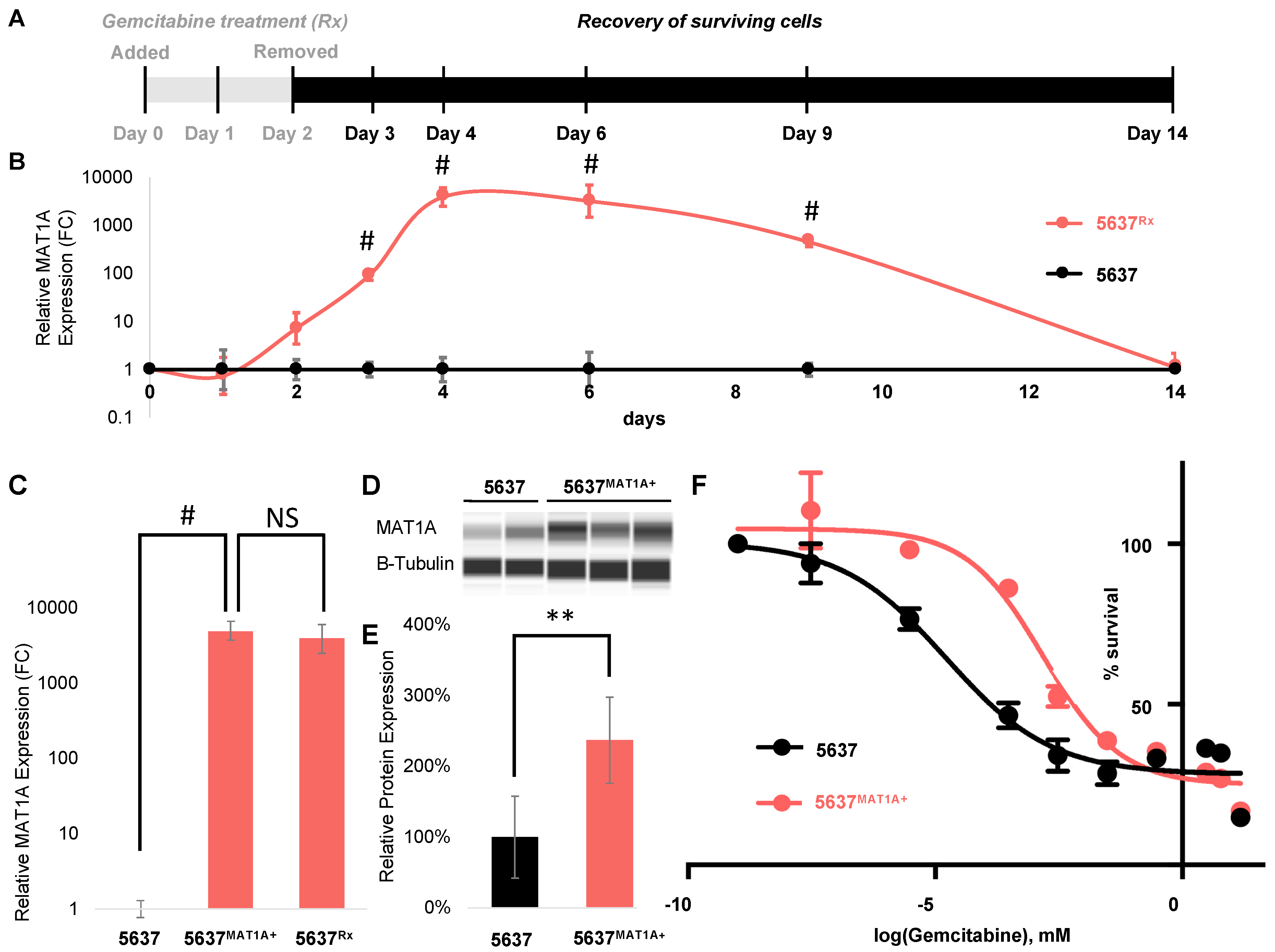
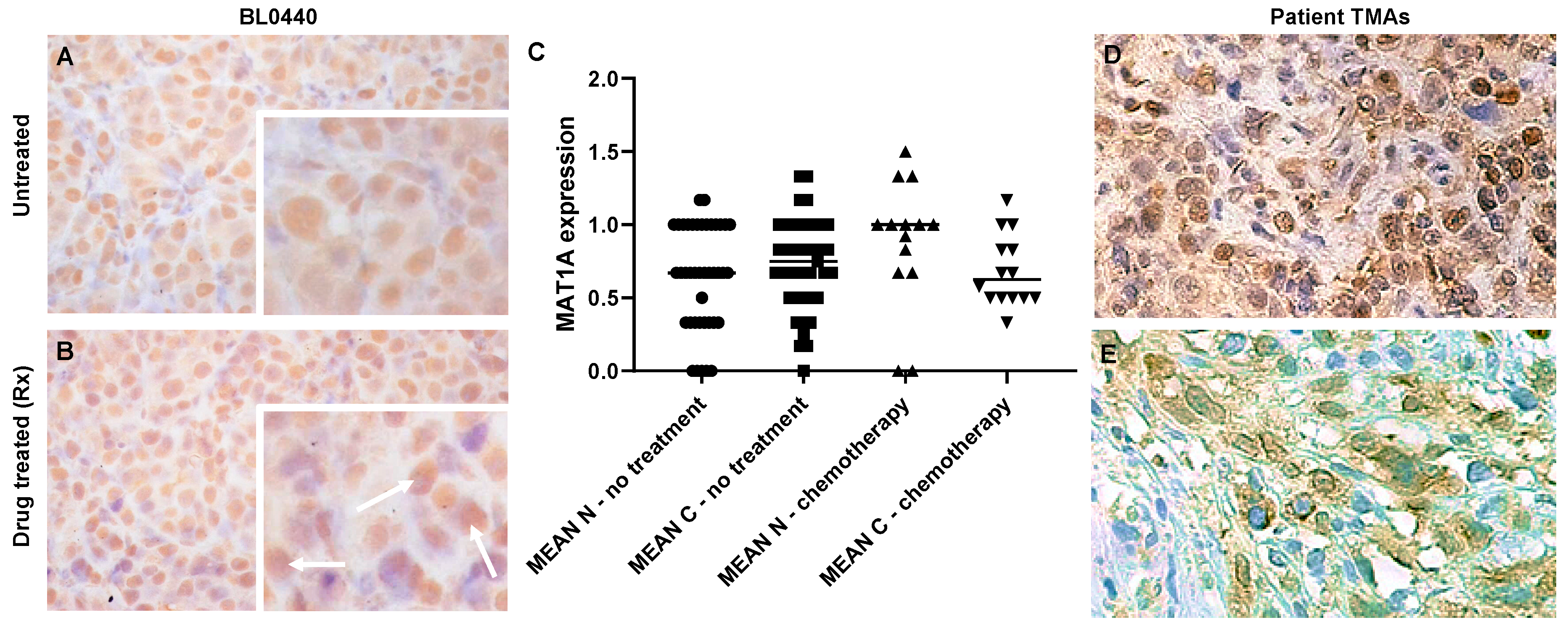
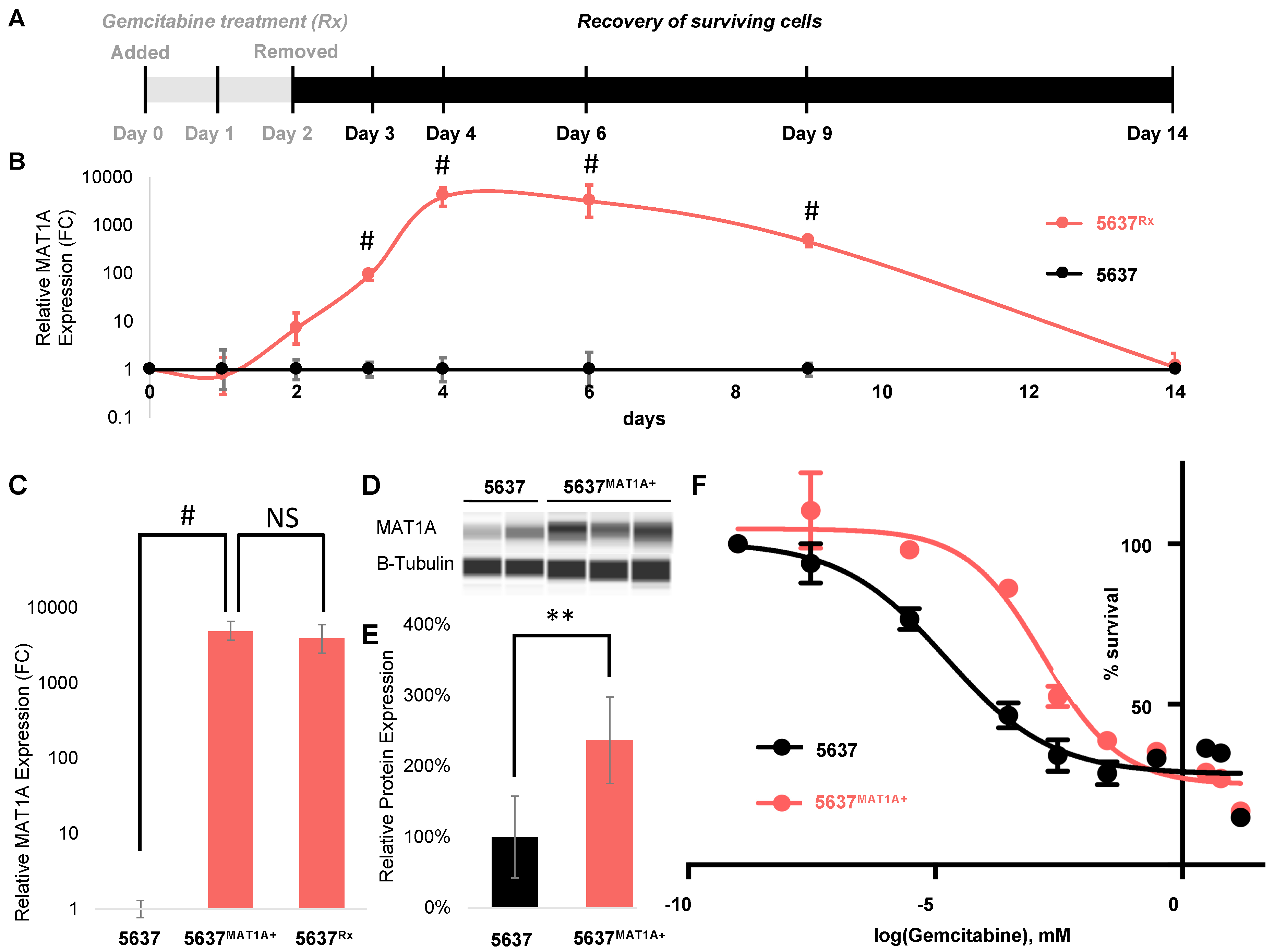
2.2. MAT1A Expression Increases in Response to Chemotherapy in Bladder Cancer and Confers a Survival Advantage
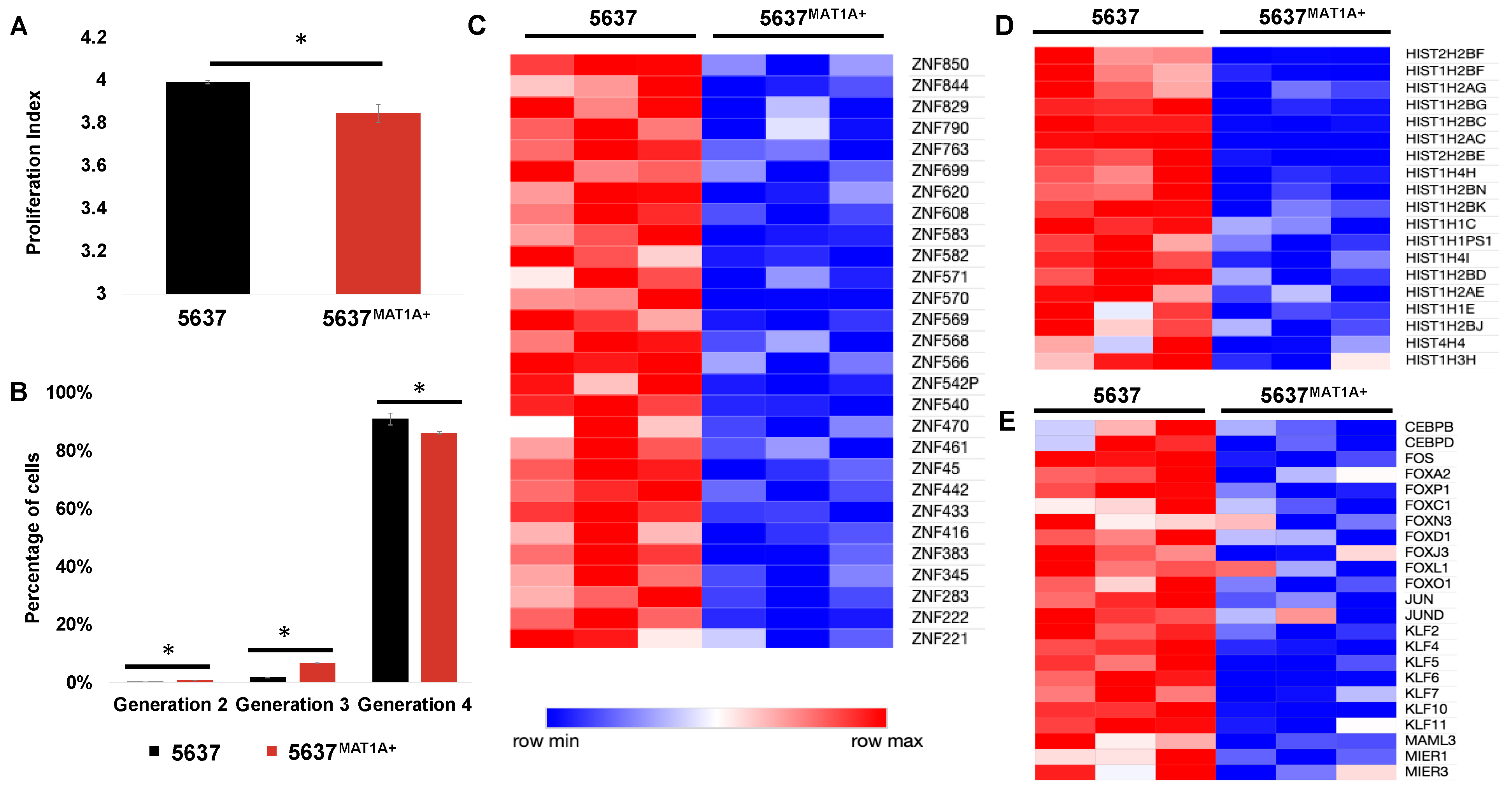
2.3. MAT1A Confers a Survival Advantage, Potentially Through Decreases in Active Transcription and Proliferation
3. Discussion
4. Materials and Methods
4.1. Bladder Cancer Patient Derived Xenografts
