Neuromuscular Junction Changes in a Mouse Model of Charcot-Marie-Tooth Disease Type 4C

 , and
, and
Abstract

1. Introduction
2. Results
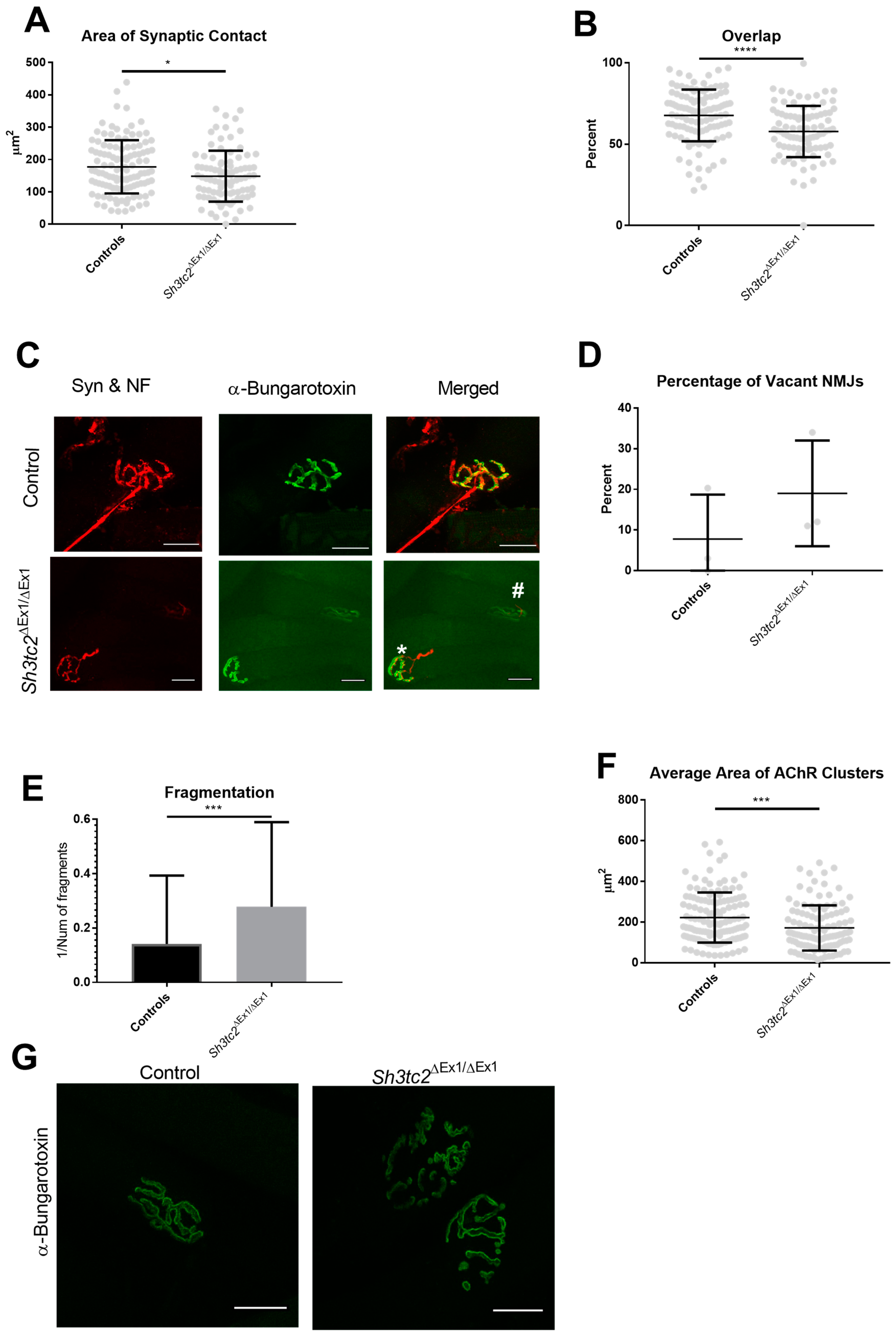
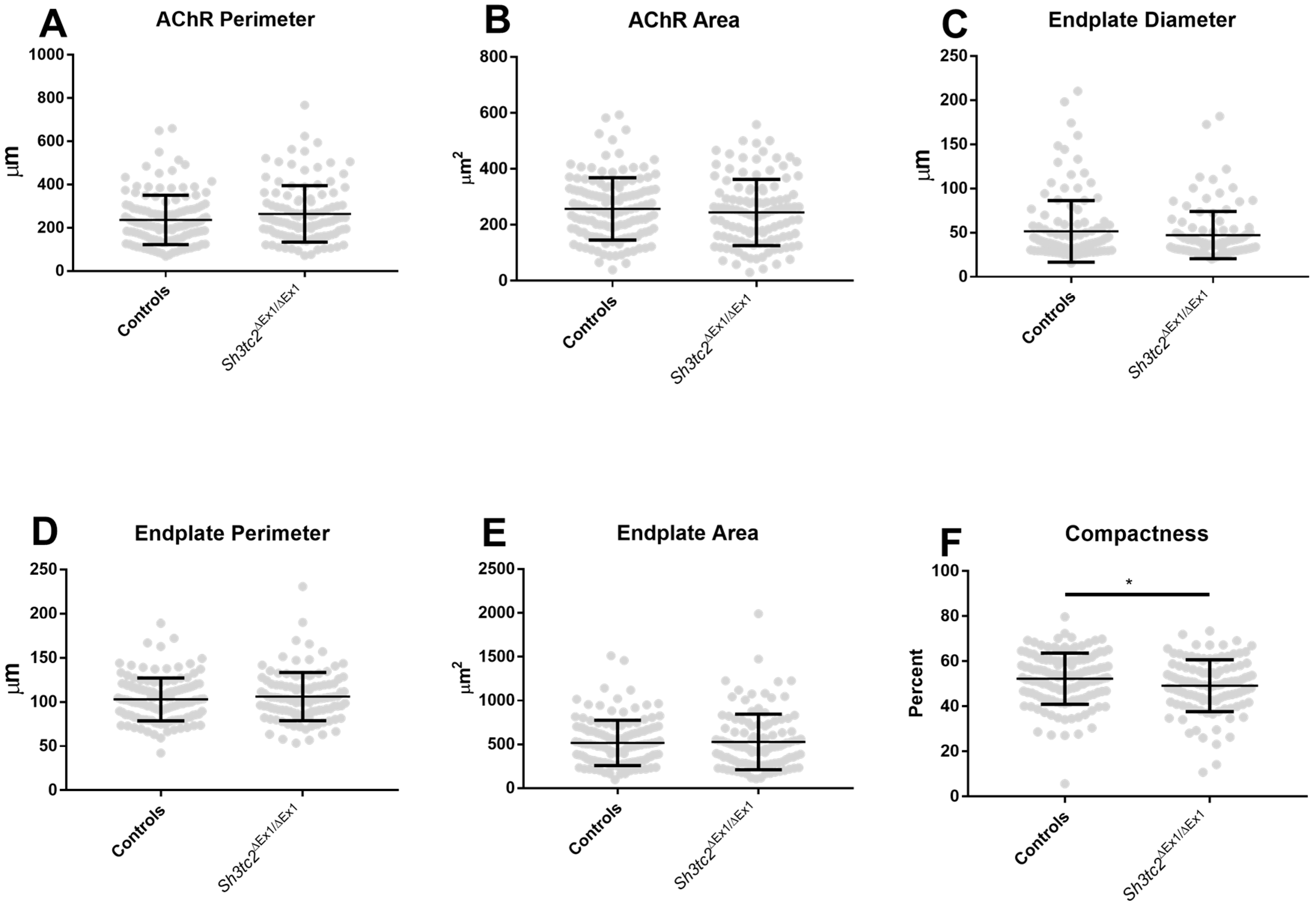
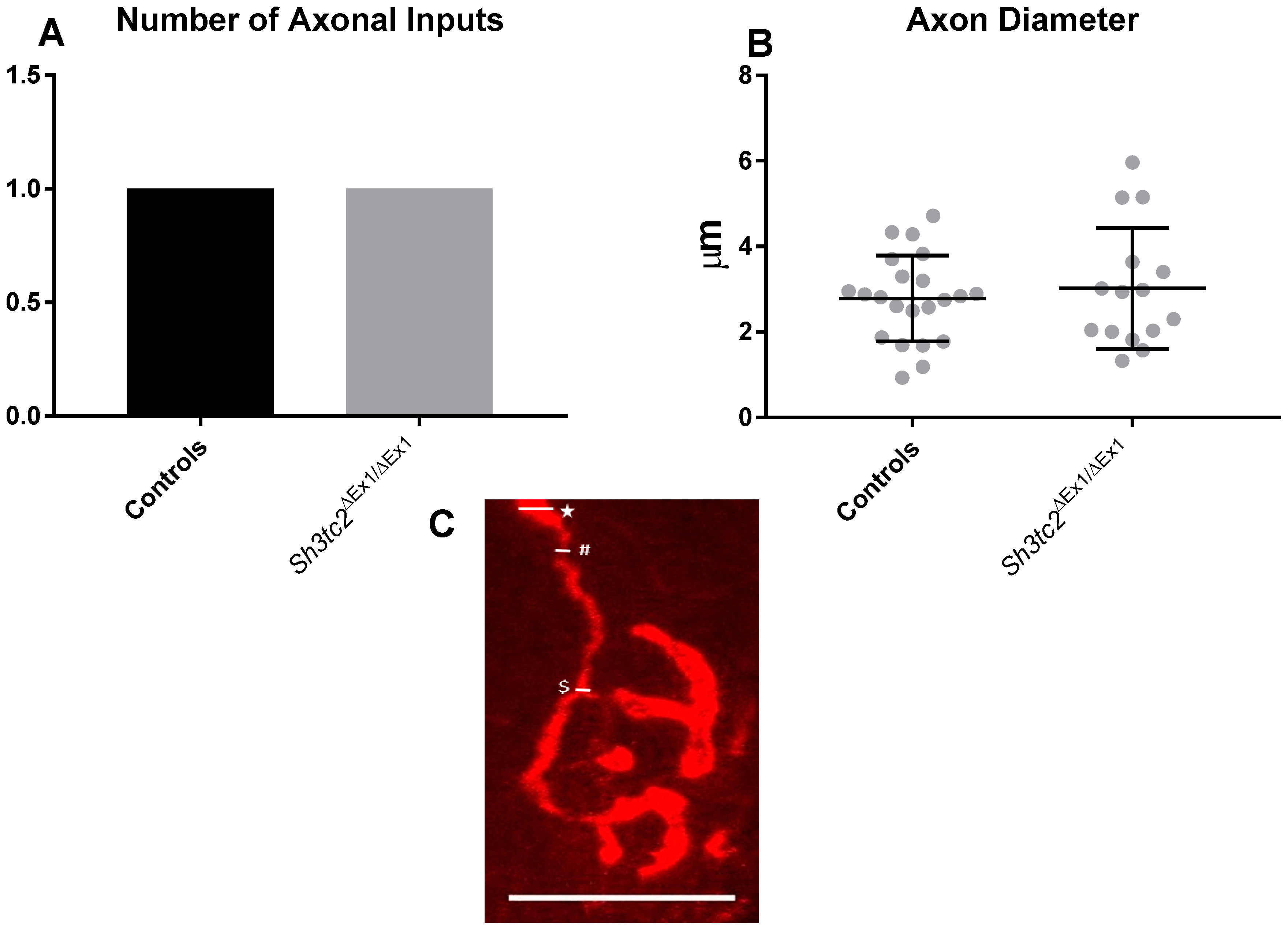
2.1. NMJ Structural Changes in Sh3tc2ΔEx1/ΔEx1 Mice
2.2. Gene Expression Analysis of Denervation and Reinnervation Markers
2.3. Relevant Findings in the Proteomic Signature of SH3TC2-deficient Nerves from Sh3tc2ΔEx1/ΔEx1 Animals
3. Discussion
4. Materials and Methods
4.1. Materials Animals
4.2. NMJ Structural Analysis Using Whole Mount Muscle
4.3. Gene Expression Studies
4.4. RNA Extraction and Conversion
4.5. Real-Time PCR (qPCR)
4.6. Sample Preparation for Proteomics
4.7. Nano LC-ddMS/MS Analysis and Label-free Protein Quantitation
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CMT | Charcot-Marie Tooth |
| SH3TC2 | SH3 domain and tetratricopeptide repeats 2 |
| Nrg1/ErbB | Neuregulin-1/ErbB |
| NMJ | Neuromuscular junction |
| tSC | Terminal Schwann cells |
| SEM | Standard Error of the Mean |
| AChRγ/α/ε | Acetylcholine Receptor gamma/alpha/epsilon |
| MuSK | Muscle Specific Kinase |
| NCAM | Neural cell adhesion molecule |
| NGF | Nerve growth factor |
| BDNF | Brain-derived neurotrophic factor |
| CNTF | Cilliary neurotrophic factor |
| GDNF | Glial cell derived neurotrophic factor |
| NTRK | Neurotrophic receptor tyrosine kinase |
