Calcium Deregulation and Mitochondrial Bioenergetics in GDAP1-Related CMT Disease

 ,
, 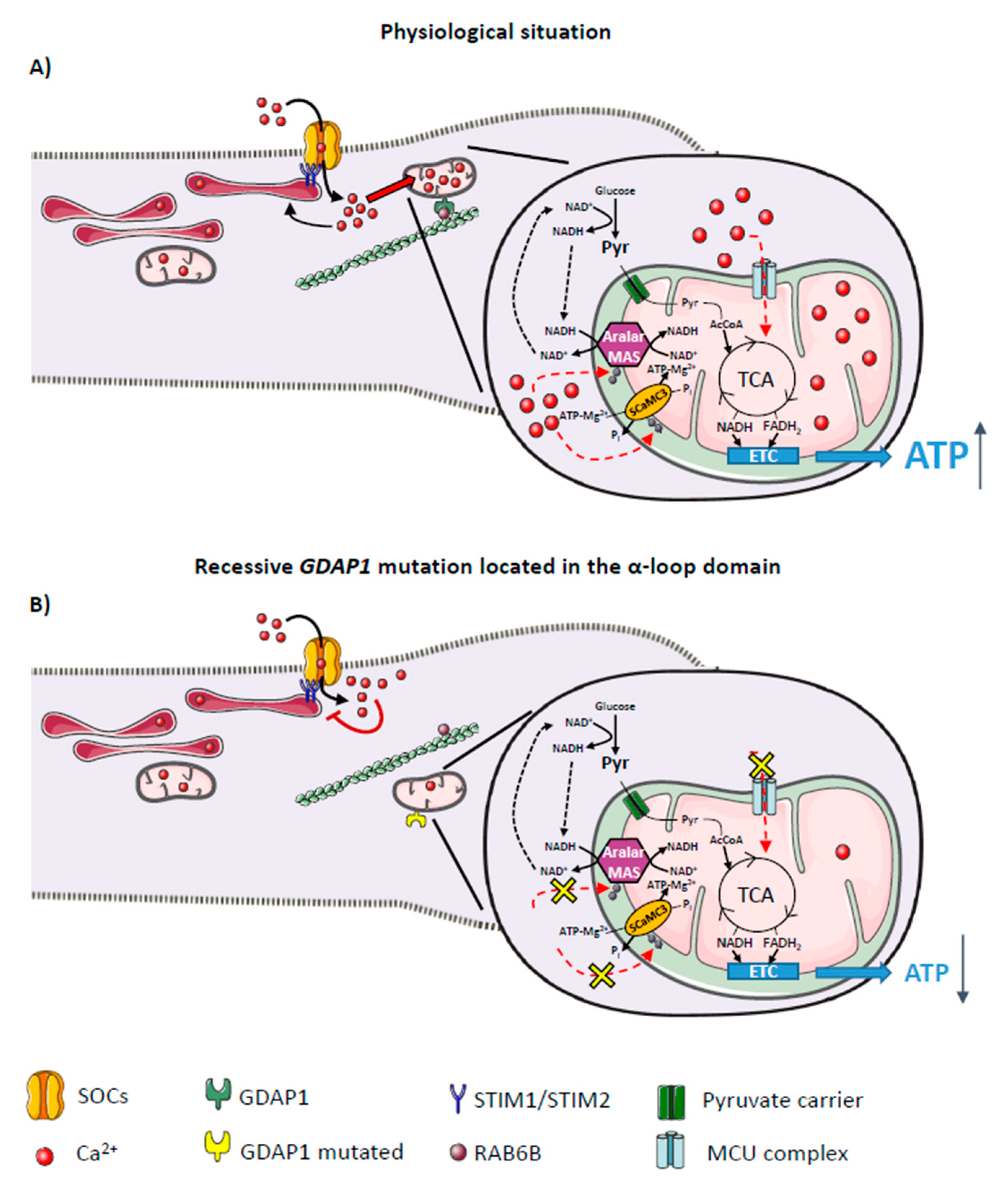{kind=link}
Abstract
1. Introduction
2. Proposed Roles of Ganglioside-Induced Differentiation-Associated Protein 1 (GDAP1) in Mitochondrial Physiology
3. Role of Mitochondrial Traffic and Location in Charcot-Marie-Tooth (CMT) Disease
4. Ca2+ Deregulation in GDAP1-Related CMT Disease
5. Neuronal Store-Operated Calcium Entry (SOCE) and Its Role in Neurodegenerative Diseases
6. Impact of Ca2+ Deregulation on Mitochondrial Bioenergetics in GDAP1-CMT Model
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Pareyson, D.; Saveri, P.; Pisciotta, C. New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr. Opin. Neurol. 2017, 30, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Juarez, P.; Palau, F. Neural and molecular features on Charcot-Marie-Tooth disease plasticity and therapy. Neural Plast. 2012, 2012, 171636. [Google Scholar] [CrossRef] [PubMed]
- Rossor, A.M.; Polke, J.M.; Houlden, H.; Reilly, M.M. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat. Rev. Neurol. 2013, 9, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Rzepnikowska, W.; Kochanski, A. A role for the GDAP1 gene in the molecular pathogenesis of Charcot Marie Tooth disease. Acta Neurobiol. Exp. 2018, 78, 1–13. [Google Scholar] [CrossRef]
- Baxter, R.V.; Ben Othmane, K.; Rochelle, J.M.; Stajich, J.E.; Hulette, C.; Dew-Knight, S.; Hentati, F.; Ben Hamida, M.; Bel, S.; Stenger, J.E.; et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat. Genet. 2002, 30, 21–22. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, A.; Pedrola, L.; Sevilla, T.; Garcia-Planells, J.; Chumillas, M.J.; Mayordomo, F.; LeGuern, E.; Marin, I.; Vilchez, J.J.; Palau, F. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat. Genet. 2002, 30, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Cassereau, J.; Chevrollier, A.; Gueguen, N.; Desquiret, V.; Verny, C.; Nicolas, G.; Dubas, F.; Amati-Bonneau, P.; Reynier, P.; Bonneau, D.; et al. Mitochondrial dysfunction and pathophysiology of Charcot-Marie-Tooth disease involving GDAP1 mutations. Exp. Neurol. 2011, 227, 31–41. [Google Scholar] [CrossRef]
- Senderek, J.; Bergmann, C.; Ramaekers, V.T.; Nelis, E.; Bernert, G.; Makowski, A.; Zuchner, S.; De Jonghe, P.; Rudnik-Schoneborn, S.; Zerres, K.; et al. Mutations in the ganglioside-induced differentiation-associated protein-1 (GDAP1) gene in intermediate type autosomal recessive Charcot-Marie-Tooth neuropathy. Brain 2003, 126, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Claramunt, R.; Pedrola, L.; Sevilla, T.; Lopez de Munain, A.; Berciano, J.; Cuesta, A.; Sanchez-Navarro, B.; Millan, J.M.; Saifi, G.M.; Lupski, J.R.; et al. Genetics of Charcot-Marie-Tooth disease type 4A: Mutations, inheritance, phenotypic variability, and founder effect. J. Med. Genet. 2005, 42, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Sivera, R.; Espinos, C.; Vilchez, J.J.; Mas, F.; Martinez-Rubio, D.; Chumillas, M.J.; Mayordomo, F.; Muelas, N.; Bataller, L.; Palau, F.; et al. Phenotypical features of the p.R120W mutation in the GDAP1 gene causing autosomal dominant Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 2010, 15, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Pedrola, L.; Espert, A.; Valdes-Sanchez, T.; Sanchez-Piris, M.; Sirkowski, E.E.; Scherer, S.S.; Farinas, I.; Palau, F. Cell expression of GDAP1 in the nervous system and pathogenesis of Charcot-Marie-Tooth type 4A disease. J. Cell. Mol. Med. 2008, 12, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Niemann, A.; Ruegg, M.; La Padula, V.; Schenone, A.; Suter, U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: New implications for Charcot-Marie-Tooth disease. J. Cell Biol. 2005, 170, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Marco, A.; Cuesta, A.; Pedrola, L.; Palau, F.; Marin, I. Evolutionary and structural analyses of GDAP1, involved in Charcot-Marie-Tooth disease, characterize a novel class of glutathione transferase-related genes. Mol. Biol. Evol. 2004, 21, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.M.; Ruegg, M.; Niemann, A.; Suter, U. Targeting and function of the mitochondrial fission factor GDAP1 are dependent on its tail-anchor. PLoS ONE 2009, 4, e5160. [Google Scholar] [CrossRef] [PubMed]