4.2. Generation of Drug Relapsed PDX Tumors
4.3. RNA Sequencing
4.4. RNA Sequencing Data Analysis
4.5. Plasmid Transfection Generating 5637MAT1A+ Cells
4.6. Immunohistochemical Analysis
4.7. Human Patient Tissue Microarray Analysis (TMA)
4.8. The Human Protein Atlas: MAT1A
4.9. Western Blotting and Protein Analysis
4.10. In Vitro Toxicity
4.11. Quantitative PCR (qPCR)
4.12. Cell Proliferation
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MAT1A | Methionine adenosyltransferase 1a |
| PDX | Patient-derived xenograft |
| SAM | S-adenosylmethionine |
| CIS | cisplatin |
| GEM | gemcitabine |
| NSG | NOD scid gamma |
| Rx | drug treated |
| PBS | phosphate buffered saline |
| FC | fold change |
| ATP | adenosine triphosphate |
| PCR | polymerase chain reaction |
| CFSE | carboxyfluorescein diacetate succinimidyl ester |
| HCC | Hepatocellular carcinoma |
| OPN | osteopontin |
| SQ | subcutaneous |
| IHC | immunohistochemical |
| TMA | tissue microarrays |
| FOLFIRINOX | Leucovorin, fluorouracil, irinotecan, oxaliplatin |
| BCG | Bacillus Calmette-Guerin therapy |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Ries, L.A.G.; Young Jr, J.L.; Keel, G.E.; Eisner, M.P.; Lin, Y.D.; Horner, M.-J.D. Cancer Survival Among Adults: US SEER Program, 1988–2001. Available online: https://seer.cancer.gov/archive/publications/survival/ (accessed on 13 September 2019).