| ECM | Extra-cellular matrix |
| MAPK | Mitogen activated protein kinases |
| GFAP | Glial fibrillary acidic protein |
| THRSP | Thyroid Hormone-Responsive |
| FIBB | Fibrinogen beta chain |
| FIBG | Fibrinogen gamma chain |
| MFAP2 | Microfibrillar-associated protein 2 |
| OLFL3 | Olfactomedin-like protein 3 |
| TGM2 | Protein-glutamine gamma-glutamyltransferase 2 |
| RABP2 | Cellular retinoic acid-binding protein 2 |
| C1QB | Complement C1q subcomponent subunit B |
| LAMA | Laminin subunit alpha |
| APOD/E | Apolipoprotein D/E |
| MATN2 | Matrilin-2 |
| SYNPO | Synaptopodin |
| pAB/mAB | Poly/mono-clonal antibody |
| IgG | Immunoglobulin g |
| PBS | Phosphate Buffered Saline |
| NTC | No template Control |
| RT | Room Temperature |
| CT | Cycle Threshold |
| BCA | bicinchoninic acid assay |
| MS | Mass spectrometry |
References
- Kazamel, M.; Boes, C.J. Charcot Marie Tooth disease (CMT): Historical perspectives and evolution. J. Neurol. 2015, 262, 801–805. [Google Scholar] [CrossRef]
- Skre, H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin. Genet. 1974, 6, 98–118. [Google Scholar] [CrossRef] [PubMed]
- Houlden, H.; Laura, M.; Ginsberg, L.; Jungbluth, H.; Robb, S.A.; Blake, J.; Robinson, S.; King, R.H.; Reilly, M.M. The phenotype of Charcot-Marie-Tooth disease type 4C due to SH3TC2 mutations and possible predisposition to an inflammatory neuropathy. Neuromuscul. Disord. NMD 2009, 19, 264–269. [Google Scholar] [CrossRef]