- Niemann, A.; Wagner, K.M.; Ruegg, M.; Suter, U. GDAP1 mutations differ in their effects on mitochondrial dynamics and apoptosis depending on the mode of inheritance. Neurobiol. Dis. 2009, 36, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Pedrola, L.; Espert, A.; Wu, X.; Claramunt, R.; Shy, M.E.; Palau, F. GDAP1, the protein causing Charcot-Marie-Tooth disease type 4A, is expressed in neurons and is associated with mitochondria. Hum. Mol. Genet. 2005, 14, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Huber, N.; Guimaraes, S.; Schrader, M.; Suter, U.; Niemann, A. Charcot-Marie-Tooth disease-associated mutants of GDAP1 dissociate its roles in peroxisomal and mitochondrial fission. EMBO Rep. 2013, 14, 545–552. [Google Scholar] [CrossRef]
- Noack, R.; Frede, S.; Albrecht, P.; Henke, N.; Pfeiffer, A.; Knoll, K.; Dehmel, T.; Meyer Zu Horste, G.; Stettner, M.; Kieseier, B.C.; et al. Charcot-Marie-Tooth disease CMT4A: GDAP1 increases cellular glutathione and the mitochondrial membrane potential. Hum. Mol. Genet. 2012, 21, 150–162. [Google Scholar] [CrossRef]
- Niemann, A.; Huber, N.; Wagner, K.M.; Somandin, C.; Horn, M.; Lebrun-Julien, F.; Angst, B.; Pereira, J.A.; Halfter, H.; Welzl, H.; et al. The Gdap1 knockout mouse mechanistically links redox control to Charcot-Marie-Tooth disease. Brain 2014, 137, 668–682. [Google Scholar] [CrossRef]
- Pla-Martin, D.; Rueda, C.B.; Estela, A.; Sanchez-Piris, M.; Gonzalez-Sanchez, P.; Traba, J.; de la Fuente, S.; Scorrano, L.; Renau-Piqueras, J.; Alvarez, J.; et al. Silencing of the Charcot-Marie-Tooth disease-associated gene GDAP1 induces abnormal mitochondrial distribution and affects Ca2+ homeostasis by reducing store-operated Ca2+ entry. Neurobiol. Dis. 2013, 55, 140–151. [Google Scholar] [CrossRef]
- Gonzalez-Sanchez, P.; Pla-Martin, D.; Martinez-Valero, P.; Rueda, C.B.; Calpena, E.; Del Arco, A.; Palau, F.; Satrustegui, J. CMT-linked loss-of-function mutations in GDAP1 impair store-operated Ca2+ entry-stimulated respiration. Sci. Rep. 2017, 7, 42993. [Google Scholar] [CrossRef] [PubMed]
- Zuchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Pathak, D.; Berthet, A.; Nakamura, K. Energy failure: Does it contribute to neurodegeneration? Ann. Neurol. 2013, 74, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Panchal, K.; Tiwari, A.K. Mitochondrial dynamics, a key executioner in neurodegenerative diseases. Mitochondrion 2018. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018. [Google Scholar] [CrossRef] [PubMed]
- Estela, A.; Pla-Martin, D.; Sanchez-Piris, M.; Sesaki, H.; Palau, F. Charcot-Marie-Tooth-related gene GDAP1 complements cell cycle delay at G2/M phase in Saccharomyces cerevisiae fis1 gene-defective cells. J. Biol. Chem. 2011, 286, 36777–36786. [Google Scholar] [CrossRef] [PubMed]
- Lopez Del Amo, V.; Seco-Cervera, M.; Garcia-Gimenez, J.L.; Whitworth, A.J.; Pallardo, F.V.; Galindo, M.I. Mitochondrial defects and neuromuscular degeneration caused by altered expression of Drosophila Gdap1: Implications for the Charcot-Marie-Tooth neuropathy. Hum. Mol. Genet. 2015, 24, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Shield, A.J.; Murray, T.P.; Board, P.G. Functional characterisation of ganglioside-induced differentiation-associated protein 1 as a glutathione transferase. Biochem. Biophys. Res. Commun. 2006, 347, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Huber, N.; Bieniossek, C.; Wagner, K.M.; Elsasser, H.P.; Suter, U.; Berger, I.; Niemann, A. Glutathione-conjugating and membrane-remodeling activity of GDAP1 relies on amphipathic C-terminal domain. Sci. Rep. 2016, 6, 36930. [Google Scholar] [CrossRef] [PubMed]
- Chahbouni, M.; del Señor-Lopez, M.; Molina-Carballo, A.; de Haro, T.; Muñoz-Hoyos, A.; Fernandez-Ortiz, M.; Guerra-Librero, A.; Acuña-Castroviejo, D. Melatonin treatment reduces oxidative damage and normalizes plasma pro-inflammatory cytokines in patients suffering from Charcot-Marie-Tooth neuropathy: A pilot study in three children. Molecules 2017, 22, 1728. [Google Scholar] [CrossRef]
- Li, W.; Zhu, H.; Zhao, X.; Brancho, D.; Liang, Y.; Zou, Y.; Bennet, C.; Chow, C.W. Dysregulated inflammatory signaling upon Charcot-Marie-Tooth type 1C mutation of SIMPLE protein. Mol. Cell. Biol. 2015, 35, 2464–2478. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Barneo-Munoz, M.; Juarez, P.; Civera-Tregon, A.; Yndriago, L.; Pla-Martin, D.; Zenker, J.; Cuevas-Martin, C.; Estela, A.; Sanchez-Arago, M.; Forteza-Vila, J.; et al. Lack of GDAP1 induces neuronal calcium and mitochondrial defects in a knockout mouse model of charcot-marie-tooth neuropathy. PLoS Genet. 2015, 11, e1005115. [Google Scholar] [CrossRef]
