Gut Microbiota and Predicted Metabolic Pathways in a Sample of Mexican Women Affected by Obesity and Obesity Plus Metabolic Syndrome

 ,
,  and
and
Abstract
1. Introduction
2. Results
2.1. Mexican Women Who Suffer from OB or OMS Exhibit Clinical Parameters in Accordance with Their Condition
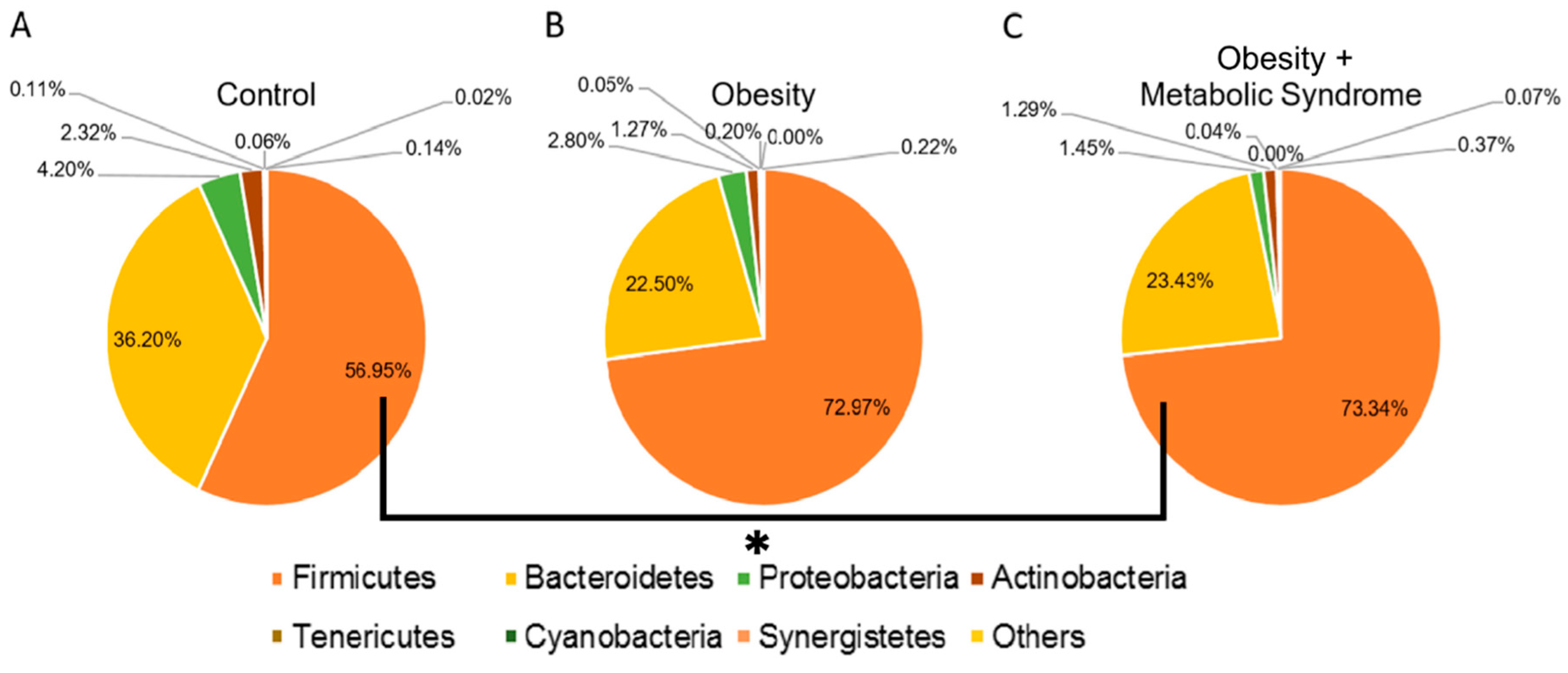
2.2. Firmicutes Is the Most Abundant Bacterial Phylum in Women Who Suffer from OB or OMS
2.3. The Relative Abundance of Some Bacteria Changed in Women with OB or OMS