- LeGuern, E.; Guilbot, A.; Kessali, M.; Ravise, N.; Tassin, J.; Maisonobe, T.; Grid, D.; Brice, A. Homozygosity mapping of an autosomal recessive form of demyelinating Charcot-Marie-Tooth disease to chromosome 5q23-q33. Hum. Mol. Genet. 1996, 5, 1685–1688. [Google Scholar] [CrossRef] [PubMed]
- Gabreels-Festen, A.; van Beersum, S.; Eshuis, L.; LeGuern, E.; Gabreels, F.; van Engelen, B.; Mariman, E. Study on the gene and phenotypic characterisation of autosomal recessive demyelinating motor and sensory neuropathy (Charcot-Marie-Tooth disease) with a gene locus on chromosome 5q23-q33. J. Neurol. Neurosurg. Psychiatry 1999, 66, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Senderek, J.; Bergmann, C.; Stendel, C.; Kirfel, J.; Verpoorten, N.; De Jonghe, P.; Timmerman, V.; Chrast, R.; Verheijen, M.H.; Lemke, G.; et al. Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy. Am. J. Hum. Genet. 2003, 73, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Arntzen, K.A.; Hoyer, H.; Orstavik, K.; Tallaksen, C.; Vedeler, C.; Ostern, R.; Nebuchennykh, M.; Braathen, G.J.; Fagerheim, T. Charcot-Marie-Tooth disease type 4C in Norway: Clinical characteristics, mutation spectrum and minimum prevalence. Neuromuscul. Disord. NMD 2018, 28, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, E.; Zenker, J.; de Preux Charles, A.S.; Stendel, C.; Roos, A.; Medard, J.J.; Tricaud, N.; Kleine, H.; Luscher, B.; Weis, J.; et