- Venturini, G.; Rose, A.M.; Shah, A.Z.; Bhattacharya, S.S.; Rivolta, C. CNOT3 is a modifier of PRPF31 mutations in retinitis pigmentosa with incomplete penetrance. PLoS Genet. 2012, 8, e1003040. [Google Scholar] [CrossRef] [PubMed]
- Lamar, K.M.; McNally, E.M. Genetic Modifiers for Neuromuscular Diseases. J. Neuromuscul. Dis. 2014, 1, 3–13. [Google Scholar] [PubMed]
- Pla-Martin, D.; Calpena, E.; Lupo, V.; Marquez, C.; Rivas, E.; Sivera, R.; Sevilla, T.; Palau, F.; Espinos, C. Junctophilin-1 is a modifier gene of GDAP1-related Charcot-Marie-Tooth disease. Hum. Mol. Genet. 2015, 24, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Rizzuto, R.; Marchi, S.; Bonora, M.; Aguiari, P.; Bononi, A.; De Stefani, D.; Giorgi, C.; Leo, S.; Rimessi, A.; Siviero, R.; et al. Ca2+ transfer from the ER to mitochondria: When, how and why. Biochim. Biophys. Acta 2009, 1787, 1342–1351. [Google Scholar] [CrossRef]
- Carafoli, E. The interplay of mitochondria with calcium: An historical appraisal. Cell Calcium 2012, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef]
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-driven local ATP synthesis is required for synaptic function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Baloh, R.H. Mitochondrial dynamics and peripheral neuropathy. Neuroscientist 2008, 14, 12–18. [Google Scholar] [CrossRef]
- Palau, F.; Estela, A.; Pla-Martin, D.; Sanchez-Piris, M. The role of mitochondrial network dynamics in the pathogenesis of Charcot-Marie-Tooth disease. Adv. Exp. Med. Biol. 2009, 652, 129–137. [Google Scholar] [PubMed]
- Pareyson, D.; Saveri, P.; Sagnelli, A.; Piscosquito, G. Mitochondrial dynamics and inherited peripheral nerve diseases. Neurosci. Lett. 2015, 596, 66–77. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Baloh, R.H.; Schmidt, R.E.; Pestronk, A.; Milbrandt, J. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J. Neurosci. 2007, 27, 422–430. [Google Scholar] [CrossRef]
- Cartoni, R.; Arnaud, E.; Medard, J.J.; Poirot, O.; Courvoisier, D.S.; Chrast, R.; Martinou, J.C. Expression of mitofusin 2(R94Q) in a transgenic mouse leads to Charcot-Marie-Tooth neuropathy type 2A. Brain 2010, 133, 1460–1469. [Google Scholar] [CrossRef]
- Vallat, J.M.; Ouvrier, R.A.; Pollard, J.D.; Magdelaine, C.; Zhu, D.; Nicholson, G.A.; Grew, S.; Ryan, M.M.; Funalot, B. Histopathological findings in hereditary motor and sensory neuropathy of axonal type with onset in early childhood associated with mitofusin 2 mutations. J. Neuropathol. Exp. Neurol. 2008, 67, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [PubMed]
- Irobi, J.; Van Impe, K.; Seeman, P.; Jordanova, A.; Dierick, I.; Verpoorten, N.; Michalik, A.; De Vriendt, E.; Jacobs, A.; Van Gerwen, V.; et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat. Genet. 2004, 36, 597–601. [Google Scholar] [CrossRef]
- Evgrafov, O.V.; Mersiyanova, I.; Irobi, J.; Van Den Bosch, L.; Dierick, I.; Leung, C.L.; Schagina, O.; Verpoorten, N.; Van Impe, K.; Fedotov, V.; et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 2004, 36, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Ackerley, S.; James, P.A.; Kalli, A.; French, S.; Davies, K.E.; Talbot, K. A mutation in the small heat-shock protein HSPB1 leading to distal hereditary motor neuronopathy disrupts neurofilament assembly and the axonal transport of specific cellular cargoes. Hum. Mol. Genet. 2006, 15, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Kalmar, B.; Innes, A.; Wanisch, K.; Kolaszynska, A.K.; Pandraud, A.; Kelly, G.; Abramov, A.Y.; Reilly, M.M.; Schiavo, G.; Greensmith, L. Mitochondrial deficits and abnormal mitochondrial retrograde axonal transport play a role in the pathogenesis of mutant Hsp27-induced Charcot Marie Tooth Disease. Hum. Mol. Genet. 2017, 26, 3313–3326. [Google Scholar] [CrossRef] [PubMed]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef]
- Kim, J.Y.; Woo, S.Y.; Hong, Y.B.; Choi, H.; Kim, J.; Choi, H.; Mook-Jung, I.; Ha, N.; Kyung, J.; Koo, S.K.; et al. HDAC6 Inhibitors Rescued the Defective Axonal Mitochondrial Movement in Motor Neurons Derived from the Induced Pluripotent Stem Cells of Peripheral Neuropathy Patients with HSPB1 Mutation. Stem Cells Int. 2016, 2016, 9475981. [Google Scholar] [CrossRef]