- Niedersüss-Beke, D.; Puntus, T.; Kunit, T.; Grünberger, B.; Lamche, M.; Loidl, W.; Böhm, R.; Kraischits, N.; Kudlacek, S.; Schramek, P.; et al. Neoadjuvant Chemotherapy with Gemcitabine plus Cisplatin in Patients with Locally Advanced Bladder Cancer. Oncology 2017, 93, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.Y.; Zu, Y.X.; Jiang, X.W.; Sun, X.T.; Liu, T.Y.; Li, R.L.; Wu, Q.; Zhang, Y.S.; Zhao, Q.C. Novel ADAM-17 inhibitor ZLDI-8 inhibits the proliferation and metastasis of chemo-resistant non-small-cell lung cancer by reversing Notch and epithelial mesenchymal transition in vitro and in vivo. Pharm. Res. 2019, 148, 104406. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Wang, S.S.; Zhang, H.; Lin, T.Y.; Malfatti, M.; Haack, K.; Ognibene, T.; Yang, H.; Airhart, S.; Turteltaub, K.W.; et al. Microdose-Induced Drug-DNA Adducts as Biomarkers of Chemotherapy Resistance in Humans and Mice. Mol. Cancer Ther. 2017, 16, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Grove, T.L.; Benner, J.S.; Radle, M.I.; Ahlum, J.H.; Landgraf, B.J.; Krebs, C.; Booker, S.J. A radically different mechanism for S-adenosylmethionine-dependent methyltransferases. Science 2011, 332, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, H.; Hoelper, D.; Jain, S.U.; Cantone, N.; Lundgren, S.M.; Poy, F.; Allis, C.D.; Cummings, R.; Bellon, S.; Lewis, P.W. S-adenosyl methionine is necessary for inhibition of the methyltransferase G9a by the lysine 9 to methionine mutation on histone H3. Proc. Natl. Acad. Sci. USA 2016, 113, 6182–6187. [Google Scholar] [CrossRef]
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 2012, 92, 1515–1542. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.X.; Zhang, H.; Tepper, C.G.; Lin, T.Y.; Davis, R.R.; Keck, J.; Ghosh, P.M.; Gill, P.; Airhart, S.; Bult, C.; et al. Development and Characterization of Bladder Cancer Patient-Derived Xenografts for Molecularly Guided Targeted Therapy. PLoS ONE 2015, 10, e0134346. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Uhlig, A.; Strauss, A.; Seif Amir Hosseini, A.; Lotz, J.; Trojan, L.; Schmid, M.; Uhlig, J. Gender-specific Differences in Recurrence of Non-muscle-invasive Bladder Cancer: A Systematic Review and Meta-analysis. Eur. Urol. Focus 2018, 4, 924–936. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.; Basson, M.D.; Ito, H. P300 inhibition enhances gemcitabine-induced apoptosis of pancreatic cancer. Oncotarget 2016, 7, 51301–51310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. New Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Adamska, A.; Elaskalani, O.; Emmanouilidi, A.; Kim, M.; Abdol Razak, N.B.; Metharom, P.; Falasca, M. Molecular and cellular mechanisms of chemoresistance in pancreatic cancer. Adv. Biol. Regul. 2018, 68, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Matossian, M.D.; Burks, H.E.; Elliott, S.; Hoang, V.T.; Bowles, A.C.; Sabol, R.A.; Wahba, B.; Anbalagan, M.; Rowan, B.; Abazeed, M.E.; et al. Drug resistance profiling of a new triple negative breast cancer patient-derived xenograft model. BMC Cancer 2019, 19, 205. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Chiang, E.P. Low-dose methotrexate inhibits methionine S-adenosyltransferase in vitro and in vivo. Mol. Med. 2012, 18, 423–432. [Google Scholar] [CrossRef]
- Murin, R.; Vidomanova, E.; Kowtharapu, B.S.; Hatok, J.; Dobrota, D. Role of S-adenosylmethionine cycle in carcinogenesis. Gen. Physiol. Biophys. 2017, 36, 513–520. [Google Scholar] [CrossRef]
- Wang, R.; Jin, Y.; Yao, X.H.; Fan, W.; Zhang, J.; Cao, Y.; Li, J. A novel mechanism of the M1-M2 methionine adenosyltransferase switch-mediated hepatocellular carcinoma metastasis. Mol. Carcinog. 2018, 57, 1201–1212. [Google Scholar] [CrossRef]