al. SH3TC2/KIAA1985 protein is required for proper myelination and the integrity of the node of Ranvier in the peripheral nervous system. Proc. Natl. Acad. Sci. USA 2009, 106, 17528–17533. [Google Scholar] [CrossRef]
- Garcia, N.; Tomas, M.; Santafe, M.M.; Lanuza, M.A.; Besalduch, N.; Tomas, J. Localization of brain-derived neurotrophic factor, neurotrophin-4, tropomyosin-related kinase b receptor, and p75 NTR receptor by high-resolution immunohistochemistry on the adult mouse neuromuscular junction. J. Peripher. Nerv. Syst. JPNS 2010, 15, 40–49. [Google Scholar] [CrossRef]
- Kessali, M.; Zemmouri, R.; Guilbot, A.; Maisonobe, T.; Brice, A.; LeGuern, E.; Grid, D. A clinical, electrophysiologic, neuropathologic, and genetic study of two large Algerian families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease. Neurology 1997, 48, 867–873. [Google Scholar] [CrossRef]
- Phan, V.; Schmidt, J.; Matyash, V.; Malchow, S.; Thanisch, M.; Lorenz, C.; Diepolder, I.; Schulz, J.B.; Stenzel, W.; Roos, A.; et al. Characterization of Naive and Vitamin C-Treated Mouse Schwann Cell Line MSC80: Induction of the Antioxidative Thioredoxin Related Transmembrane Protein 1. J. Proteome Res. 2018, 17, 2925–2936. [Google Scholar] [CrossRef] [PubMed]