- Shen, S.; Benoy, V.; Bergman, J.A.; Kalin, J.H.; Frojuello, M.; Vistoli, G.; Haeck, W.; Van Den Bosch, L.; Kozikowski, A.P. Bicyclic-Capped Histone Deacetylase 6 Inhibitors with Improved Activity in a Model of Axonal Charcot-Marie-Tooth Disease. ACS Chem. Neurosci. 2016, 7, 240–258. [Google Scholar] [CrossRef]
- Benoy, V.; Vanden Berghe, P.; Jarpe, M.; Van Damme, P.; Robberecht, W.; Van Den Bosch, L. Development of Improved HDAC6 Inhibitors as Pharmacological Therapy for Axonal Charcot-Marie-Tooth Disease. Neurotherapeutics 2017, 14, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Roos, J.; Digregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef]
- Rosado, J.A.; Diez, R.; Smani, T.; Jardin, I. STIM and Orai1 Variants in Store-Operated Calcium Entry. Front. Pharmacol. 2015, 6, 325. [Google Scholar] [CrossRef] [PubMed]
- Worley, P.F.; Zeng, W.; Huang, G.N.; Yuan, J.P.; Kim, J.Y.; Lee, M.G.; Muallem, S. TRPC channels as STIM1-regulated store-operated channels. Cell Calcium 2007, 42, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.S. The molecular choreography of a store-operated calcium channel. Nature 2007, 446, 284–287. [Google Scholar] [CrossRef]
- Parekh, A.B. Regulation of CRAC channels by Ca2+-dependent inactivation. Cell Calcium 2017, 63, 20–23. [Google Scholar] [CrossRef]
- Varadi, A.; Cirulli, V.; Rutter, G.A. Mitochondrial localization as a determinant of capacitative Ca2+ entry in HeLa cells. Cell Calcium 2004, 36, 499–508. [Google Scholar] [CrossRef]
- Nunez, L.; Valero, R.A.; Senovilla, L.; Sanz-Blasco, S.; Garcia-Sancho, J.; Villalobos, C. Cell proliferation depends on mitochondrial Ca2+ uptake: Inhibition by salicylate. J. Physiol. 2006, 571, 57–73. [Google Scholar] [CrossRef]
- Quintana, A.; Schwarz, E.C.; Schwindling, C.; Lipp, P.; Kaestner, L.; Hoth, M. Sustained activity of calcium release-activated calcium channels requires translocation of mitochondria to the plasma membrane. J. Biol. Chem. 2006, 281, 40302–40309. [Google Scholar] [CrossRef] [PubMed]
- Quintana, A.; Schwindling, C.; Wenning, A.S.; Becherer, U.; Rettig, J.; Schwarz, E.C.; Hoth, M. T cell activation requires mitochondrial translocation to the immunological synapse. Proc. Natl. Acad. Sci. USA 2007, 104, 14418–14423. [Google Scholar] [CrossRef] [PubMed]
- Samanta, K.; Douglas, S.; Parekh, A.B. Mitochondrial calcium uniporter MCU supports cytoplasmic Ca2+ oscillations, store-operated Ca2+ entry and Ca2+-dependent gene expression in response to receptor stimulation. PLoS ONE 2014, 9, e101188. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Segal, M.; Korkotian, E. Roles of Calcium Stores and Store-Operated Channels in Plasticity of Dendritic Spines. Neuroscientist 2016, 22, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Emptage, N.J.; Reid, C.A.; Fine, A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron 2001, 29, 197–208. [Google Scholar] [CrossRef]
- de Juan-Sanz, J.; Holt, G.T.; Schreiter, E.R.; de Juan, F.; Kim, D.S.; Ryan, T.A. Axonal Endoplasmic Reticulum Ca2+ Content Controls Release Probability in CNS Nerve Terminals. Neuron 2017, 93, 867–881.e6. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Karl, R.M.; Alexander, R.P.; Adelsberger, H.; Brill, M.S.; Ruhlmann, C.; Ansel, A.; Sakimura, K.; Baba, Y.; Kurosaki, T.; et al. STIM1 controls neuronal Ca2+ signaling, mGluR1-dependent synaptic transmission, and cerebellar motor behavior. Neuron 2014, 82, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca2+ homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef]
- Shore, G.C.; Papa, F.R.; Oakes, S.A. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 2011, 23, 143–149. [Google Scholar] [CrossRef]
- Kawamata, H.; Ng, S.K.; Diaz, N.; Burstein, S.; Morel, L.; Osgood, A.; Sider, B.; Higashimori, H.; Haydon, P.G.; Manfredi, G.; et al. Abnormal intracellular calcium signaling and SNARE-dependent exocytosis contributes to SOD1G93A astrocyte-mediated toxicity in amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 2331–2348. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ryskamp, D.A.; Liang, X.; Egorova, P.; Zakharova, O.; Hung, G.; Bezprozvanny, I. Enhanced Store-Operated Calcium Entry Leads to Striatal Synaptic Loss in a Huntington’s Disease Mouse Model. J. Neurosci. 2016, 36, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Intracellular calcium homeostasis and signaling. Met. Ions Life Sci. 2013, 12, 119–168. [Google Scholar] [PubMed]