- Maldonado, L.Y.; Arsene, D.; Mato, J.M.; Lu, S.C. Methionine adenosyltransferases in cancers: Mechanisms of dysregulation and implications for therapy. Exp. Biol. Med. 2018, 243, 107–117. [Google Scholar] [CrossRef]
- Brocard, M.; Ruggieri, A.; Locker, N. m6A RNA methylation, a new hallmark in virus-host interactions. J. Gen. Virol. 2017, 98, 2207–2214. [Google Scholar] [CrossRef]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Da Silveira, W.A.; Hazard, E.S.; Chung, D.; Hardiman, G. Molecular Profiling of RNA Tumors Using High-Throughput RNA Sequencing: From Raw Data to Systems Level Analyses. Methods Mol. Biol. 2019, 1908, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ranking | Gene Symbol | Gene Name | BL0293_FC | BL0440_FC |
|---|---|---|---|---|
| 1 | MAGEA11 | Melanoma-Associated Antigen 11 | 2.75 | 8.50 |
| 2 | MAT1A | Methionine Adenosyltransferase 1A | 6.66 | 3.40 |
| 3 | DIO2 | Iodothyronine Deiodinase 2 | 3.04 | 6.72 |
| 4 | PWWP2B | PWWP Domain-Containing Protein 2B | 4.91 | 3.67 |
| 5 | PAGE2 | Prostate-Associated Gene 2 Protein | 4.68 | 2.67 |
| 6 | FAM133A | Family With Sequence Similarity 133 Member A | 4.25 | 2.81 |
| 7 | ABHD17C | Abhydrolase Domain Containing 17C | 2.37 | 4.53 |
| 8 | LINC00839 | Long Intergenic Non-Protein Coding RNA 839 | 2.75 | 3.62 |
| 9 | OAS1 | 2′-5′-Oligoadenylate Synthetase 1 | 1.63 | 4.56 |
| 10 | RSAD2 | Radical S-Adenosyl Methionine Domain Containing 2 | 2.49 | 3.28 |
| 11 | CMPK2 | Cytidine/Uridine Monophosphate Kinase 2 | 1.80 | 3.67 |
| 12 | HMGN5 | High Mobility Group Nucleosome Binding Domain 5 | 2.80 | 2.49 |
| 13 | ZNF300P1 | Zinc Finger Protein 300 Pseudogene 1 | 2.64 | 1.91 |
| 14 | SEPT14 | Septin 14 | 1.75 | 2.57 |
| 15 | CEBPA | CCAAT Enhancer Binding Protein Alpha | 2.09 | 1.81 |
| 16 | KCNC4 | Potassium Voltage-Gated Channel Subfamily C Member 4 | 1.66 | 2.19 |
| 17 | NNMT | Nicotinamide N-Methyltransferase | 1.52 | 2.23 |
| 18 | MPC1 | Mitochondrial Pyruvate Carrier 1 | 1.69 | 1.71 |
| 19 | ZNF385B | Zinc Finger Protein 385B | 1.70 | 1.52 |
| 20 | CEBPD | CCAAT Enhancer Binding Protein Delta | 1.57 | 1.60 |
| Reactome Identifier | Name | p-Value |
|---|---|---|
| R-HSA-909733 | Interferon alpha/beta signaling | 1.324 × 10-5 |
| R-HSA-2408508 | Metabolism of ingested SeMet, Sec, MeSec into H2Se | 2.084 × 10-5 |
| R-HSA-381340 | Transcriptional regulation of white adipocyte differentiation | 2.142 × 10-5 |
| R-HSA-156581 | Methylation | 6.927 × 10-5 |
| R-HSA-71291 | Metabolism of amino acids and derivatives | 2.613 × 10-3 |
| R-HSA-156580 | Phase II conjugation | 3.383 × 10-3 |
| R-HSA-2408522 | Selenoamino acid metabolism | 4.137 × 10-3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, K.A.; Hum, N.R.; Sebastian, A.; He, W.; Siddiqui, S.; Ghosh, P.M.; Pan, C.-x.; de Vere White, R.; Loots, G.G. Methionine Adenosyltransferase 1a (MAT1A) Enhances Cell Survival During Chemotherapy Treatment and is Associated with Drug Resistance in Bladder Cancer PDX Mice. Int. J. Mol. Sci. 2019, 20, 4983. https://doi.org/10.3390/ijms20204983
Martin KA, Hum NR, Sebastian A, He W, Siddiqui S, Ghosh PM, Pan C-x, de Vere White R, Loots GG. Methionine Adenosyltransferase 1a (MAT1A) Enhances Cell Survival During Chemotherapy Treatment and is Associated with Drug Resistance in Bladder Cancer PDX Mice. International Journal of Molecular Sciences. 2019; 20(20):4983. https://doi.org/10.3390/ijms20204983
Chicago/Turabian StyleMartin, Kelly A., Nicholas R. Hum, Aimy Sebastian, Wei He, Salma Siddiqui, Paramita M. Ghosh, Chong-xian Pan, Ralph de Vere White, and Gabriela G. Loots. 2019. "Methionine Adenosyltransferase 1a (MAT1A) Enhances Cell Survival During Chemotherapy Treatment and is Associated with Drug Resistance in Bladder Cancer PDX Mice" International Journal of Molecular Sciences 20, no. 20: 4983. https://doi.org/10.3390/ijms20204983