- Lupo, V.; Galindo, M.I.; Martinez-Rubio, D.; Sevilla, T.; Vilchez, J.J.; Palau, F.; Espinos, C. Missense mutations in the SH3TC2 protein causing Charcot-Marie-Tooth disease type 4C affect its localization in the plasma membrane and endocytic pathway. Hum. Mol. Genet. 2009, 18, 4603–4614. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.C.; Peden, A.A.; Buss, F.; Bright, N.A.; Latouche, M.; Reilly, M.M.; Kendrick-Jones, J.; Luzio, J.P. Mistargeting of SH3TC2 away from the recycling endosome causes Charcot-Marie-Tooth disease type 4C. Hum. Mol. Genet. 2010, 19, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Stendel, C.; Roos, A.; Kleine, H.; Arnaud, E.; Ozcelik, M.; Sidiropoulos, P.N.; Zenker, J.; Schupfer, F.; Lehmann, U.; Sobota, R.M.; et al. SH3TC2, a protein mutant in Charcot-Marie-Tooth neuropathy, links peripheral nerve myelination to endosomal recycling. Brain 2010, 133, 2462–2474. [Google Scholar] [CrossRef] [PubMed]
- Gouttenoire, E.A.; Lupo, V.; Calpena, E.; Bartesaghi, L.; Schupfer, F.; Medard, J.J.; Maurer, F.; Beckmann, J.S.; Senderek, J.; Palau, F.; et al. Sh3tc2 deficiency affects neuregulin-1/ErbB signaling. Glia 2013, 61, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, S.E.; Warrington, A.E.; Bansal, R. The oligodendrocyte and its many cellular processes. Trends Cell Boil. 1993, 3, 191–197. [Google Scholar] [CrossRef]
- Hamid, A.; Eric LeGuern, M.D.; Mustafa ASalih, M.D. Charcot-Marie-Tooth Neuropathy Type 4C. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1340/ (accessed on 26 September 2018).