- Pfeiffer, R.F. Parkinson disease: Calcium channel blockers and Parkinson disease. Nat. Rev. Neurol. 2010, 6, 188–189. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Fedrizzi, L.; Tartari, M.; Zuccato, C.; Cattaneo, E.; Brini, M.; Carafoli, E. Calcium homeostasis and mitochondrial dysfunction in striatal neurons of Huntington disease. J. Biol. Chem. 2008, 283, 5780–5789. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Calcium regulation of neural rhythms, memory and Alzheimer’s disease. J. Physiol. 2014, 592, 281–293. [Google Scholar] [CrossRef]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I. Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease—A therapeutic opportunity? Biochem. Biophys. Res. Commun. 2017, 483, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Leal, S.S.; Gomes, C.M. Calcium dysregulation links ALS defective proteins and motor neuron selective vulnerability. Front. Cell. Neurosci. 2015, 9, 225. [Google Scholar] [CrossRef] [PubMed]
- Grienberger, C.; Konnerth, A. Imaging calcium in neurons. Neuron 2012, 73, 862–885. [Google Scholar] [CrossRef]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W., Jr. Capacitative calcium entry in the nervous system. Cell Calcium 2003, 34, 339–344. [Google Scholar] [CrossRef]
- Lu, B.; Fivaz, M. Neuronal SOCE: Myth or Reality? Trends Cell Biol. 2016, 26, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sanchez, P.; Del Arco, A.; Esteban, J.A.; Satrustegui, J. Store-Operated Calcium Entry Is Required for mGluR-Dependent Long Term Depression in Cortical Neurons. Front. Cell. Neurosci. 2017, 11, 363. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Taubenberger, N.; Huchzermeyer, C.; Papageorgiou, I.E.; Benninger, F.; Heinemann, U.; Kovacs, R. Muscarinic receptor activation determines the effects of store-operated Ca2+-entry on excitability and energy metabolism in pyramidal neurons. Cell Calcium 2012, 51, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Samtleben, S.; Wachter, B.; Blum, R. Store-operated calcium entry compensates fast ER calcium loss in resting hippocampal neurons. Cell Calcium 2015, 58, 147–159. [Google Scholar] [CrossRef]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2009, 2, ra67. [Google Scholar] [CrossRef]
- Xia, J.; Pan, R.; Gao, X.; Meucci, O.; Hu, H. Native store-operated calcium channels are functionally expressed in mouse spinal cord dorsal horn neurons and regulate resting calcium homeostasis. J. Physiol. 2014, 592, 3443–3461. [Google Scholar] [CrossRef]
- Gemes, G.; Bangaru, M.L.; Wu, H.E.; Tang, Q.; Weihrauch, D.; Koopmeiners, A.S.; Cruikshank, J.M.; Kwok, W.M.; Hogan, Q.H. Store-operated Ca2+ entry in sensory neurons: Functional role and the effect of painful nerve injury. J. Neurosci. 2011, 31, 3536–3549. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, H.; Liu, J.; Popugaeva, E.; Xu, N.J.; Feske, S.; White, C.L., 3rd; Bezprozvanny, I. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 2014, 82, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Korkotian, E.; Segal, M. Orai1 regulates calcium entry into dendritic spines. Channels 2017, 11, 99–100. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, J.; Saia, G.; Gill, G. Store-operated calcium entry promotes the degradation of the transcription factor Sp4 in resting neurons. Sci. Signal. 2014, 7, ra51. [Google Scholar] [CrossRef] [PubMed]
- Baba, A.; Yasui, T.; Fujisawa, S.; Yamada, R.X.; Yamada, M.K.; Nishiyama, N.; Matsuki, N.; Ikegaya, Y. Activity-evoked capacitative Ca2+ entry: Implications in synaptic plasticity. J. Neurosci. 2003, 23, 7737–7741. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.F.; Liu, Z.H.; Li, N.; Cheng, W.J.; Guo, S.W. Knockdown of STIM1 improves neuronal survival after traumatic neuronal injury through regulating mGluR1-dependent Ca2+ signaling in mouse cortical neurons. Cell. Mol. Neurobiol. 2015, 35, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Gruszczynska-Biegala, J.; Sladowska, M.; Kuznicki, J. AMPA Receptors Are Involved in Store-Operated Calcium Entry and Interact with STIM Proteins in Rat Primary Cortical Neurons. Front. Cell. Neurosci. 2016, 10, 251. [Google Scholar] [CrossRef] [PubMed]
- Majewski, L.; Kuznicki, J. SOCE in neurons: Signaling or just refilling? Biochim. Biophys. Acta 2015, 1853, 1940–1952. [Google Scholar] [CrossRef]
- Moccia, F.; Zuccolo, E.; Soda, T.; Tanzi, F.; Guerra, G.; Mapelli, L.; Lodola, F.; D’Angelo, E. Stim and Orai proteins in neuronal Ca2+ signaling and excitability. Front. Cell. Neurosci. 2015, 9, 153. [Google Scholar] [CrossRef]
- Wegierski, T.; Kuznicki, J. Neuronal calcium signaling via store-operated channels in health and disease. Cell Calcium 2018, 74, 102–111. [Google Scholar] [CrossRef]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef]
- Herms, J.; Schneider, I.; Dewachter, I.; Caluwaerts, N.; Kretzschmar, H.; Van Leuven, F. Capacitive calcium entry is directly attenuated by mutant presenilin-1, independent of the expression of the amyloid precursor protein. J. Biol. Chem. 2003, 278, 2484–2489. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A.; Akbari, Y.; Fanger, C.M.; Cahalan, M.D.; Mattson, M.P.; LaFerla, F.M. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J. Cell Biol. 2000, 149, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Yoo, A.S.; Cheng, I.; Chung, S.; Grenfell, T.Z.; Lee, H.; Pack-Chung, E.; Handler, M.; Shen, J.; Xia, W.; Tesco, G.; et al. Presenilin-mediated modulation of capacitative calcium entry. Neuron 2000, 27, 561–572. [Google Scholar] [CrossRef]
- Tong, B.C.; Lee, C.S.; Cheng, W.H.; Lai, K.O.; Foskett, J.K.; Cheung, K.H. Familial Alzheimer’s disease-associated presenilin 1 mutants promote gamma-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci. Signal. 2016, 9, ra89. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Caro, C.; Berrocal, M.; Lopez-Guerrero, A.M.; Alvarez-Barrientos, A.; Pozo-Guisado, E.; Gutierrez-Merino, C.; Mata, A.M.; Martin-Romero, F.J. STIM1 deficiency is linked to Alzheimer’s disease and triggers cell death in SH-SY5Y cells by upregulation of L-type voltage-operated Ca2+ entry. J. Mol. Med. 2018, 96, 1061–1079. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Saito, T.; Saido, T.; Bezprozvanny, I. Neuronal Store-Operated Calcium Entry and Mushroom Spine Loss in Amyloid Precursor Protein Knock-In Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 13275–13286. [Google Scholar] [CrossRef]
- Park, C.Y.; Shcheglovitov, A.; Dolmetsch, R. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 2010, 330, 101–105. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, X.; Mancarella, S.; Hendron, E.; Eguchi, S.; Soboloff, J.; Tang, X.D.; Gill, D.L. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 2010, 330, 105–109. [Google Scholar] [CrossRef]
- Arshad, A.; Chen, X.; Cong, Z.; Qing, H.; Deng, Y. TRPC1 protects dopaminergic SH-SY5Y cells from MPP+, salsolinol, and N-methyl-(R)-salsolinol-induced cytotoxicity. Acta Biochim. Biophys. Sin. 2014, 46, 22–30. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, H.; Selvaraj, S.; Sukumaran, P.; Lei, S.; Birnbaumer, L.; Singh, B.B. Inhibition of L-Type Ca2+ Channels by TRPC1-STIM1 Complex Is Essential for the Protection of Dopaminergic Neurons. J. Neurosci. 2017, 37, 3364–3377. [Google Scholar] [CrossRef]
- Zhou, Q.; Yen, A.; Rymarczyk, G.; Asai, H.; Trengrove, C.; Aziz, N.; Kirber, M.T.; Mostoslavsky, G.; Ikezu, T.; Wolozin, B.; et al. Impairment of PARK14-dependent Ca2+ signalling is a novel determinant of Parkinson’s disease. Nat. Commun. 2016, 7, 10332. [Google Scholar] [CrossRef]
- Wu, J.; Shih, H.P.; Vigont, V.; Hrdlicka, L.; Diggins, L.; Singh, C.; Mahoney, M.; Chesworth, R.; Shapiro, G.; Zimina, O.; et al. Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington’s disease treatment. Chem. Biol. 2011, 18, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Vigont, V.; Kolobkova, Y.; Skopin, A.; Zimina, O.; Zenin, V.; Glushankova, L.; Kaznacheyeva, E. Both Orai1 and TRPC1 are Involved in Excessive Store-Operated Calcium Entry in Striatal Neurons Expressing Mutant Huntingtin Exon 1. Front. Physiol. 2015, 6, 337. [Google Scholar] [CrossRef]
- Czeredys, M.; Maciag, F.; Methner, A.; Kuznicki, J. Tetrahydrocarbazoles decrease elevated SOCE in medium spiny neurons from transgenic YAC128 mice, a model of Huntington’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ryskamp, D.; Birnbaumer, L.; Bezprozvanny, I. Inhibition of TRPC1-Dependent Store-Operated Calcium Entry Improves Synaptic Stability and Motor Performance in a Mouse Model of Huntington’s Disease. J. Huntington’s Dis. 2018, 7, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, J.A.; Henke, N.; Opatz, J.; Gruszczynska-Biegala, J.; Schneider, L.; Theiss, S.; Hamacher, N.; Steinfarz, B.; Golz, S.; Brustle, O.; et al. Store-operated calcium entry modulates neuronal network activity in a model of chronic epilepsy. Exp. Neurol. 2011, 232, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Xia, J.; Gao, R.; Gao, X.; Munoz, F.M.; Wei, D.; Tian, Y.; Barrett, J.E.; Ajit, S.; Meucci, O.; et al. Orai1 Plays a Crucial Role in Central Sensitization by Modulating Neuronal Excitability. J. Neurosci. 2018, 38, 887–900. [Google Scholar] [CrossRef]