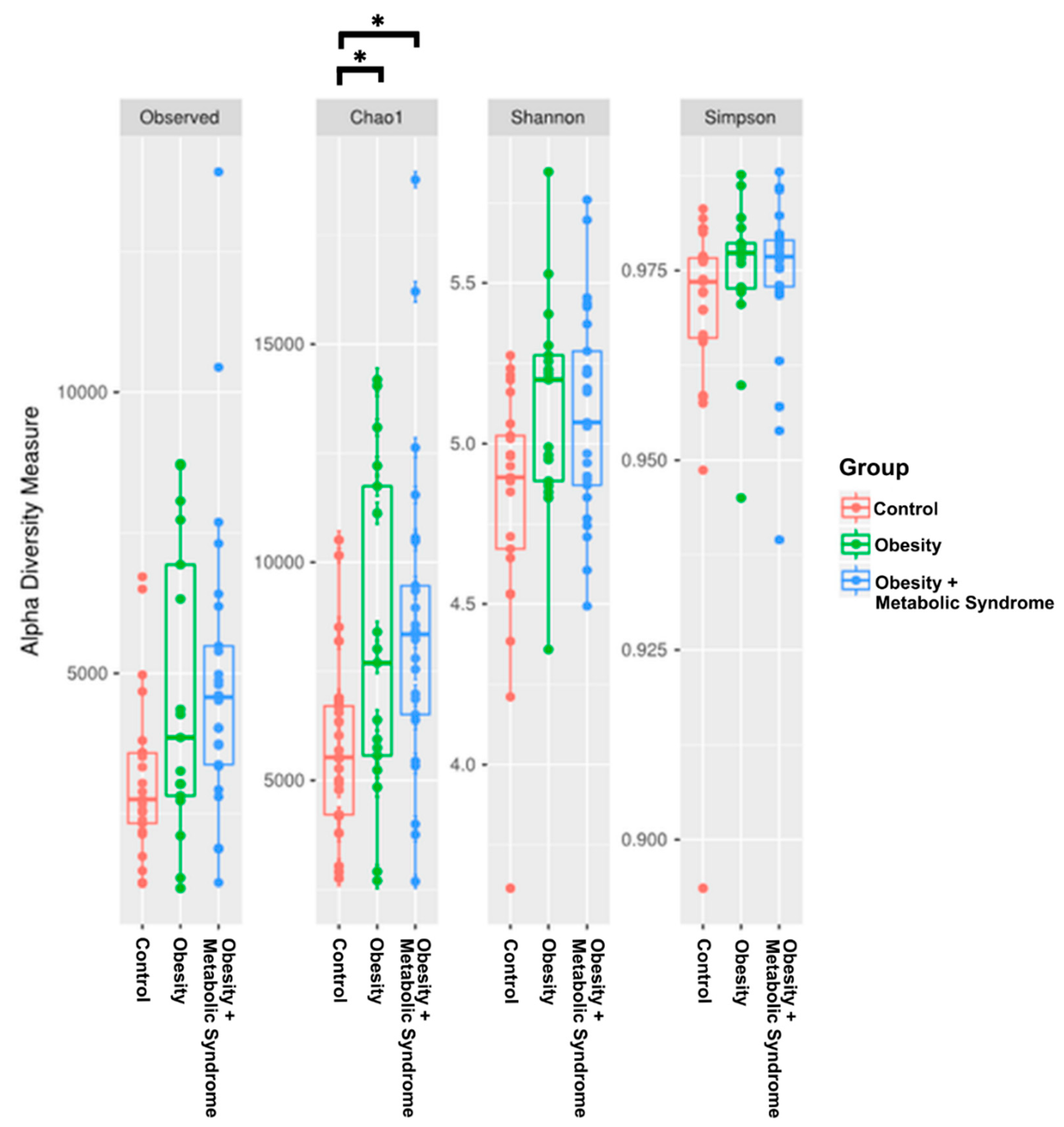
2.4. The Gut Microbiota Alpha Diversity of Women with OMS Is Higher than the Diversity in OB and CO
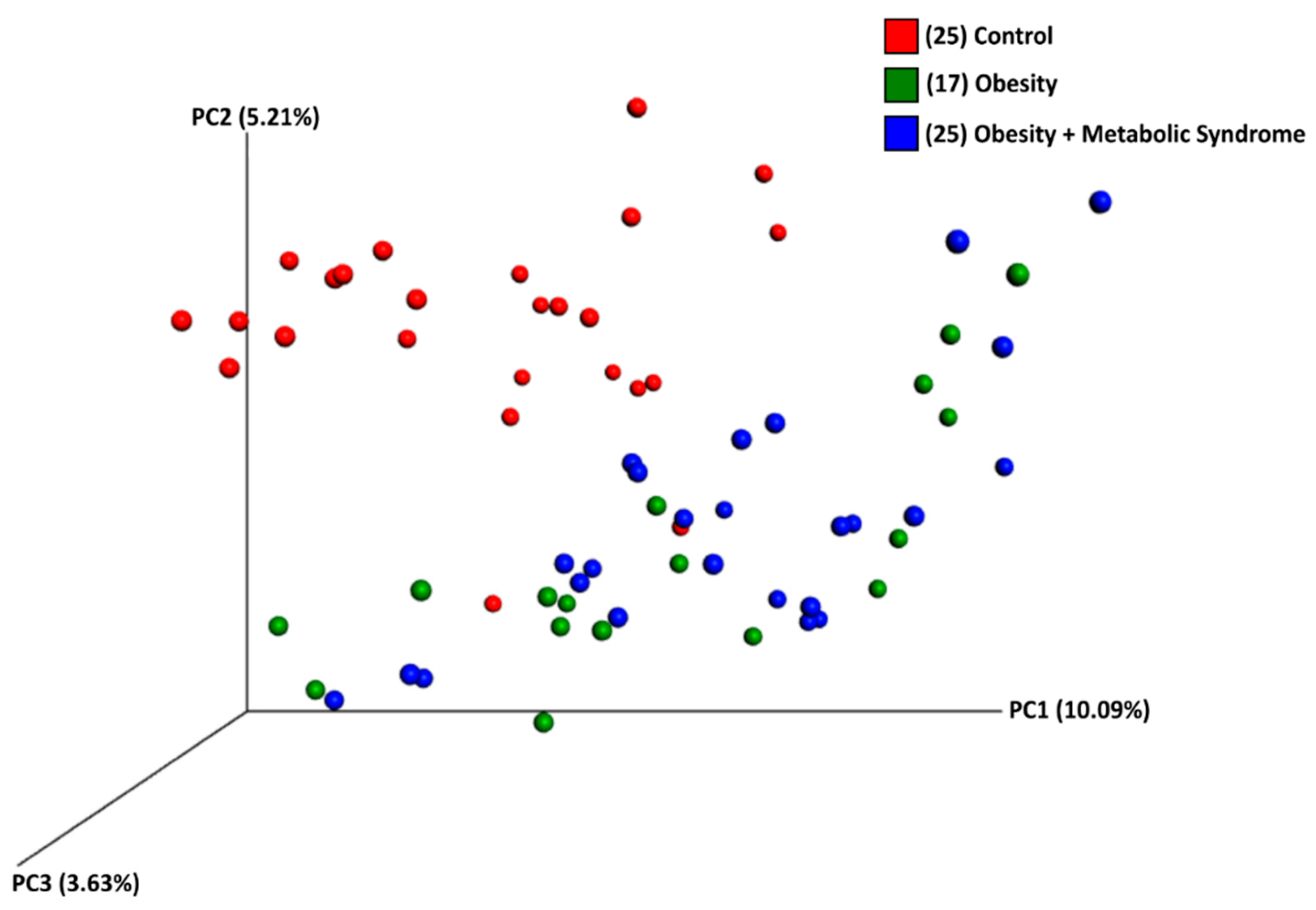
2.5. The Beta Diversity of the Gut Microbiota of Women with OB or OMS Is Similar to Each Other but Different from the CO Group
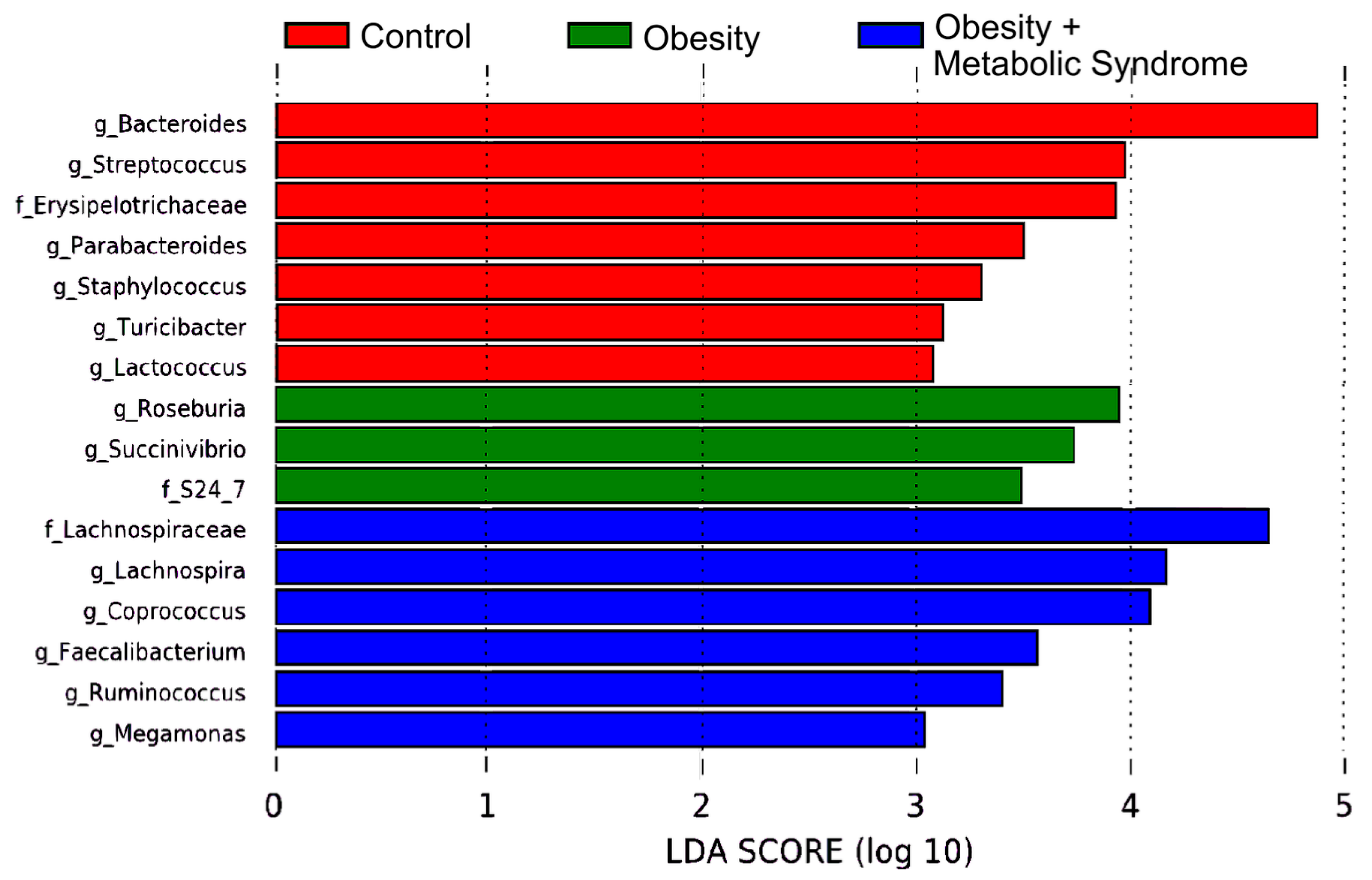
2.6. Gut Bacteria from 16 Different Taxa Might Explain the Observed Metabolic Differences Among the CO, OB, and OMS Groups
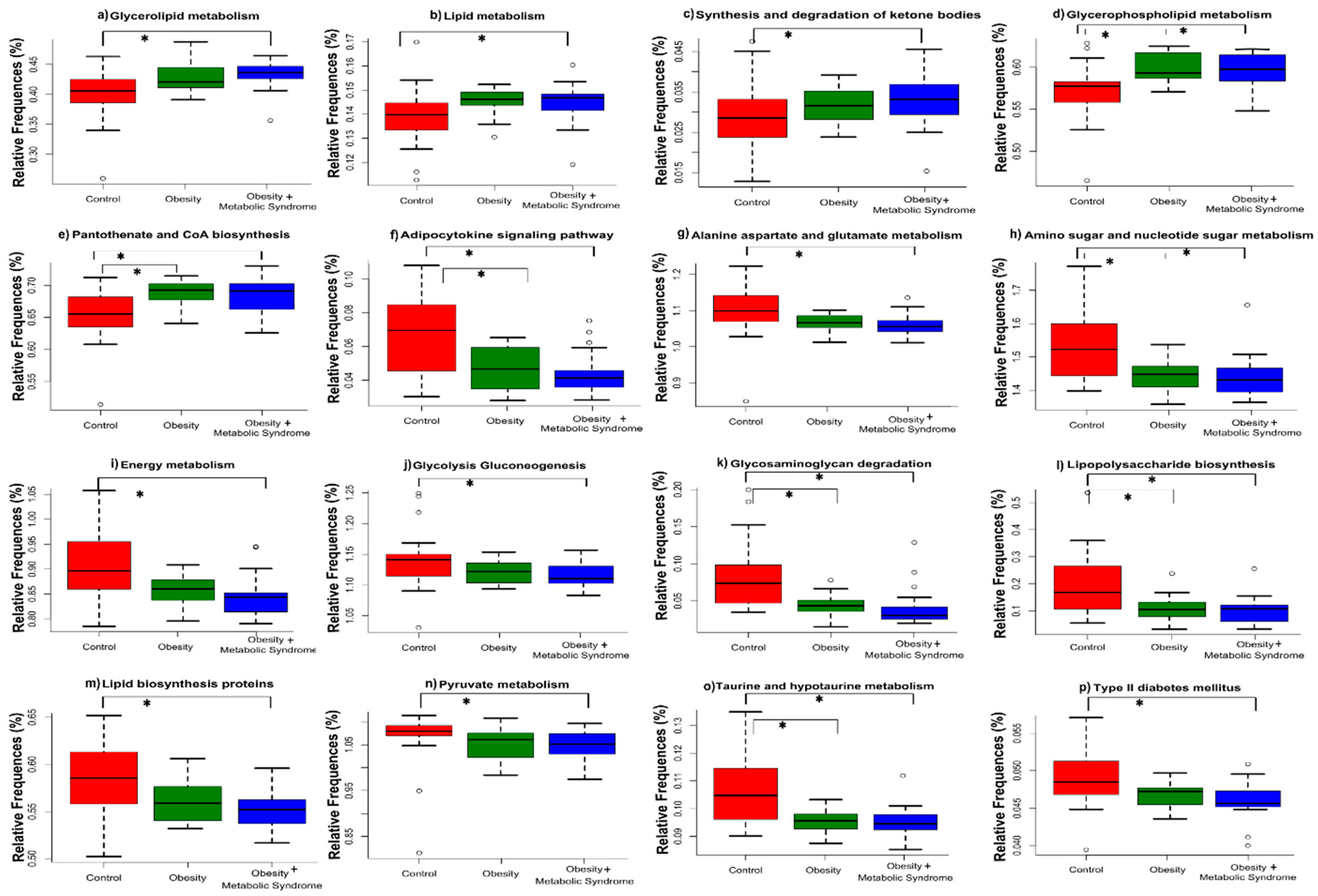
2.7. The Predicted Functional Metagenome of the Gut Microbiota Shows Differences among the Groups
3. Discussion
4. Materials and Methods
4.1. Study Subjects
4.2. Anthropometric Evaluation
4.3. Dietary Assessment
4.4. Biochemical Studies
4.5. DNA Extraction from Feces
4.6. Construction of the V3-16S rDNA Library and High-Throughput DNA Sequencing
4.7. Microbial Diversity Analysis
4.8. Analysis of Enrichment of Bacteria in Each Study Group
4.9. Predictive Functional Genes of the Gut Microbiota Using PICRUSt
4.10. qPCR Assessment of the Bacterial Load and Composition
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| LEfSe | Linear Discriminant Analysis Effect Size |
| MaAsLin | Multivariate Association with Linear Models |
| PICRUSt | Phylogenetic Investigation of Communities by Reconstruction of Unobserved States |
| SCFAs | Short-Chain Fatty Acids |
References
- World Health Organization. Obesity 2017 [Web Page]. Available online: http://www.who.int/topics/obesity/en/ (accessed on 20 November 2018).