- Poort, J.E.; Rheuben, M.B.; Breedlove, S.M.; Jordan, C.L. Neuromuscular junctions are pathological but not denervated in two mouse models of spinal bulbar muscular atrophy. Hum. Mol. Genet. 2016, 25, 3768–3783. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.A.; Southam, K.A.; Blizzard, C.A.; King, A.E.; Dickson, T.C. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Chem. Neuroanat. 2016, 76, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Plomp, J.J.; Molenaar, P.C.; O’Hanlon, G.M.; Jacobs, B.C.; Veitch, J.; Daha, M.R.; van Doorn, P.A.; van der Meche, F.G.; Vincent, A.; Morgan, B.P.; et al. Miller Fisher anti-GQ1b antibodies: α-latrotoxin-like effects on motor end plates. Ann. Neurol. 1999, 45, 189–199. [Google Scholar] [CrossRef]
- Ang, E.T.; Schafer, R.; Baltensperger, R.; Wernig, A.; Celio, M.; Oliver, S.S. Motor axonal sprouting and neuromuscular junction loss in an animal model of Charcot-Marie-Tooth disease. J. Neuropathol. Exp. Neurol. 2010, 69, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Scurry, A.N.; Heredia, D.J.; Feng, C.Y.; Gephart, G.B.; Hennig, G.W.; Gould, T.W. Structural and Functional Abnormalities of the Neuromuscular Junction in the Trembler-J Homozygote Mouse Model of Congenital Hypomyelinating Neuropathy. J. Neuropathol. Exp. Neurol. 2016, 75, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Sleigh, J.N.; Grice, S.J.; Burgess, R.W.; Talbot, K.; Cader, M.Z. Neuromuscular junction maturation defects precede impaired lower motor neuron connectivity in Charcot-Marie-Tooth type 2D mice. Hum. Mol. Genet. 2014, 23, 2639–2650. [Google Scholar] [CrossRef] [PubMed]
- Spaulding, E.L.; Sleigh, J.N.; Morelli, K.H.; Pinter, M.J.; Burgess, R.W.; Seburn, K.L. Synaptic Deficits at Neuromuscular Junctions in Two Mouse Models of Charcot-Marie-Tooth Type 2d. J. Neurosci. 2016, 36, 3254–3267. [Google Scholar] [CrossRef] [PubMed]
- McMacken, G.; Abicht, A.; Evangelista, T.; Spendiff, S.; Lochmuller, H. The Increasing Genetic and Phenotypical Diversity of Congenital Myasthenic Syndromes. Neuropediatrics 2017, 48, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Santosa, K.B.; Keane, A.M.; Jablonka-Shariff, A.; Vannucci, B.; Snyder-Warwick, A.K. Clinical relevance of terminal Schwann cells: An overlooked component of the neuromuscular junction. J. Neurosci. Res. 2018, 96, 1125–1135. [Google Scholar] [CrossRef]
- Jones, R.A.; Reich, C.D.; Dissanayake, K.N.; Kristmundsdottir, F.; Findlater, G.S.; Ribchester, R.R.; Simmen, M.W.; Gillingwater, T.H. NMJ-morph reveals principal components of synaptic morphology influencing structure-function relationships at the neuromuscular junction. Open Boil. 2016, 6. [Google Scholar] [CrossRef]
- Roos, A.; Thompson, R.; Horvath, R.; Lochmuller, H.; Sickmann, A. Intersection of Proteomics and Genomics to “Solve the Unsolved” in Rare Disorders such as Neurodegenerative and Neuromuscular Diseases. Proteom. Clin. Appl. 2018, 12. [Google Scholar] [CrossRef]