- Rao, W.; Zhang, L.; Peng, C.; Hui, H.; Wang, K.; Su, N.; Wang, L.; Dai, S.H.; Yang, Y.F.; Chen, T.; et al. Downregulation of STIM2 improves neuronal survival after traumatic brain injury by alleviating calcium overload and mitochondrial dysfunction. Biochim. Biophys. Acta 2015, 1852, 2402–2413. [Google Scholar] [CrossRef]
- Fonteriz, R.; Matesanz-Isabel, J.; Arias-Del-Val, J.; Alvarez-Illera, P.; Montero, M.; Alvarez, J. Modulation of Calcium Entry by Mitochondria. Adv. Exp. Med. Biol. 2016, 898, 405–421. [Google Scholar]
- Szabadkai, G.; Duchen, M.R. Mitochondria: The hub of cellular Ca2+ signaling. Physiology 2008, 23, 84–94. [Google Scholar] [CrossRef]
- Blachly-Dyson, E.; Forte, M. VDAC channels. IUBMB Life 2001, 52, 113–118. [Google Scholar] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; Mccombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabo, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2018, 26, 179–195. [Google Scholar] [CrossRef]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovacs-Bogdan, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [PubMed]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabo, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef]
- Mallilankaraman, K.; Cardenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenar, T.; Csordas, G.; Madireddi, P.; Yang, J.; Muller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Elustondo, P.A.; Nichols, M.; Robertson, G.S.; Pavlov, E.V. Mitochondrial Ca2+ uptake pathways. J. Bioenerg. Biomembr. 2017, 49, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Llorente-Folch, I.; Rueda, C.B.; Pardo, B.; Szabadkai, G.; Duchen, M.R.; Satrustegui, J. The regulation of neuronal mitochondrial metabolism by calcium. J. Physiol. 2015, 593, 3447–3462. [Google Scholar] [CrossRef] [PubMed]
- Olianas, M.C.; Dedoni, S.; Onali, P. Involvement of store-operated Ca2+ entry in activation of AMP-activated protein kinase and stimulation of glucose uptake by M3 muscarinic acetylcholine receptors in human neuroblastoma cells. Biochim. Biophys. Acta 2014, 1843, 3004–3017. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M.; McCormack, J.G. The calcium sensitive dehydrogenases of vertebrate mitochondria. Cell Calcium 1986, 7, 377–386. [Google Scholar] [CrossRef]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Nemoto, T.; Iino, M.; Kasai, H. Rapid Ca2+-dependent increase in oxygen consumption by mitochondria in single mammalian central neurons. Cell Calcium 2005, 37, 359–370. [Google Scholar] [CrossRef]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef]
- Llorente-Folch, I.; Rueda, C.B.; Amigo, I.; del Arco, A.; Saheki, T.; Pardo, B.; Satrustegui, J. Calcium-regulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1-malate aspartate shuttle in intact cortical neurons. J. Neurosci. 2013, 33, 13957–13971. [Google Scholar] [CrossRef]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef]
- Balaban, R.S. The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim. Biophys. Acta 2009, 1787, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- del Arco, A.; Satrustegui, J. Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain. J. Biol. Chem. 1998, 273, 23327–23334. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, L.; Pardo, B.; Lasorsa, F.M.; del Arco, A.; Kobayashi, K.; Iijima, M.; Runswick, M.J.; Walker, J.E.; Saheki, T.; Satrustegui, J.; et al. Citrin and aralar1 are Ca2+-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001, 20, 5060–5069. [Google Scholar] [CrossRef] [PubMed]
- del Arco, A.; Satrustegui, J. Identification of a novel human subfamily of mitochondrial carriers with calcium-binding domains. J. Biol. Chem. 2004, 279, 24701–24713. [Google Scholar] [CrossRef] [PubMed]
- Fiermonte, G.; De Leonardis, F.; Todisco, S.; Palmieri, L.; Lasorsa, F.M.; Palmieri, F. Identification of the mitochondrial ATP-Mg/Pi transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution. J. Biol. Chem. 2004, 279, 30722–30730. [Google Scholar] [CrossRef] [PubMed]