- Martin-Rodriguez, E.; Guillen-Grima, F.; Martí, A.; Brugos-Larumbe, A. Comorbidity associated with obesity in a large population: The APNA study. Obes. Res. Clin. Pract. 2015, 9, 435–447. [Google Scholar] [CrossRef]
- Di Cesare, M.; Bentham, J.; Stevens, G.A.; Zhou, B.; Danaei, G.; Lu, Y.; Bixby, H.; Cowan, M.J.; Riley, L.M.; Hajifathalian, K.; et al. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- Encuesta Nacional de Salud y Nutrición de Medio Camino 2016. Available online: https://www.gob.mx/cms/uploads/attachment/file/209093/ENSANUT.pdf (accessed on 20 November 2018).
- Meldrum, D.R.; Morris, M.A.; Gambone, J.C. Obesity pandemic: Causes, consequences, and solutions—But do we have the will? Fertil. Steril. 2017, 107, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Fabbiano, S.; Suárez-Zamorano, N.; Trajkovski, M. Host-microbiota mutualism in metabolic diseases. Front. Endocrinol. (Lausanne) 2017, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Nirmalkar, K.; Murugesan, S.; Pizano-Zárate, M.L.; Villalobos-Flores, L.E.; García-González, C.; Morales-Hernández, R.M.; Nuñez-Hernández, J.A.; Hernández-Quiroz, F.; Romero-Figueroa, M.S.; Hernández-Guerrero, C.; et al. Gut Microbiota and Endothelial Dysfunction Markers in Obese Mexican Children and Adolescents. Nutrients 2018, 10, 2009. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. NIH Public Access. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef]
- John, G.K.; Mullin, G.E. The Gut Microbiome and Obesity. Curr. Oncol. Rep. 2016, 18, 45. [Google Scholar] [CrossRef]
- Quigley, E.M.M. Gut bacteria in health and disease. Gastroenterol. Hepatol. 2013, 9, 560–569. [Google Scholar]
- Lone, S.; Lone, K.; Khan, S.; Pampori, R.A. Assessment of metabolic syndrome in Kashmiri population with type 2 diabetes employing the standard criteria’s given by WHO, NCEPATP III and IDF. J. Epidemiol. Glob. Health 2017, 7, 235–239. [Google Scholar] [CrossRef]
- Heiss, G.; Snyder, M.L.; Teng, Y.; Schneiderman, N.; Llabre, M.M.; Cowie, C.; Carnethon, M.; Kaplan, R.; Giachello, A.; Gallo, L.; et al. Prevalence of metabolic syndrome among Hispanics/Latinos of diverse background: The Hispanic Community Health Study/Study of Latinos. Diabetes Care 2014, 37, 2391–2399. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.M.M.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.T.; Loria, C.M.; Smith, S.C. Harmonizing the metabolic syndrome: A joint interim statement of the international diabetes federation task force on epidemiology and prevention; National heart, lung, and blood institute; American heart association; World heart federation; International. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef]
- Ussar, S.; Griffin, N.W.; Bezy, O.; Fujisaka, S.; Vienberg, S.; Softic, S.; Deng, L.; Bry, L.; Gordon, J.I.; Kahn, C.R. Interactions between Gut Microbiota, Host Genetics and Diet Modulate the Predisposition to Obesity and Metabolic Syndrome. Cell Metab. 2015, 22, 516–530. [Google Scholar] [CrossRef]
- Lippert, K.; Kedenko, L.; Antonielli, L.; Kedenko, I.; Gemeier, C.; Leitner, M.; Kautzky-Willer, A.; Paulweber, B.; Hackl, E. Gut microbiota dysbiosis associated with glucose metabolism disorders and the metabolic syndrome in older adults. Benef. Microbes 2017, 8, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, S.; Ulloa-Martínez, M.; Martínez-Rojano, H.; Galván-Rodríguez, F.M.; Miranda-Brito, C.; Romano, M.C.; Piña-Escobedo, A.; Pizano-Zárate, M.L.; Hoyo-Vadillo, C.; García-Mena, J. Study of the diversity and short-chain fatty acids production by the bacterial community in overweight and obese Mexican children. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 1337–1346. [Google Scholar] [CrossRef]
- Nguyen, T.T.B.; Jin, Y.Y.; Chung, H.J.; Hong, S.T. Pharmabiotics as an emerging medication for metabolic syndrome and its related diseases. Molecules 2017, 22, 1795. [Google Scholar] [CrossRef]
- Cani, P.D. Microbiota and metabolites in metabolic diseases. Nat. Rev. Endocrinol. 2019. [Google Scholar] [CrossRef]
- American Diabetes Association. Standards of Medical Care in Diabetes-2017: Summary of Revisions. Diabetes Care 2017, 40, S12–S14. [Google Scholar]
- Millán, J.; Pintó, X.; Muñoz, A.; Zúñiga, M.; Rubiés-Prat, J.; Pallardo, L.F.; Masana, L.; Mangas, A.; Hernández-Mijares, A.; González-Santos, P.; et al. Lipoprotein ratios: Physiological significance and clinical usefulness in cardiovascular prevention. Vasc. Health Risk Manag. 2009, 5, 757–765. [Google Scholar] [PubMed]
- Norma Oficial Mexicana NOM-030-SSA2-2009, Para la Prevención, Detección, Diagnóstico, Tratamiento y Control de la Hipertensión Arterial Sistémica [Web Page]. Available online: http://dof.gob.mx/nota_detalle.php?codigo=5144642&fecha=31/05/2010 (accessed on 20 November 2018).