- Boido, M.; De Amicis, E.; Valsecchi, V.; Trevisan, M.; Ala, U.; Ruegg, M.A.; Hettwer, S.; Vercelli, A. Increasing Agrin Function Antagonizes Muscle Atrophy and Motor Impairment in Spinal Muscular Atrophy. Front. Cell. Neurosci. 2018, 12, 17. [Google Scholar] [CrossRef]
- Miyoshi, S.; Tezuka, T.; Arimura, S.; Tomono, T.; Okada, T.; Yamanashi, Y. DOK7 gene therapy enhances motor activity and life span in ALS model mice. EMBO Mol. Med. 2017, 9, 880–889. [Google Scholar] [CrossRef]
- Willadt, S.; Nash, M.; Slater, C.R. Age-related fragmentation of the motor endplate is not associated with impaired neuromuscular transmission in the mouse diaphragm. Sci. Rep. 2016, 6, 24849. [Google Scholar] [CrossRef]
- Ling, K.K.; Gibbs, R.M.; Feng, Z.; Ko, C.P. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2012, 21, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Michalski, B.; Bain, J.R.; Fahnestock, M. Long-term changes in neurotrophic factor expression in distal nerve stump following denervation and reinnervation with motor or sensory nerve. J. Neurochem. 2008, 105, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Aare, S.; Spendiff, S.; Vuda, M.; Elkrief, D.; Perez, A.; Wu, Q.; Mayaki, D.; Hussain, S.N.; Hettwer, S.; Hepple, R.T. Failed reinnervation in aging skeletal muscle. Skelet. Muscle 2016, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Freeman, R.S.; Burch, R.L.; Crowder, R.J.; Lomb, D.J.; Schoell, M.C.; Straub, J.A.; Xie, L. NGF deprivation-induced gene expression: After ten years, where do we stand? Prog. Brain Res. 2004, 146, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Heumann, R.; Korsching, S.; Bandtlow, C.; Thoenen, H. Changes of nerve growth factor synthesis in nonneuronal cells in response to sciatic nerve transection. J. Cell Boil. 1987, 104, 1623–1631. [Google Scholar] [CrossRef]
- Mancardi, G.L.; Cadoni, A.; Tabaton, M.; Schenone, A.; Zicca, A.; De Martini, I.; Bianchini, D.; Damiani, G.; Zaccheo, D. Schwann cell GFAP expression increases in axonal neuropathies. J. Neurol. Sci. 1991, 102, 177–183. [Google Scholar] [CrossRef]
- Triolo, D.; Dina, G.; Lorenzetti, I.; Malaguti, M.; Morana, P.; Del Carro, U.; Comi, G.; Messing, A.; Quattrini, A.; Previtali, S.C. Loss of glial fibrillary acidic protein (GFAP) impairs Schwann cell proliferation and delays nerve regeneration after damage. J. Cell Sci. 2006, 119, 3981–3993. [Google Scholar] [CrossRef]
- Saller, M.M.; Huettl, R.E.; Mayer, J.M.; Feuchtinger, A.; Krug, C.; Holzbach, T.; Volkmer, E. Validation of a novel animal model for sciatic nerve repair with an adipose-derived stem cell loaded fibrin conduit. Neural Regen. Res. 2018, 13, 854–861. [Google Scholar] [CrossRef]
- Anholt, R.R. Olfactomedin proteins: Central players in development and disease. Front. Cell Dev. Boil. 2014, 2, 6. [Google Scholar] [CrossRef]
- Algarni, A.S.; Hargreaves, A.J.; Dickenson, J.M. Activation of transglutaminase 2 by nerve growth factor in differentiating neuroblastoma cells: A role in cell survival and neurite outgrowth. Eur. J. Pharmacol. 2018, 820, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Ganfornina, M.D.; Do Carmo, S.; Martinez, E.; Tolivia, J.; Navarro, A.; Rassart, E.; Sanchez, D. ApoD, a glia-derived apolipoprotein, is required for peripheral nerve functional integrity and a timely response to injury. Glia 2010, 58, 1320–1334. [Google Scholar] [CrossRef] [PubMed]