- Satrustegui, J.; Pardo, B.; Del Arco, A. Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol. Rev. 2007, 87, 29–67. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Contreras, L.; Serrano, A.; Ramos, M.; Kobayashi, K.; Iijima, M.; Saheki, T.; Satrustegui, J. Essential role of aralar in the transduction of small Ca2+ signals to neuronal mitochondria. J. Biol. Chem. 2006, 281, 1039–1047. [Google Scholar] [CrossRef]
- Contreras, L.; Gomez-Puertas, P.; Iijima, M.; Kobayashi, K.; Saheki, T.; Satrustegui, J. Ca2+ Activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: Role in the heart malate-aspartate NADH shuttle. J. Biol. Chem. 2007, 282, 7098–7106. [Google Scholar] [CrossRef]
- Traba, J.; Froschauer, E.M.; Wiesenberger, G.; Satrustegui, J.; Del Arco, A. Yeast mitochondria import ATP through the calcium-dependent ATP-Mg/Pi carrier Sal1p, and are ATP consumers during aerobic growth in glucose. Mol. Microbiol. 2008, 69, 570–585. [Google Scholar] [CrossRef]
- Traba, J.; Del Arco, A.; Duchen, M.R.; Szabadkai, G.; Satrustegui, J. SCaMC-1 promotes cancer cell survival by desensitizing mitochondrial permeability transition via ATP/ADP-mediated matrix Ca2+ buffering. Cell Death Differ. 2012, 19, 650–660. [Google Scholar] [CrossRef]
- Luongo, T.S.; Lambert, J.P.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q.; Lu, X.; Correll, R.N.; Schwanekamp, J.A.; Vagnozzi, R.J.; Sargent, M.A.; York, A.J.; Zhang, J.; Bers, D.M.; Molkentin, J.D. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015, 12, 15–22. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Huo, J.; Bround, M.J.; Boyer, J.G.; Schwanekamp, J.A.; Ghazal, N.; Maxwell, J.T.; Jang, Y.C.; Khuchua, Z.; Shi, K.; et al. The mitochondrial calcium uniporter underlies metabolic fuel preference in skeletal muscle. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, G.; Nogara, L.; Ciciliot, S.; Fadini, G.P.; Blaauw, B.; Braghetta, P.; Bonaldo, P.; De Stefani, D.; Rizzuto, R.; Mammucari, C. Loss of mitochondrial calcium uniporter rewires skeletal muscle metabolism and substrate preference. Cell Death Differ. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rueda, C.B.; Llorente-Folch, I.; Amigo, I.; Contreras, L.; Gonzalez-Sanchez, P.; Martinez-Valero, P.; Juaristi, I.; Pardo, B.; del Arco, A.; Satrustegui, J. Ca2+ regulation of mitochondrial function in neurons. Biochim. Biophys. Acta 2014, 1837, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Requejo-Aguilar, R.; Bolanos, J.P. Mitochondrial control of cell bioenergetics in Parkinson’s disease. Free Radic. Biol. Med. 2016, 100, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Verstreken, P.; Ly, C.V.; Venken, K.J.; Koh, T.W.; Zhou, Y.; Bellen, H.J. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef]
- Trotta, N.; Rodesch, C.K.; Fergestad, T.; Broadie, K. Cellular bases of activity-dependent paralysis in Drosophila stress-sensitive mutants. J. Neurobiol. 2004, 60, 328–347. [Google Scholar] [CrossRef]
- Guo, X.; Macleod, G.T.; Wellington, A.; Hu, F.; Panchumarthi, S.; Schoenfield, M.; Marin, L.; Charlton, M.P.; Atwood, H.L.; Zinsmaier, K.E. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 2005, 47, 379–393. [Google Scholar] [CrossRef]
- Ly, C.V.; Verstreken, P. Mitochondria at the synapse. Neuroscientist 2006, 12, 291–299. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Sánchez, P.; Satrústegui, J.; Palau, F.; del Arco, A. Calcium Deregulation and Mitochondrial Bioenergetics in GDAP1-Related CMT Disease. Int. J. Mol. Sci. 2019, 20, 403. https://doi.org/10.3390/ijms20020403
González-Sánchez P, Satrústegui J, Palau F, del Arco A. Calcium Deregulation and Mitochondrial Bioenergetics in GDAP1-Related CMT Disease. International Journal of Molecular Sciences. 2019; 20(2):403. https://doi.org/10.3390/ijms20020403
Chicago/Turabian StyleGonzález-Sánchez, Paloma, Jorgina Satrústegui, Francesc Palau, and Araceli del Arco. 2019. "Calcium Deregulation and Mitochondrial Bioenergetics in GDAP1-Related CMT Disease" International Journal of Molecular Sciences 20, no. 2: 403. https://doi.org/10.3390/ijms20020403
APA StyleGonzález-Sánchez, P., Satrústegui, J., Palau, F., & del Arco, A. (2019). Calcium Deregulation and Mitochondrial Bioenergetics in GDAP1-Related CMT Disease. International Journal of Molecular Sciences, 20(2), 403. https://doi.org/10.3390/ijms20020403