- Barczynska, R.; Kapusniak, J.; Litwin, M.; Slizewska, K.; Szalecki, M. Dextrins from Maize Starch as Substances Activating the Growth of Bacteroidetes and Actinobacteria Simultaneously Inhibiting the Growth of Firmicutes, Responsible for the Occurrence of Obesity. Plant. Foods Hum. Nutr. 2016, 71, 190–196. [Google Scholar] [CrossRef]
- Feng, J.; Tang, H.; Li, M.; Pang, X.; Wang, L.; Zhang, M.; Zhao, Y.; Zhang, X.; Shen, J. The abundance of fecal Faecalibacterium prausnitzii in relation to obesity and gender in Chinese adults. Arch. Microbiol. 2014, 196, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Salonen, A.; Lahti, L.; Salojärvi, J.; Holtrop, G.; Korpela, K.; Duncan, S.H.; Date, P.; Farquharson, F.; Johnstone, A.M.; Lobley, G.E.; et al. Impact of diet and individual variation on intestinal microbiota composition and fermentation products in obese men. ISME J. 2014, 8, 2218–2230. [Google Scholar] [CrossRef] [PubMed]
- Biddle, A.; Stewart, L.; Blanchard, J.; Leschine, S. Untangling the genetic basis of fibrolytic specialization by lachnospiraceae and ruminococcaceae in diverse gut communities. Diversity 2013, 5, 627–640. [Google Scholar] [CrossRef]
- Kaakoush, N.O. Insights into the Role of Erysipelotrichaceae in the Human Host. Front. Cell. Infect. Microbiol. 2015, 5, 1–4. [Google Scholar] [CrossRef]
- Zhang, H.; Dibaise, J.K.; Zuccolo, A.; Kudrna, D.; Braidotti, M.; Yu, Y.; Parameswaran, P.; Crowell, M.D.; Wing, R.; Rittmann, B.E.; Krajmalnik-brown, R. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA 2009, 106, 2365–2370. [Google Scholar] [CrossRef]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.W.M.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef]
- Davis, S.C.; Yadav, J.S.; Barrow, S.D.; Robertson, B.K. Gut microbiome diversity influenced more by the Westernized dietary regime than the body mass index as assessed using effect size statistic. Microbiologyopen 2017, 6, 1–17. [Google Scholar] [CrossRef]
- Franco-De-Moraes, A.C.; De Almeida-Pititto, B.; Da Rocha Fernandes, G.; Gomes, E.P.; Da Costa Pereira, A.; Ferreira, S.R.G. Worse inflammatory profile in omnivores than in vegetarians associates with the gut microbiota composition. Diabetol. Metab. Syndr. 2017, 9, 1–8. [Google Scholar] [CrossRef]
- Kang, C.; Wang, B.; Kaliannan, K.; Wang, X.; Lang, H.; Hui, S.; Huang, L.; Zhang, Y.; Zhou, M.; Chen, M.; et al. Gut microbiota mediates the protective effects of dietary capsaicin against chronic low-grade inflammation and associated obesity induced by high-fat diet. MBio 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kverka, M.; Zakostelska, Z.; Klimesova, K.; Sokol, D.; Hudcovic, T.; Hrncir, T.; Rossmann, P.; Mrazek, J.; Kopecny, J.; Verdu, E.F.; et al. Oral administration of Parabacteroides distasonis antigens attenuates experimental murine colitis through modulation of immunity and microbiota composition. Clin. Exp. Immunol. 2011, 163, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Yoda, K.; Sun, X.; Kawase, M.; Kubota, A.; Miyazawa, K.; Harata, G.; Hosoda, M.; Hiramatsu, M.; He, F.; Zemel, M.B. A combination of probiotics and whey proteins enhances anti-obesity effects of calcium and dairy products during nutritional energy restriction in aP2-agouti transgenic mice. Br. J. Nutr. 2015, 113, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Tamanai-Shacoori, Z.; Smida, I.; Bousarghin, L.; Loreal, O.; Meuric, V.; Fong, S.B.; Bonnaure-Mallet, M.; Jolivet-Gougeon, A. Roseburia spp.: A marker of health? Future Microbiol. 2017, 12, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Graessler, J.; Qin, Y.; Zhong, H.; Zhang, J.; Licinio, J.; Wong, M.L.; Xu, A.; Chavakis, T.; Bornstein, A.B.; Ehrhart-Bornstein, M.; et al. Metagenomic sequencing of the human gut microbiome before and after bariatric surgery in obese patients with type 2 diabetes: Correlation with inflammatory and metabolic parameters. Pharmacogenomics J. 2013, 13, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Heiss, C.N.; Olofsson, L.E. Gut Microbiota-Dependent Modulation of Energy Metabolism. J. Innate Immun. 2018, 10, 163–171. [Google Scholar] [CrossRef]
- Fetissov, S.O. Role of the gut microbiota in host appetite control: Bacterial growth to animal feeding behaviour. Nat. Rev. Endocrinol. 2017, 13, 11–25. [Google Scholar] [CrossRef]
- Maya-Lucas, O.; Murugesan, S.; Nirmalkar, K.; Alcaraz, L.D.; Hoyo-Vadillo, C.; Pizano-Zárate, M.L.; García-Mena, J. The gut microbiome of Mexican children affected by obesity. Anaerobe 2019, 55, 11–23. [Google Scholar] [CrossRef]
- Del Chierico, F.; Abbatini, F.; Russo, A.; Quagliariello, A.; Reddel, S.; Capoccia, D.; Caccamo, R.; Corradini, S.G.; Nobili, V.; De Peppo, F.; et al. Gut microbiota markers in obese adolescent and adult patients: Age-dependent differential patterns. Front. Microbiol. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Bairey Merz, C.N.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults. Circulation 2014, 129, S1–S45. [Google Scholar] [CrossRef]
- Haro, C.; Rangel-zú, O.A.; Alcalá-díaz, J.F.; Gómez-Delgado, F.; Pérez-martínez, P.; Delgado-lista, J.; Quintana-Navarro, G.M. Intestinal Microbiota Is Influenced by Gender and Body Mass Index. PLoS ONE 2016, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Avila, M.; Romieu, I.; Parra, S.; Hernández-Avila, J.; Madrigal, H.; Willett, W. Validity and reproducibility of a food frequency questionnaire to assess dietary intake of women living in Mexico City. Salud Publica Mex. 1998, 40, 133–140. [Google Scholar] [CrossRef]
- García-Mena, J.; Murugesan, S.; Pérez-Muñoz, A.A.; García-Espitia, M.; Maya, O.; Jacinto-Montiel, M.; Monsalvo-Ponce, G.; Piña-Escobedo, A.; Domínguez-Malfavón, L.; Gómez-Ramírez, M.; et al. Airborne Bacterial Diversity from the Low Atmosphere of Greater Mexico City. Microb. Ecol. 2016, 72, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, J.; Brejnrod, A.; Mortensen, M.; Rasmussen, M.A.; Stokholm, J.; Al-Soud, W.A.; Sørensen, S.; Bisgaard, H.; Waage, J. Large-scale benchmarking reveals false discoveries and count transformation sensitivity in 16S rRNA gene amplicon data analysis methods used in microbiome studies. Microbiome 2016, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Hawinkel, S.; Mattiello, F.; Bijnens, L.; Thas, O. A broken promise: Microbiome differential abundance methods do not control the false discovery rate. Brief. Bioinform. 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.F.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 1814–1821. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Bacchetti De Gregoris, T.; Aldred, N.; Clare, A.S.; Burgess, J.G. Improvement of phylum- and class-specific primers for real-time PCR quantification of bacterial taxa. J. Microbiol. Methods 2011, 86, 351–356. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Obesity | Obesity + Metabolic Syndrome | p-Value | |
|---|---|---|---|---|
| Number of subjects | 25 | 17 | 25 | |
| Age (years) | 23.3 ± 3.1 | 38.8 ± 8.4 * | 40.5 ± 10.3 * | <0.001 |
| Age range | 18 to 30 | 22 to 51 | 22 to 59 | nd |
| Anthropometric data | ||||
| Height (m) | 1.61 ± 0.05 | 1.55 ± 0.08 * | 1.56 ± 0.06 * | 0.002 |
| Weight (kg) | 55.3 ± 6.4 | 83.2 ± 14.1 * | 86.8 ± 15.1 * | <0.001 |
| BMI (kg/m2) | 21.4 ± 1.9 | 34.8 ± 6.1 * | 35.8 ± 5.1 * | <0.001 |
| WC (cm) | 71.4 ± 7.0 | 96.9 ± 11.1 * | 99.6 ± 10.2 * | <0.001 a |
| HC (cm) | 92.0 ± 6.7 | 114.6 ± 11.1 * | 115.7 ± 11.8 * | <0.001 a |
| W/H ratio | 0.78 ± 0.6 | 0.85 ± 0.08 * | 0.86 ± 0.05 * | <0.001 a |
| Biochemical data | ||||
| Fasting Glucose (mg/dL) | 84.2 ± 5.5 | 91.6 ± 05.6 | 104.6 ± 17.5 * | <0.001 |
| Triglycerides (mg/dL) | 109.4 ± 29.1 | 102.2 ± 27.0 | 177.2 ± 69.6 * | <0.001 |
| HDL (mg/dL) | 60.6 ± 12.4 | 45.6 ± 11.5 * | 42.4 ± 8.4 * | <0.001 |
| LDL (mg/dL) | 87.4 ± 24.2 | 102.0 ± 21.1 | 116.4 ± 23.3 * | <0.001 a |
| Cholesterol (mg/dL) | 170.0 ± 26.2 | 168.0 ± 26.2 | 194.3 ± 29.0 * | 0.002 |
| LDL-c/HDL-c ratio | 1.5 ± 0.6 | 2.5 ± 1.0 * | 2.9 ± 0.8 * | <0.001 a |
| C/HDL-c ratio | 2.9 ± 0.7 | 3.9 ± 1.1 * | 4.7 ± 1.1 * | <0.001 |
| Blood pressure | ||||
| SBP (mm Hg) | 109.3 ± 10.7 | 122.7 ± 16.6 * | 125.4 ± 13.1 * | <0.001 |
| DBP (mm Hg) | 71.9 ± 10.9 | 80.2 ± 13.7 | 85.5 ± 12.7 * | 0.003 |
| Taxa | This Work | Other Reports | Reference |
|---|---|---|---|
| Phylum Proteobacteria | |||
| Succinivibrio spp. | 3-fold more abundant in OB than CO and OMS | Succinivibrio spp. and Halomonas spp. overrepresented in Brazilian subjects with overweight and omnivores, this group showed higher values of insulin and HOMA-IR and a worse lipid profile. | [33] |
| Phylum Bacteroidetes | |||
| Family S24–7 | 3-fold more abundant in OB than CO and OMS | LEfSe analysis indicated that members of the LPS-producing family S24–7 were 5-fold more abundant in mice on a high-fat diet versus a high-fat diet plus capsaicin (potent anti-obesity function). | [34] |
| Bacteroides spp. | 4-fold more abundant in CO than OB and OMS | In women from Austria, the relative abundance of Bacteroidaceae family (genera from Bacteroides) was significantly decreased in obese but not non-obese women. | [18] |
| Parabacteroides spp. | 3-fold more abundant in CO than OB and OMS | Oral administration of Parabacteroides distasonis antigens attenuates experimental murine colitis through modulation of immunity and microbiota composition. | [35] |
| Phylum Firmicutes | |||
| Streptococcus spp. | 3-fold more abundant in CO than OB and OMS | S. thermophilus, a probiotic, is associated with reduction of body weight, fat accumulation, and fatty acid synthase activity in adipocytes in mice. | [36] |
| Roseburia spp. | 3-fold more abundant in OB than CO and OMS | Commensal bacteria producing short-chain fatty acids, especially butyrate. It is associated with several diseases including irritable bowel syndrome, obesity, type 2 diabetes, nervous system conditions, and allergies. | [37] |
| Family Erysipelotrichaceae | 3-fold more abundant in CO than OB and OMS | High relative abundances of Erispielotrichaceae have been reported in women from Austria with impaired fasting glucose vs. normal glucose. | [18] |
| Strongest evidence for a role for Erysipelotrichaceae in human disease comes from studies related to metabolic disorders. | [29] | ||
| Coprococcus spp. | 4-fold more abundant in OMS than OB and CO | C. comes abundance decreases along with BMI, serum triglycerides, cholesterol, and LDL-cholesterol in German adults affected with T2D/OB after Roux-en-Y gastric-bypass surgery. | [38] |
| Family Lachnospiraceae & Lachnospira spp. | 4-fold more abundant in OMS than OB and CO | Lachnospiraceae were more abundant in Austrian subjects with impaired fasting glucose and impaired glucose tolerance, and in women with MS. | [18] |
| Turicibacter spp. | 3-fold more abundant in CO than OB and OMS | Turicibacter spp. had a negative association with clinical indicators of metabolic disorder such as insulin and HbA1c in Austrian population. | [18] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chávez-Carbajal, A.; Nirmalkar, K.; Pérez-Lizaur, A.; Hernández-Quiroz, F.; Ramírez-del-Alto, S.; García-Mena, J.; Hernández-Guerrero, C. Gut Microbiota and Predicted Metabolic Pathways in a Sample of Mexican Women Affected by Obesity and Obesity Plus Metabolic Syndrome. Int. J. Mol. Sci. 2019, 20, 438. https://doi.org/10.3390/ijms20020438
Chávez-Carbajal A, Nirmalkar K, Pérez-Lizaur A, Hernández-Quiroz F, Ramírez-del-Alto S, García-Mena J, Hernández-Guerrero C. Gut Microbiota and Predicted Metabolic Pathways in a Sample of Mexican Women Affected by Obesity and Obesity Plus Metabolic Syndrome. International Journal of Molecular Sciences. 2019; 20(2):438. https://doi.org/10.3390/ijms20020438
Chicago/Turabian StyleChávez-Carbajal, Alejandra, Khemlal Nirmalkar, Ana Pérez-Lizaur, Fernando Hernández-Quiroz, Silvia Ramírez-del-Alto, Jaime García-Mena, and César Hernández-Guerrero. 2019. "Gut Microbiota and Predicted Metabolic Pathways in a Sample of Mexican Women Affected by Obesity and Obesity Plus Metabolic Syndrome" International Journal of Molecular Sciences 20, no. 2: 438. https://doi.org/10.3390/ijms20020438
APA StyleChávez-Carbajal, A., Nirmalkar, K., Pérez-Lizaur, A., Hernández-Quiroz, F., Ramírez-del-Alto, S., García-Mena, J., & Hernández-Guerrero, C. (2019). Gut Microbiota and Predicted Metabolic Pathways in a Sample of Mexican Women Affected by Obesity and Obesity Plus Metabolic Syndrome. International Journal of Molecular Sciences, 20(2), 438. https://doi.org/10.3390/ijms20020438