- Comley, L.H.; Fuller, H.R.; Wishart, T.M.; Mutsaers, C.A.; Thomson, D.; Wright, A.K.; Ribchester, R.R.; Morris, G.E.; Parson, S.H.; Horsburgh, K.; et al. ApoE isoform-specific regulation of regeneration in the peripheral nervous system. Hum. Mol. Genet. 2011, 20, 2406–2421. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, A.; Ikenberg, B.; Lenz, M.; Becker, D.; Reifenberg, K.; Bas-Orth, C.; Deller, T. Synaptopodin regulates denervation-induced homeostatic synaptic plasticity. Proc. Natl. Acad. Sci. USA 2013, 110, 8242–8247. [Google Scholar] [CrossRef] [PubMed]
- Zhelyaznik, N.; Schrage, K.; McCaffery, P.; Mey, J. Activation of retinoic acid signalling after sciatic nerve injury: Up-regulation of cellular retinoid binding proteins. Eur. J. Neurosci. 2003, 18, 1033–1040. [Google Scholar] [CrossRef]
- Cescon, M.; Gregorio, I.; Eiber, N.; Borgia, D.; Fusto, A.; Sabatelli, P.; Scorzeto, M.; Megighian, A.; Pegoraro, E.; Hashemolhosseini, S.; et al. Collagen VI is required for the structural and functional integrity of the neuromuscular junction. Acta Neuropathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zainul, Z.; Heikkinen, A.; Koivisto, H.; Rautalahti, I.; Kallio, M.; Lin, S.; Haronen, H.; Norman, O.; Ruegg, M.A.; Tanila, H.; et al. Collagen XIII Is Required for Neuromuscular Synapse Regeneration and Functional Recovery after Peripheral Nerve Injury. J. Neurosci. 2018, 38, 4243–4258. [Google Scholar] [CrossRef] [PubMed]
- Kung, F.H.; Sillitti, D.; Shrirao, A.B.; Shreiber, D.I.; Firestein, B.L. Collagen nanofiber anisotropy induces myotube differentiation and acetylcholine receptor clustering. J. Tissue Eng. Regen. Med. 2018, 12, e2010–e2019. [Google Scholar] [CrossRef] [PubMed]
- Bahia El Idrissi, N.; Bosch, S.; Ramaglia, V.; Aronica, E.; Baas, F.; Troost, D. Complement activation at the motor end-plates in amyotrophic lateral sclerosis. J. Neuroinflamm. 2016, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Maselli, R.A.; Arredondo, J.; Vazquez, J.; Chong, J.X.; Bamshad, M.J.; Nickerson, D.A.; Lara, M.; Ng, F.; Lo, V.L.; Pytel, P.; et al. Presynaptic congenital myasthenic syndrome with a homozygous sequence variant in LAMA5 combines myopia, facial tics, and failure of neuromuscular transmission. Am. J. Med. Genet. Part A 2017, 173, 2240–2245. [Google Scholar] [CrossRef]
- Malin, D.; Sonnenberg-Riethmacher, E.; Guseva, D.; Wagener, R.; Aszodi, A.; Irintchev, A.; Riethmacher, D. The extracellular-matrix protein matrilin 2 participates in peripheral nerve regeneration. J. Cell Sci. 2009, 122, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Cheng, H.J. Functions of axon guidance molecules in synapse formation. Curr. Opin. Neurobiol. 2009, 19, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Leal, M.F.; Belangero, P.S.; Cohen, C.; Figueiredo, E.A.; Loyola, L.C.; Pochini, A.C.; Smith, M.C.; Andreoli, C.V.; