WRKYs, the Jack-of-various-Trades, Modulate Dehydration Stress in Populus davidiana—A Transcriptomic Approach

 , , ,
, , , 
Abstract
1. Introduction
2. Results
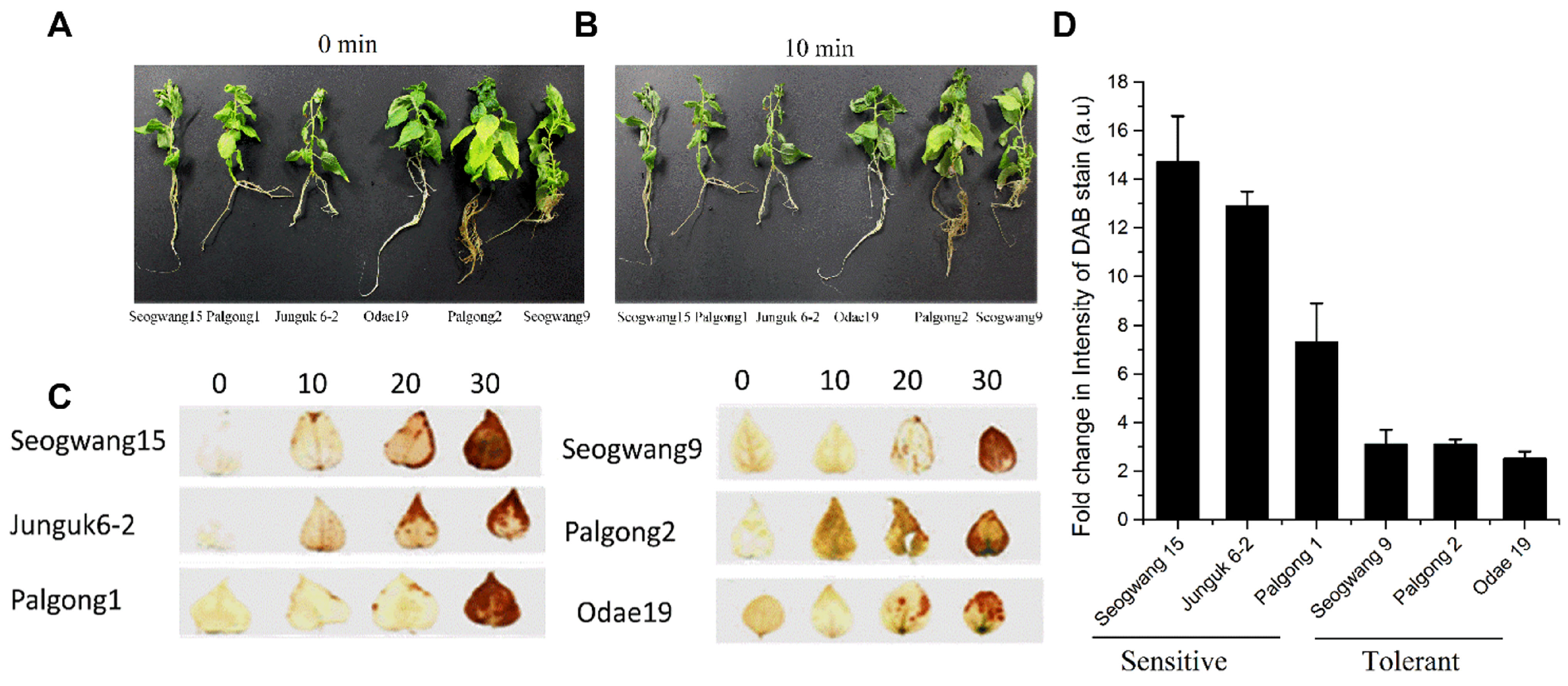
2.1. Selection of Drought Sensitive and Tolerant Cultivars
2.2. Transcriptome-Wide Identification of PopdaWRKYs
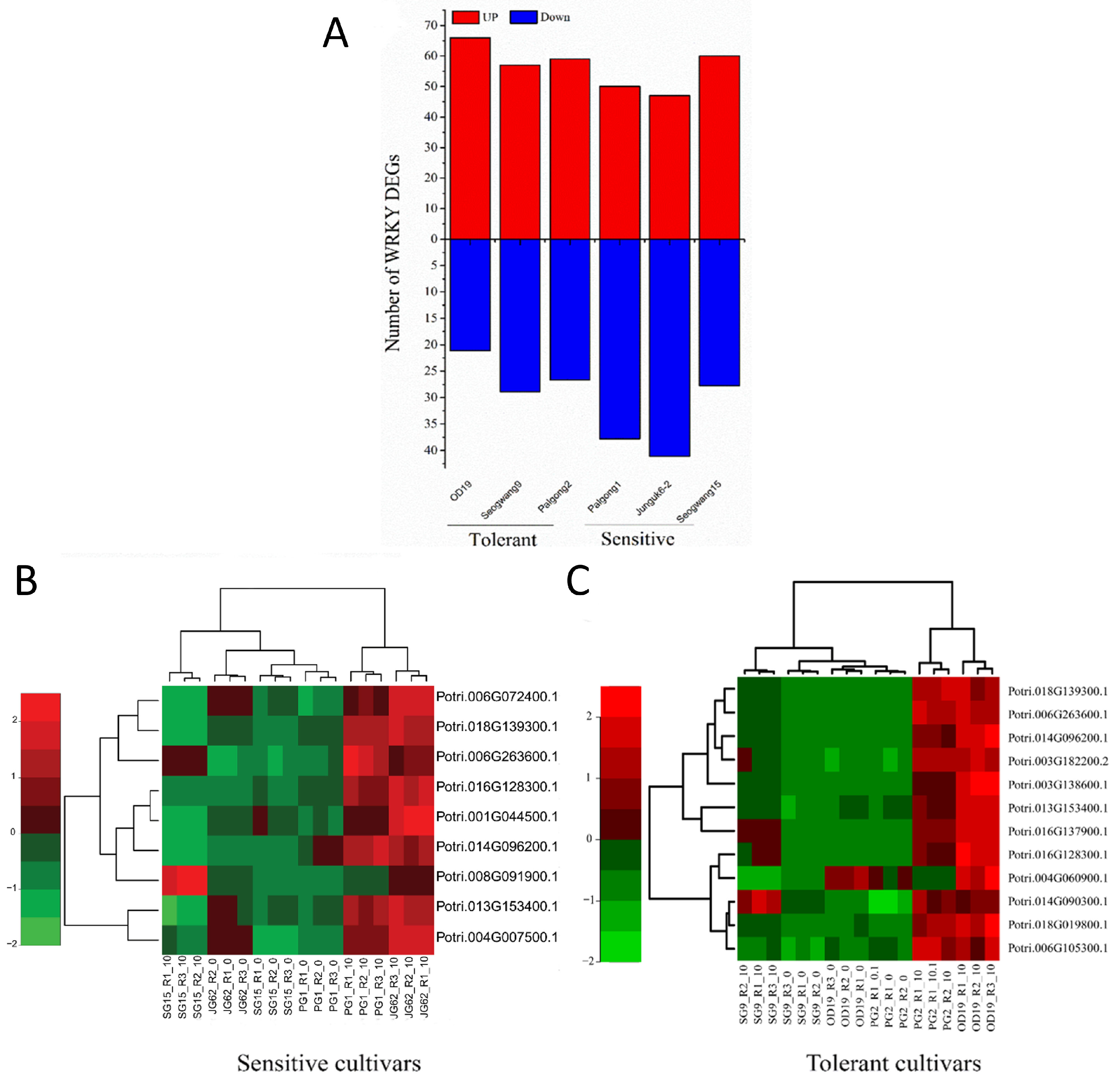
2.3. Global Gene Regulation Is Different in Sensitive and Tolerant P. davidiana Cultivars
2.4. Chromosomal Location of PopdaWRKYs and Their Orthologs in Other Species
2.5. Fold Enrichment of GO Terms
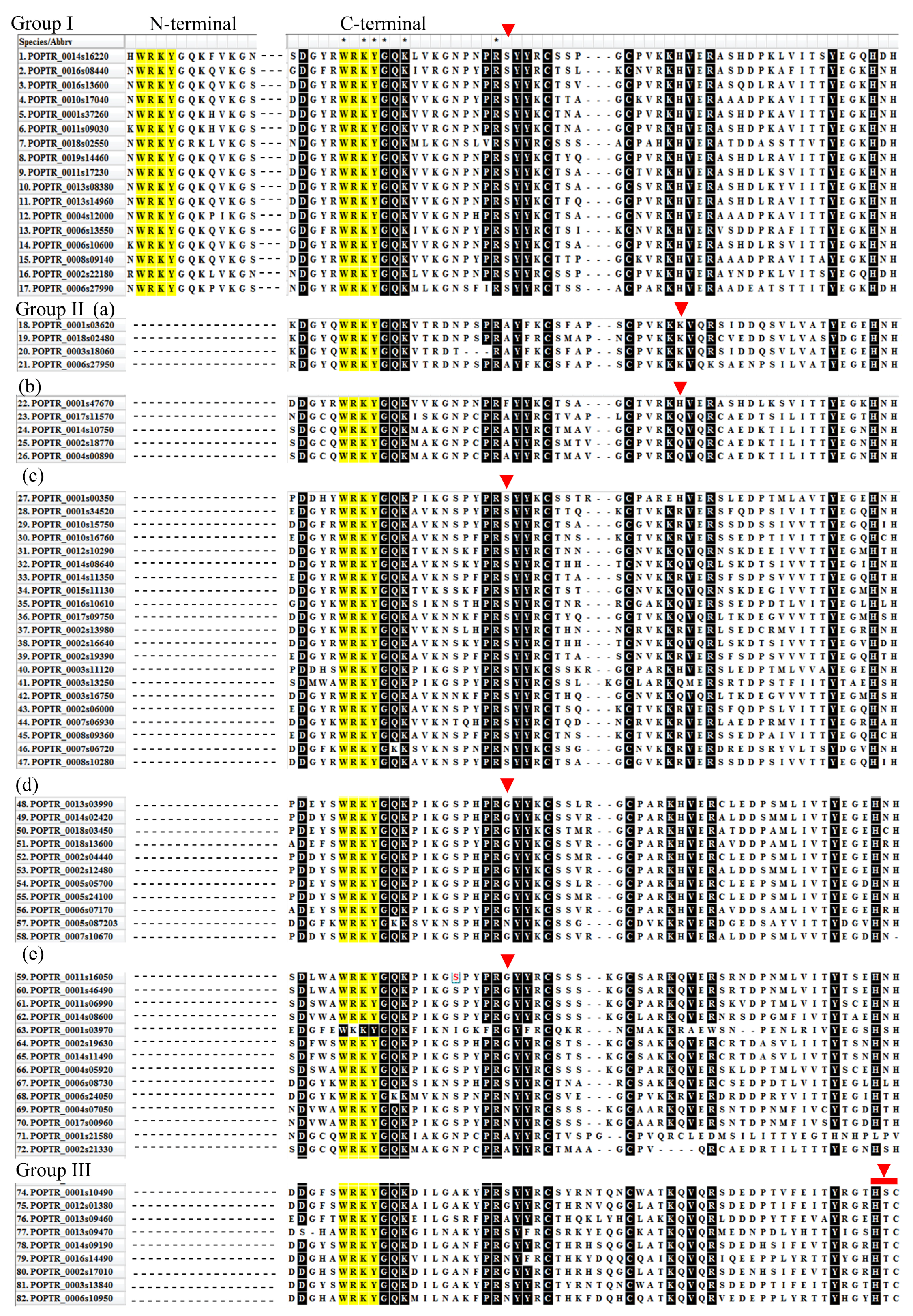
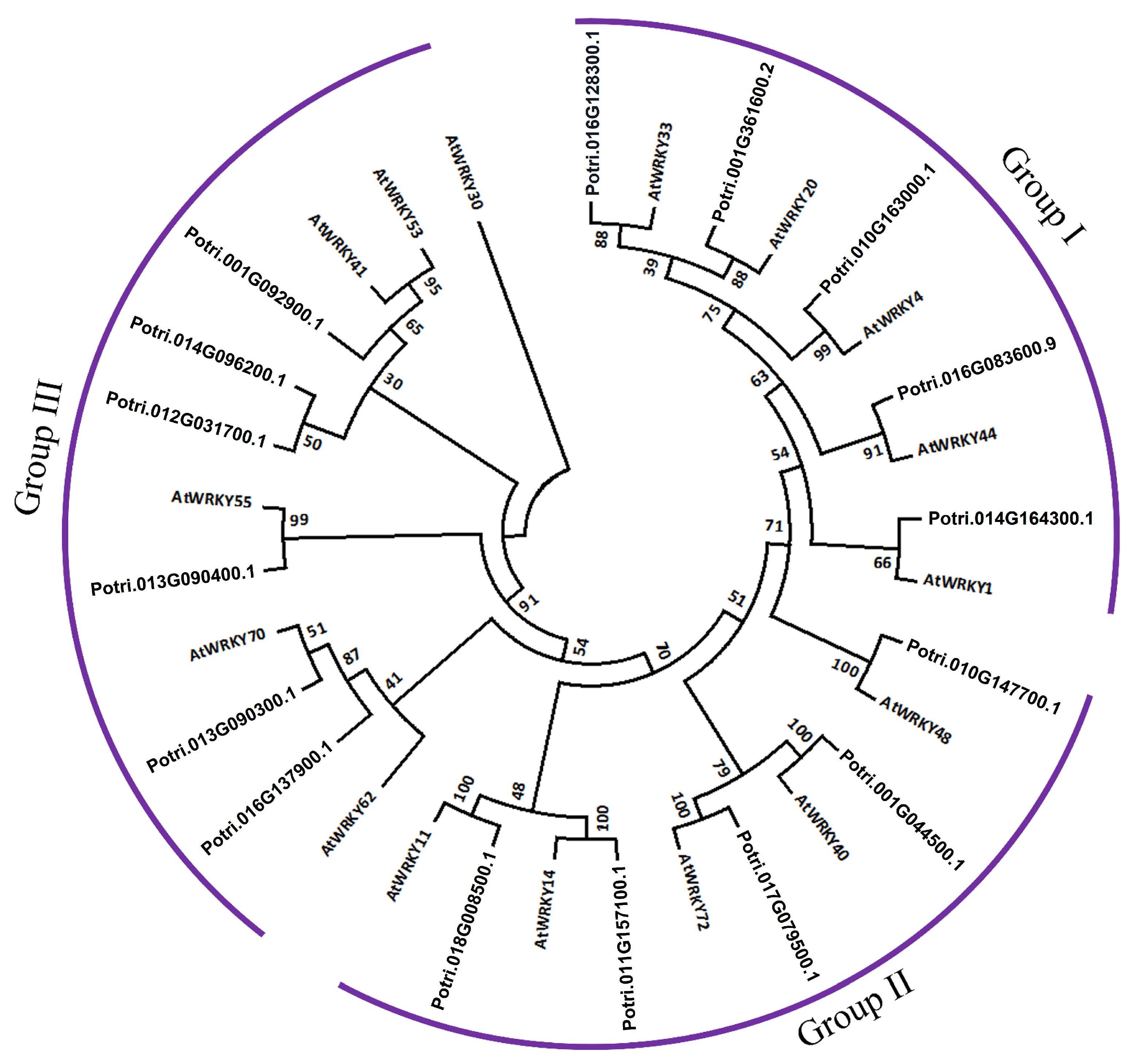
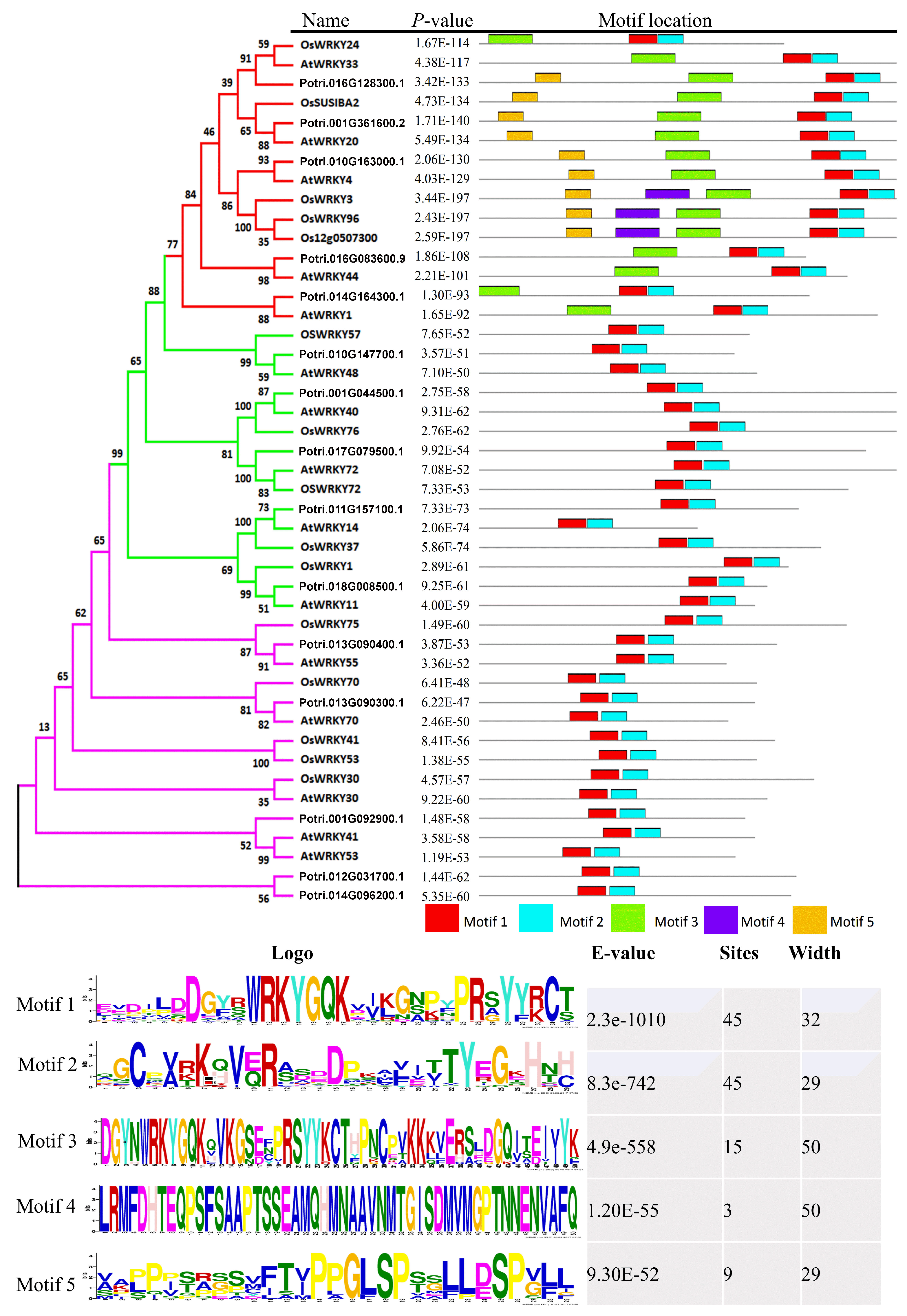
2.6. Classification and Phylogenetic Analysis
2.7. Structural Divergence in Motif Composition of WRKYs in P. davidiana and Its Orthologs in Other Species
2.8. Involvement of Dehydration-Responsive WRKYs in Different Metabolic Pathways
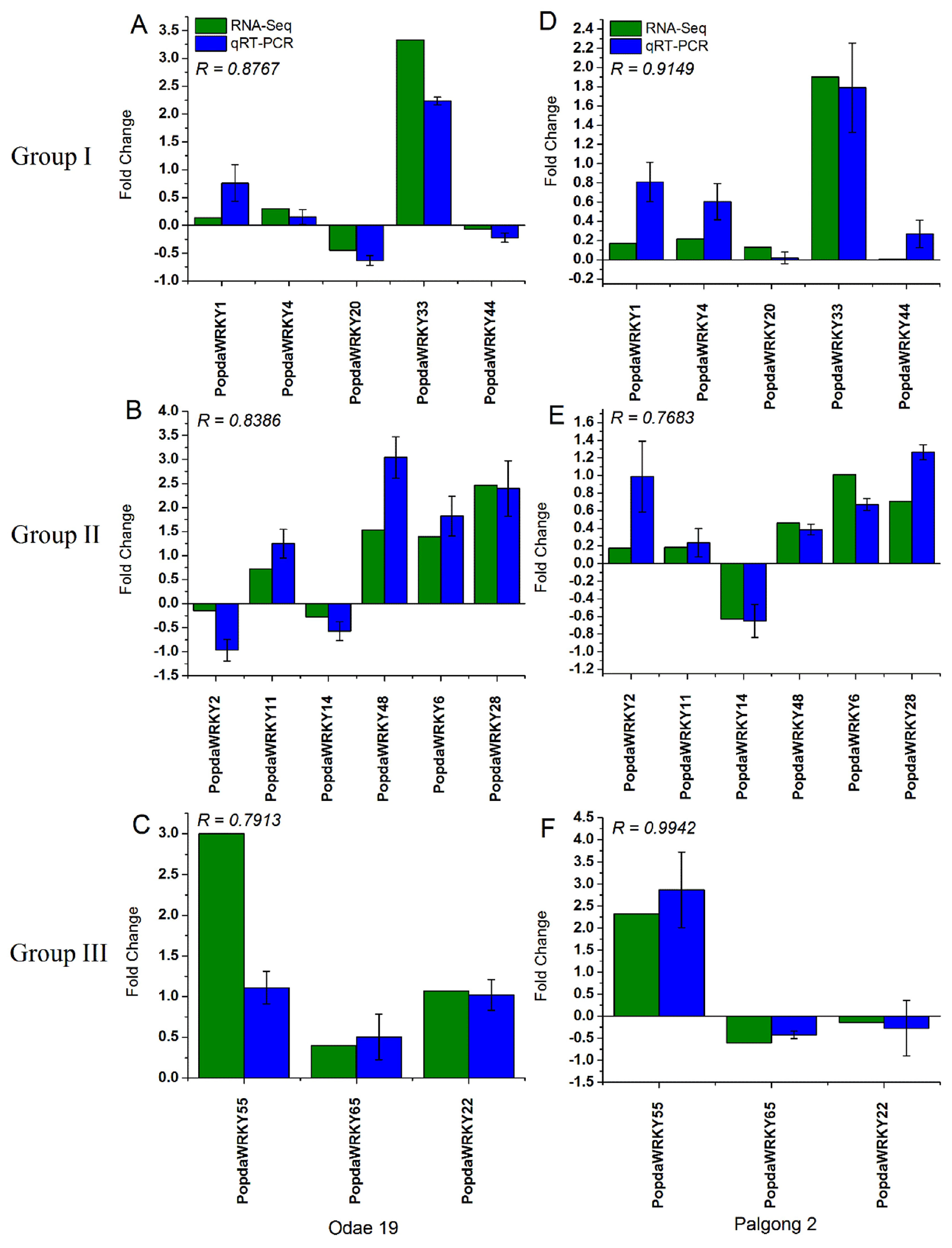
2.9. RNA-seq and qRT-PCR Analysis Are Highly Correlated with Each Other
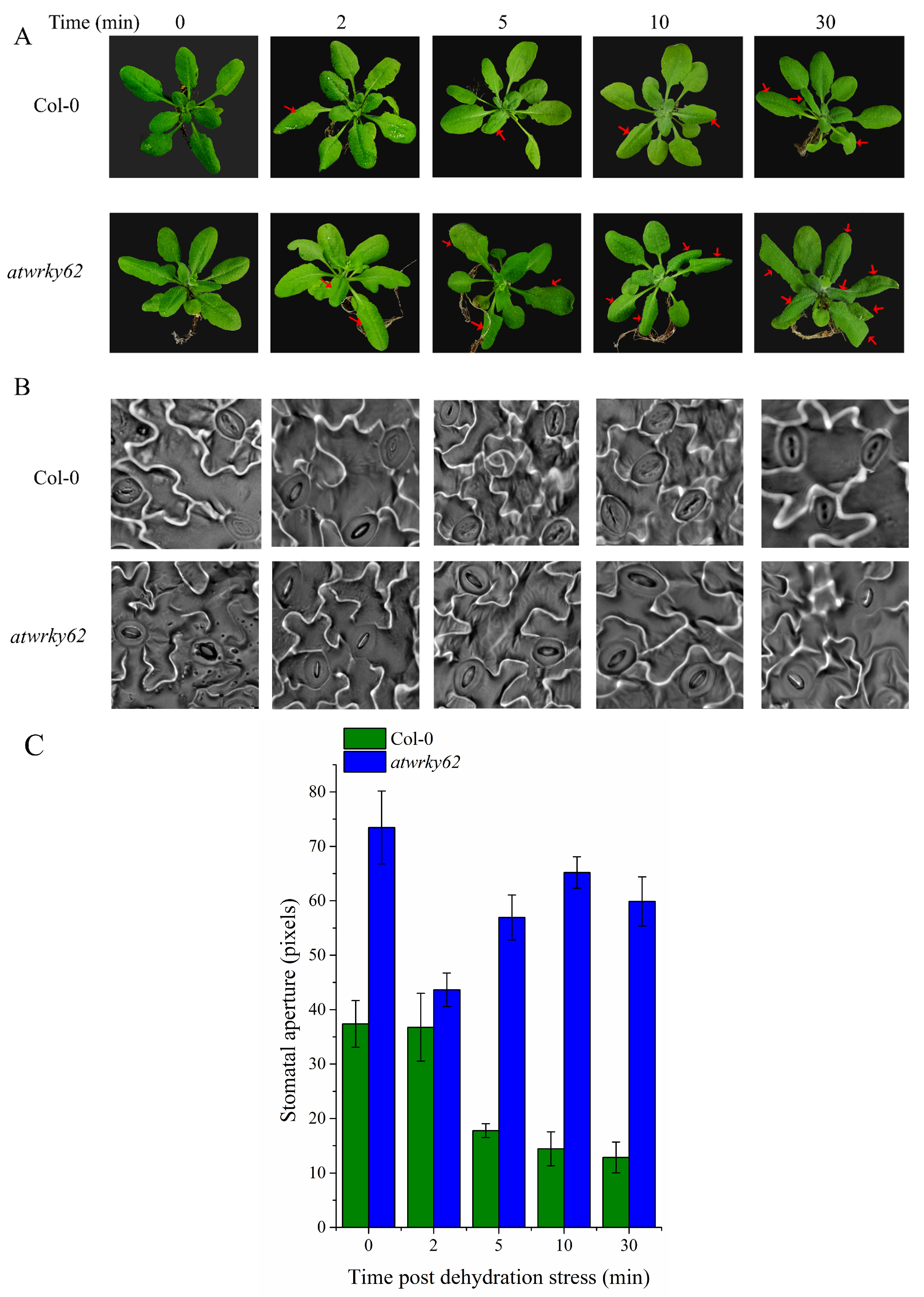
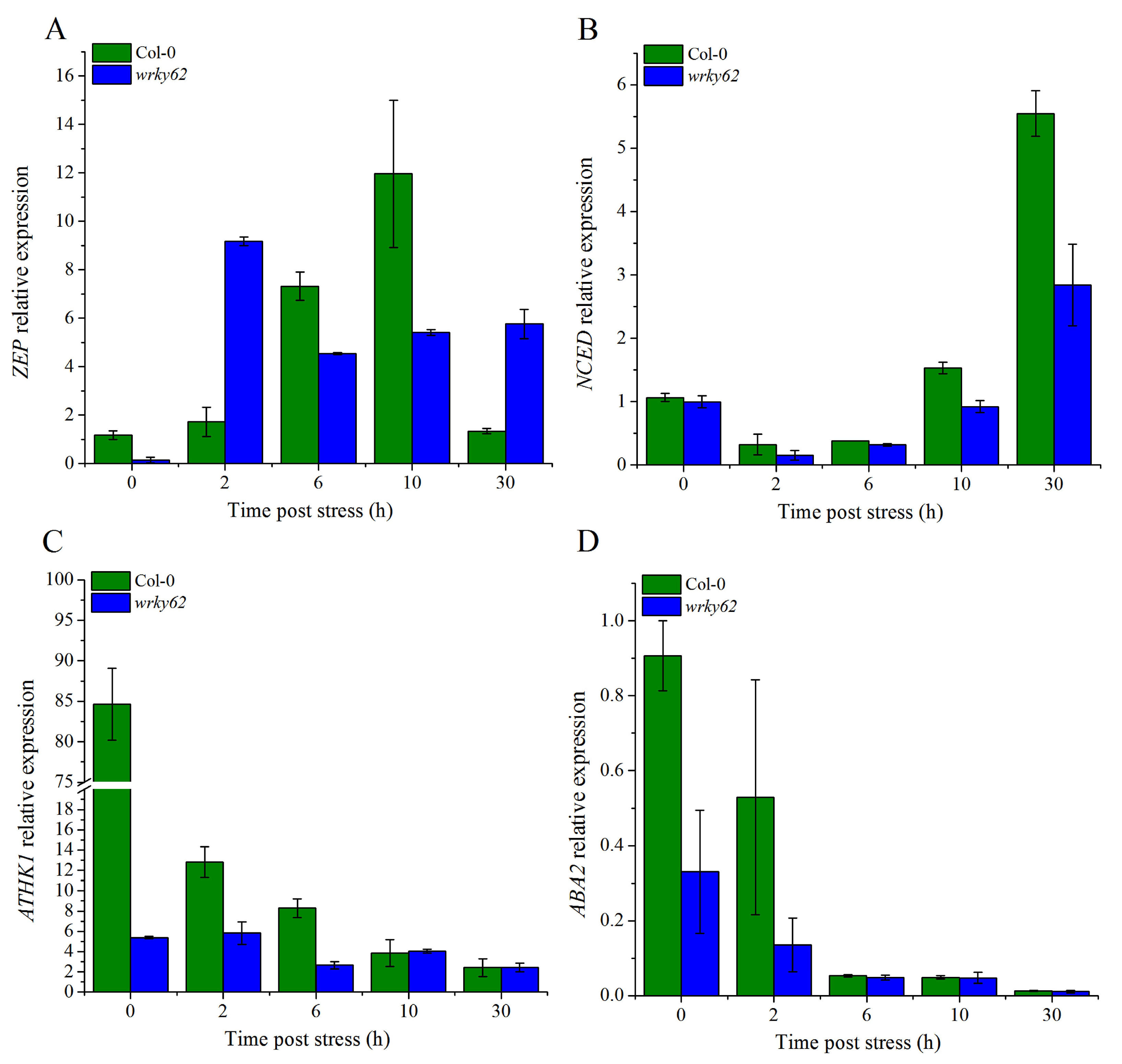
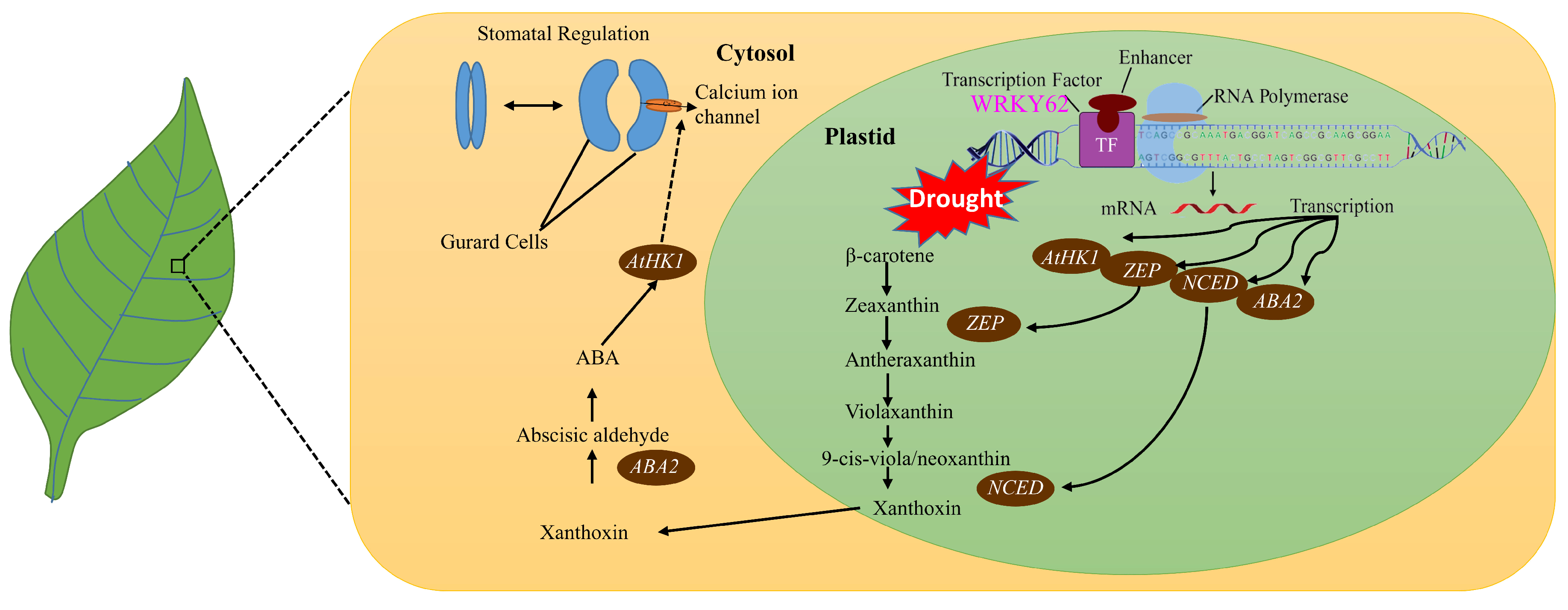
2.10. AtWRKY62 Modulates Dehydration Stress via Stomatal Regulation and Transcriptional Regulation of the ABA Pathway in Arabidopsis thaliana
3. Discussion
4. Materials and Methods
4.1. Plant Material and Induction of Dehydration Stress
4.2. RNA Extraction and RNA Sequencing
4.3. Identification and Classification of WRKY TFs in Transcriptome Data
4.4. Identification of Common DEGs among Tolerant and Sensitive P. davidiana Cultivars
4.5. Chromosomal Location of P. davidiana WRKYs and Their Orthologs in Other Plant Species
4.6. Classification, Phylogenetic Analysis, and Motif Composition of PopdaWRKYs
4.7. Fold Enrichment Analysis
4.8. KEGG Pathway Enrichment Analysis
4.9. In Vivo Analysis of Stomatal Regulation by WRKY TF in Arabidopsis
4.10. Expression Analysis of ABA Biosynthesis and Signaling Genes
4.11. Validation through Real-Time Reverse Transcription PCR (qRT-PCR)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ROS | reactive oxygen species |
| RNS | reactive nitrogen species |
| ABA | abscisic acid |
| TF | transcription factor |
| DEG | differential expressed gene |
| MDS | multi dimensional scatter plot |
| FPKM | fragment per kilobase million |
| GO | gene ontology |
| BLAST | basic local alignment search tool |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MAPK | mitogen-activated protein kinase |
| PAD3 | phytoalexin deficient3 |
| FLS2 | flagellin sensitive2 |
| PR5.1 | pathogenesis-related protein5.1 |
| SA | salicylic acid |
| Pst | Pseudomonas syringae pv. tomato |
| KO | knock out |
| KFRI | Korea Forest Research Institute |
| NAA | naphthalene acetic acid |
| MS | Murashige and Skoog |
| NCBI | National Center for Biotechnology Information. |
References
- Fedoroff, N.V.; Battisti, D.S.; Beachy, R.N.; Cooper, P.J.; Fischhoff, D.A.; Hodges, C.; Knauf, V.C.; Lobell, D.; Mazur, B.J.; Molden, D.; et al. Radically rethinking agriculture for the 21st century. Science 2010, 327, 833–834. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.J.; Park, W.; Bauer, P.J.; Udall, J.A.; Page, J.T.; Raney, J.; Scheffler, B.E.; Jones, D.C.; Campbell, B.T. RNA-Seq transcriptome profiling of upland cotton (Gossypium hirsutum L.) root tissue under water-deficit stress. PLoS ONE 2013, 8, e82634. [Google Scholar] [CrossRef] [PubMed]
- Deeba, F.; Pandey, A.K.; Ranjan, S.; Mishra, A.; Singh, R.; Sharma, Y.; Shirke, P.A.; Pandey, V. Physiological and proteomic responses of cotton (Gossypium herbaceum L.) to drought stress. Plant Physiol. Biochem. 2012, 53, 6–18. [Google Scholar] [CrossRef]
- Lawlor, D.W.; Cornic, G. Photosynthetic carbon assimilation and associated metabolism in relation to water deficits in higher plants. Plant Cell Environ. 2002, 25, 275–294. [Google Scholar] [CrossRef] [PubMed]
- Ackerson, R.C. Stomatal response of cotton to water stress and abscisic acid as affected by water stress history. Plant Physiol. 1980, 65, 455–459. [Google Scholar] [CrossRef]
- Aranjuelo, I.; Molero, G.; Erice, G.; Avice, J.C.; Nogués, S. Plant physiology and proteomics reveals the leaf response to drought in alfalfa (Medicago sativa L.). J. Exp. Bot. 2010, 62, 111–123. [Google Scholar] [CrossRef]
- Jubany-Marí, T.; Munné-Bosch, S.; Alegre, L. Redox regulation of water stress responses in field-grown plants. Role of hydrogen peroxide and ascorbate. Plant Physiol. Biochem. 2010, 48, 351–358. [Google Scholar] [CrossRef]
- Zhu, J.K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef]
- Gewin, V. Food: An underground revolution. Nat. News 2010, 466, 552–553. [Google Scholar] [CrossRef]
- Deak, K.I.; Malamy, J. Osmotic regulation of root system architecture. Plant J. 2005, 43, 17–28. [Google Scholar] [CrossRef]
- Takahashi, N.; Yamazaki, Y.; Kobayashi, A.; Higashitani, A.; Takahashi, H. Hydrotropism interacts with gravitropism by degrading amyloplasts in seedling roots of Arabidopsis and radish. Plant Physiol. 2003, 132, 805–810. [Google Scholar] [CrossRef]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Malamy, J. Intrinsic and environmental response pathways that regulate root system architecture. Plant Cell Environ. 2005, 28, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Moumeni, A.; Satoh, K.; Kondoh, H.; Asano, T.; Hosaka, A.; Venuprasad, R.; Serraj, R.; Kumar, A.; Leung, H.; Kikuchi, S. Comparative analysis of root transcriptome profiles of two pairs of drought-tolerant and susceptible rice near-isogenic lines under different drought stress. BMC Plant Biol. 2011, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- Bruce, W.B.; Edmeades, G.O.; Barker, T.C. Molecular and physiological approaches to maize improvement for drought tolerance. J. Exp. Bot. 2002, 53, 13–25. [Google Scholar] [CrossRef]
- Dubos, C.; Plomion, C. Identification of water-deficit responsive genes in maritime pine (Pinus pinaster Ait.) roots. Plant Mol. Biol. 2003, 51, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Provart, N.J.; Glazebrook, J.; Katagiri, F.; Chang, H.S.; Eulgem, T.; Mauch, F.; Luan, S.; Zou, G.; Whitham, S.A.; et al. Expression profile matrix of Arabidopsis transcription factor genes suggests their putative functions in response to environmental stresses. Plant Cell 2002, 14, 559–574. [Google Scholar] [CrossRef]
- Kalde, M.; Barth, M.; Somssich, I.E.; Lippok, B. Members of the Arabidopsis WRKY group III transcription factors are part of different plant defense signaling pathways. Mol. Plant-Microbe Interact. 2003, 16, 295–305. [Google Scholar] [CrossRef]
- Pabo, C.O.; Sauer, R.T. Transcription factors: Structural families and principles of DNA recognition. Annu. Rev. Biochem. 1992, 61, 1053–1095. [Google Scholar] [CrossRef]
- Bakshi, M.; Oelmüller, R. WRKY transcription factors: Jack of many trades in plants. Plant Signal. Behav. 2014, 9, e27700. [Google Scholar] [CrossRef]
- Cheong, Y.H.; Chang, H.S.; Gupta, R.; Wang, X.; Zhu, T.; Luan, S. Transcriptional profiling reveals novel interactions between wounding, pathogen, abiotic stress, and hormonal responses in Arabidopsis. Plant Physiol. 2002, 129, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Huaxia, Y.; Lu, W.; Wu, C.; Cao, X.; Guo, X. GhWRKY15, a member of the WRKY transcription factor family identified from cotton (Gossypium hirsutum L.), is involved in disease resistance and plant development. BMC Plant Biol. 2012, 12, 144. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Narusaka, M.; Ishida, J.; Nanjo, T.; Fujita, M.; Oono, Y.; Kamiya, A.; Nakajima, M.; Enju, A.; Sakurai, T.; et al. Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 2002, 31, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Azeem, F.; Nawaz, M.A.; Acet, T.; Abbas, A.; Imran, Q.M.; Shah, K.H.; Rehman, H.M.; Chung, G.; Yang, S.H.; et al. Transcription factors WRKY11 and WRKY17 are involved in abiotic stress responses in Arabidopsis. J. Plant Physiol. 2018, 226, 12–21. [Google Scholar] [CrossRef]
- Imran, Q.M.; Hussain, A.; Mun, B.G.; Lee, S.U.; Asaf, S.; Ali, M.A.; Lee, I.J.; Yun, B.W. Transcriptome wide identification and characterization of NO-responsive WRKY transcription factors in Arabidopsis thaliana L. Environ. Exp. Bot. 2018, 148, 128–143. [Google Scholar] [CrossRef]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Pandey, S.P.; Somssich, I.E. The role of WRKY transcription factors in plant immunity. Plant Physiol. 2009, 150, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef]
- Liu, J.J.; Ekramoddoullah, A.K. Identification and characterization of the WRKY transcription factor family in Pinus monticola. Genome 2009, 52, 77–88. [Google Scholar] [CrossRef]
- Mangelsen, E.; Kilian, J.; Berendzen, K.W.; Kolukisaoglu, Ü.H.; Harter, K.; Jansson, C.; Wanke, D. Phylogenetic and comparative gene expression analysis of barley (Hordeum vulgare) WRKY transcription factor family reveals putatively retained functions between monocots and dicots. BMC Genom. 2008, 9, 194. [Google Scholar] [CrossRef]
- Agarwal, P.; Reddy, M.; Chikara, J. WRKY: Its structure, evolutionary relationship, DNA-binding selectivity, role in stress tolerance and development of plants. Mol. Biol. Rep. 2011, 38, 3883–3896. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Ai, C.R.; Jing, S.J.; Yu, D.Q. Research progress on functional analysis of rice WRKY genes. Rice Sci. 2010, 17, 60–72. [Google Scholar]
- Breshears, D.D.; Cobb, N.S.; Rich, P.M.; Price, K.P.; Allen, C.D.; Balice, R.G.; Romme, W.H.; Kastens, J.H.; Floyd, M.L.; Belnap, J.; et al. Regional vegetation die-off in response to global-change-type drought. Proc. Natl. Acad. Sci. USA 2005, 102, 15144–15148. [Google Scholar] [CrossRef] [PubMed]
- Hicke, J.A.; Zeppel, M.J. Climate-driven tree mortality: Insights from the piñon pine die-off in the United States. New Phytol. 2013, 200, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [PubMed]
- Qiu, Q.; Ma, T.; Hu, Q.; Liu, B.; Wu, Y.; Zhou, H.; Wang, Q.; Wang, J.; Liu, J. Genome-scale transcriptome analysis of the desert poplar, Populus euphratica. Tree Physiol. 2011, 31, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Zhou, T.; Bo, W.; Xu, F.; Wu, R. Genome-wide analysis of salt-responsive and novel microRNAs in Populus euphratica by deep sequencing. BMC Genet. 2014, 15 (Suppl. 1), S6. [Google Scholar] [CrossRef]
- Yan, D.H.; Fenning, T.; Tang, S.; Xia, X.; Yin, W. Genome-wide transcriptional response of Populus euphratica to long-term drought stress. Plant Sci. 2012, 195, 24–35. [Google Scholar] [CrossRef]
- Rockman, M.V.; Skrovanek, S.S.; Kruglyak, L. Selection at linked sites shapes heritable phenotypic variation in C. elegans. Science 2010, 330, 372–376. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Marin, E.; Nussaume, L.; Quesada, A.; Gonneau, M.; Sotta, B.; Hugueney, P.; Frey, A.; Marion-Poll, A. Molecular identification of zeaxanthin epoxidase of Nicotiana plumbaginifolia, a gene involved in abscisic acid biosynthesis and corresponding to the ABA locus of Arabidopsis thaliana. EMBO J. 1996, 15, 2331–2342. [Google Scholar] [CrossRef] [PubMed]
- North, H.M.; Almeida, A.D.; Boutin, J.P.; Frey, A.; To, A.; Botran, L.; Sotta, B.; Marion-Poll, A. The Arabidopsis ABA-deficient mutant aba4 demonstrates that the major route for stress-induced ABA accumulation is via neoxanthin isomers. Plant J. 2007, 50, 810–824. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.L.; Benning, G.; Grill, E. ABI2, a second protein phosphatase 2C involved in abscisic acid signal transduction in Arabidopsis. FEBS Lett. 1998, 421, 185–190. [Google Scholar] [CrossRef]
- Lü, D.; Wang, W.; Miao, C. ATHK1 acts downstream of hydrogen peroxide to mediate ABA signaling through regulation of calcium channel activity in Arabidopsis guard cells. Chin. Sci. Bull. 2013, 58, 336–343. [Google Scholar] [CrossRef]
- Li, B.; Qin, Y.; Duan, H.; Yin, W.; Xia, X. Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J. Exp. Bot. 2011, 62, 3765–3779. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, M.; Uma, S.; Backiyarani, S.; Saraswathi, M.S.; Chandrasekar, A. Transcriptomic changes of drought-tolerant and sensitive banana cultivars exposed to drought stress. Front. Plant Sci. 2016, 7, 1609. [Google Scholar] [CrossRef]
- Maleck, K.; Levine, A.; Eulgem, T.; Morgan, A.; Schmid, J.; Lawton, K.A.; Dangl, J.L.; Dietrich, R.A. The transcriptome of Arabidopsis thaliana during systemic acquired resistance. Nat. Genet. 2000, 26, 403–410. [Google Scholar] [CrossRef]
- Fan, X.; Guo, Q.; Xu, P.; Gong, Y.; Shu, H.; Yang, Y.; Ni, W.; Zhang, X.; Shen, X. Transcriptome-wide identification of salt-responsive members of the WRKY gene family in Gossypium aridum. PLoS ONE 2015, 10, e0126148. [Google Scholar] [CrossRef]
- Jiang, Y.; Duan, Y.; Yin, J.; Ye, S.; Zhu, J.; Zhang, F.; Lu, W.; Fan, D.; Luo, K. Genome-wide identification and characterization of the Populus WRKY transcription factor family and analysis of their expression in response to biotic and abiotic stresses. J. Exp. Bot. 2014, 65, 6629–6644. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K. Plant Amino Acids: Biochemistry and Biotechnology; CRC Press: Boca Raton, FL, USA, 1998. [Google Scholar]
- Joshi, V.; Joung, J.G.; Fei, Z.; Jander, G. Interdependence of threonine, methionine and isoleucine metabolism in plants: Accumulation and transcriptional regulation under abiotic stress. Amino Acids 2010, 39, 933–947. [Google Scholar] [CrossRef]
- Corpas, F.J.; Leterrier, M.; Valderrama, R.; Airaki, M.; Chaki, M.; Palma, J.M.; Barroso, J.B. Nitric oxide imbalance provokes a nitrosative response in plants under abiotic stress. Plant Sci. 2011, 181, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Cristina, M.S.; Petersen, M.; Mundy, J. Mitogen-activated protein kinase signaling in plants. Annu. Rev. Plant Biol. 2010, 61, 621–649. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Xiao, J.; Xie, W.; Liu, H.; Li, X.; Xiong, L.; Wang, S. Rice gene network inferred from expression profiling of plants overexpressing OsWRKY13, a positive regulator of disease resistance. Mol. Plant 2008, 1, 538–551. [Google Scholar]
- Ye, S.; Jiang, Y.; Duan, Y.; Karim, A.; Fan, D.; Yang, L.; Zhao, X.; Yin, J.; Luo, K. Constitutive expression of the poplar WRKY transcription factor PtoWRKY60 enhances resistance to Dothiorella gregaria Sacc. in transgenic plants. Tree Physiol. 2014, 34, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Jiang, Y.; Ye, S.; Karim, A.; Ling, Z.; He, Y.; Yang, S.; Luo, K. PtrWRKY73, a salicylic acid-inducible poplar WRKY transcription factor, is involved in disease resistance in Arabidopsis thaliana. Plant Cell Rep. 2015, 34, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Li, C.L.; Zhang, W. WRKY1 regulates stomatal movement in drought-stressed Arabidopsis thaliana. Plant Mol. Biol. 2016, 91, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Liu, N.; Virlouvet, L.; Riethoven, J.J.; Fromm, M.; Avramova, Z. Four distinct types of dehydration stress memory genes in Arabidopsis thaliana. BMC Plant Biol. 2013, 13, 229. [Google Scholar] [CrossRef] [PubMed]
- Apuya, N.R.; Park, J.H.; Zhang, L.; Ahyow, M.; Davidow, P.; Van Fleet, J.; Rarang, J.C.; Hippley, M.; Johnson, T.W.; Yoo, H.D.; et al. Enhancement of alkaloid production in opium and California poppy by transactivation using heterologous regulatory factors. Plant Biotechnol. J. 2008, 6, 160–175. [Google Scholar] [CrossRef]
- Ding, Z.J.; Yan, J.Y.; Xu, X.Y.; Yu, D.Q.; Li, G.X.; Zhang, S.Q.; Zheng, S.J. Transcription factor WRKY 46 regulates osmotic stress responses and stomatal movement independently in A rabidopsis. Plant J. 2014, 79, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Wang, C.; Chen, X.; Lu, W.; Li, H.; Wang, X.; Hao, L.; Guo, X. The cotton WRKY gene GhWRKY41 positively regulates salt and drought stress tolerance in transgenic Nicotiana benthamiana. PLoS ONE 2015, 10, e0143022. [Google Scholar] [CrossRef] [PubMed]
- Li, J.B.; Luan, Y.S.; Liu, Z. Overexpression of SpWRKY1 promotes resistance to Phytophthora nicotianae and tolerance to salt and drought stress in transgenic tobacco. Physiol. Plant. 2015, 155, 248–266. [Google Scholar] [CrossRef] [PubMed]
- He, G.H.; Xu, J.Y.; Wang, Y.X.; Liu, J.M.; Li, P.S.; Chen, M.; Ma, Y.Z.; Xu, Z.S. Drought-responsive WRKY transcription factor genes TaWRKY1 and TaWRKY33 from wheat confer drought and/or heat resistance in Arabidopsis. BMC Plant Biol. 2016, 16, 116. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Chen, J.; Jiang, Y.; Lin, L.; Cao, Y.; Wang, M.; Zhang, Y.; Rong, J.; Ye, W. Genome-wide investigation and transcriptome analysis of the WRKY gene family in Gossypium. Mol. Genet. Genom. 2015, 290, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Qin, G. EXB1/WRKY71 transcription factor regulates both shoot branching and responses to abiotic stresses. Plant Signal. Behav. 2016, 11, e1150404. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, N.; Hu, R.; Xiang, F. Genome-wide identification of soybean WRKY transcription factors in response to salt stress. SpringerPlus 2016, 5, 920. [Google Scholar] [CrossRef]
- Zou, Z.; Yang, L.; Wang, D.; Huang, Q.; Mo, Y.; Xie, G. Gene structures, evolution and transcriptional profiling of the WRKY gene family in castor bean (Ricinus communis L.). PLoS ONE 2016, 11, e0148243. [Google Scholar] [CrossRef]
- Zhou, H.; Li, Y.; Zhang, Q.; Ren, S.; Shen, Y.; Qin, L.; Xing, Y. Genome-wide analysis of the expression of WRKY family genes in different developmental stages of wild strawberry (Fragaria vesca) fruit. PLoS ONE 2016, 11, e0154312. [Google Scholar] [CrossRef]
- He, Y.; Mao, S.; Gao, Y.; Zhu, L.; Wu, D.; Cui, Y.; Li, J.; Qian, W. Genome-wide identification and expression analysis of WRKY transcription factors under multiple stresses in Brassica napus. PLoS ONE 2016, 11, e0157558. [Google Scholar] [CrossRef]
- Eulgem, T.; Somssich, I.E. Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 2007, 10, 366–371. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Dong, Q.; Shao, Y.; Jiang, H.; Zhu, S.; Cheng, B.; Xiang, Y. Genome-wide survey and characterization of the WRKY gene family in Populus trichocarpa. Plant Cell Rep. 2012, 31, 1199–1217. [Google Scholar] [CrossRef] [PubMed]
- Mun, B.G.; Lee, S.U.; Park, E.J.; Kim, H.H.; Hussain, A.; Imran, Q.M.; Lee, I.J.; Yun, B.W. Analysis of transcription factors among differentially expressed genes induced by drought stress in Populus davidiana. 3 Biotech 2017, 7, 209. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Mun, B.G.; Imran, Q.M.; Lee, S.U.; Adamu, T.A.; Shahid, M.; Kim, K.M.; Yun, B.W. Nitric oxide mediated transcriptome profiling reveals activation of multiple regulatory pathways in Arabidopsis thaliana. Front. Plant Sci. 2016, 7, 975. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in bipolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar] [PubMed]
- Shahzad, R.; Waqas, M.; Khan, A.L.; Hamayun, M.; Kang, S.M.; Lee, I.J. Foliar application of methyl jasmonate induced physio-hormonal changes in Pisum sativum under diverse temperature regimes. Plant Physiol. Biochem. 2015, 96, 406–416. [Google Scholar] [CrossRef]
- Imran, Q.M.; Falak, N.; Hussain, A.; Mun, B.G.; Sharma, A.; Lee, S.U.; Kim, K.M.; Yun, B.W. Nitric oxide responsive heavy metal-associated gene AtHMAD1 contributes to development and disease resistance in Arabidopsis thaliana. Front. Plant Sci. 2016, 7, 1712. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Names | Total | Elements |
|---|---|---|
| Junguk62 Odae19 Palgong1 Palgong2 Seogwang15 Seogwang9 | 5 | Potri.018G139300.1 Potri.016G128300.1 Potri.014G096200.1 Potri.006G263600.1 Potri.013G153400.1 |
| Junguk62 Odae19 Palgong1 Palgong2 Seogwang9 | 1 | Potri.004G060900.1 |
| Junguk62 Odae19 Palgong2 Seogwang15 Seogwang9 | 5 | Potri.006G105300.1 Potri.014G090300.1 Potri.003G182200.2 Potri.003G138600.1 Potri.018G019800.1 |
| Junguk62 Odae19 Palgong1 Seogwang15 Seogwang9 | 1 | Potri.004G007500.1 |
| Junguk62 Odae19 Palgong1 Palgong2 Seogwang15 | 2 | Potri.006G072400.1 Potri.001G044500.1 |
| Junguk62 Odae19 Palgong2 Seogwang15 | 2 | Potri.011G007800.1 Potri.006G109100.1 |
| Junguk62 Palgong1 Seogwang15 Seogwang9 | 1 | Potri.008G091900.1 |
| Odae19 Palgong1 Seogwang9 | 1 | Potri.008G103300.1 |
| Odae19 Seogwang15 Seogwang9 | 1 | Potri.014G024200.2 |
| Junguk62 Odae19 Palgong2 | 3 | Potri.007G047400.1 Potri.005G085200.1 Potri.016G137900.1 |
| Odae19 Palgong1 Seogwang15 | 1 | Potri.008G094000.1 |
| Junguk62 Palgong1 Palgong2 | 1 | Potri.006G133200.4 |
| Odae19 Seogwang9 | 1 | Potri.010G147700.1 |
| Odae19 Palgong2 | 1 | Potri.010G160100.3 |
| Odae19 Palgong1 | 3 | Potri.003G132700.1 Potri.002G168700.1 Potri.019G123500.1 |
| Junguk62 Odae19 | 1 | Potri.018G008500.1 |
| Seogwang15 Seogwang9 | 1 | Potri.010G163000.1 |
| Junguk62 Seogwang15 | 1 | Potri.005G141400.1 |
| Odae19 | 1 | Potri.014G111900.1 |
| Seogwang9 | 4 | Potri.002G059100.1 Potri.015G099200.1 Potri.005G203200.1 Potri.004G072000.1 |
| Seogwang15 | 1 | Potri.014G119800.1 |
| Junguk62 | 1 | Potri.005G055300.1 Potri.002G221600.1 Potri.016G083600.9 |
| GO Terms | Reference List | P. davidiana WRKYs | Fold Enrichment | p-Value |
|---|---|---|---|---|
| Biological Processes | ||||
| Regulation of transcription | 2484 | 80 | 16.69 | 2.37 × 10 |
| Regulation of RNA biosynthetic process | 2524 | 80 | 16.43 | 8.50 × 10 |
| Regulation of RNA metabolic process | 2543 | 80 | 16.3 | 1.55 × 10 |
| Macromolecule biosynthetic process | 2607 | 80 | 15.9 | 1.13 × 10 |
| Cellular biosynthetic process | 2654 | 80 | 15.62 | 4.73 × 10 |
| Regulation of biosynthetic process | 2654 | 80 | 15.62 | 4.73 × 10 |
| Nitrogen compound metabolic process | 2681 | 80 | 15.47 | 1.06 × 10 |
| Regulation of gene expression | 2790 | 80 | 14.86 | 2.57 × 10 |
| Regulation of cellular metabolic process | 2881 | 80 | 14.39 | 3.36 × 10 |
| macromolecular metabolic process | 3071 | 80 | 13.5 | 5.56 × 10 |
| Regulation of metabolic process | 3103 | 80 | 13.36 | 1.27 × 10 |
| Regulation of cellular process | 5120 | 80 | 8.1 | 3.19 × 10 |
| Regulation of biological process | 5389 | 80 | 7.69 | 1.92 × 10 |
| Molecular Function | ||||
| Sequence-specific DNA binding | 1057 | 80 | 39.23 | 4.01× 10 |
| Transcription factor activity | 1239 | 80 | 33.46 | 1.33 × 10 |
| Nucleic acid binding activity | 1239 | 80 | 33.46 | 1.33 × 10 |
| DNA binding | 2753 | 80 | 15.06 | 7.28 × 10 |
| Heterocyclic compound binding | 10024 | 80 | 4.14 | 5.77 × 10 |
| Binding | 14108 | 80 | 2.94 | 4.31 × 10 |
| Molecular function | 22374 | 80 | 1.85 | 4.54 × 10 |
| Sequence-specific DNA binding | 1057 | 80 | 39.23 | 4.01 × 10 |
| Transcription factor activity | 1239 | 80 | 33.46 | 1.33 × 10 |
| Nucleic acid binding | 4983 | 80 | 8.32 | 3.00 × 10 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imran, Q.M.; Lee, S.-U.; Mun, B.-G.; Hussain, A.; Asaf, S.; Lee, I.-J.; Yun, B.-W. WRKYs, the Jack-of-various-Trades, Modulate Dehydration Stress in Populus davidiana—A Transcriptomic Approach. Int. J. Mol. Sci. 2019, 20, 414. https://doi.org/10.3390/ijms20020414
Imran QM, Lee S-U, Mun B-G, Hussain A, Asaf S, Lee I-J, Yun B-W. WRKYs, the Jack-of-various-Trades, Modulate Dehydration Stress in Populus davidiana—A Transcriptomic Approach. International Journal of Molecular Sciences. 2019; 20(2):414. https://doi.org/10.3390/ijms20020414
Chicago/Turabian StyleImran, Qari Muhammad, Sang-Uk Lee, Bong-Gyu Mun, Adil Hussain, Sajjad Asaf, In-Jung Lee, and Byung-Wook Yun. 2019. "WRKYs, the Jack-of-various-Trades, Modulate Dehydration Stress in Populus davidiana—A Transcriptomic Approach" International Journal of Molecular Sciences 20, no. 2: 414. https://doi.org/10.3390/ijms20020414
APA StyleImran, Q. M., Lee, S.-U., Mun, B.-G., Hussain, A., Asaf, S., Lee, I.-J., & Yun, B.-W. (2019). WRKYs, the Jack-of-various-Trades, Modulate Dehydration Stress in Populus davidiana—A Transcriptomic Approach. International Journal of Molecular Sciences, 20(2), 414. https://doi.org/10.3390/ijms20020414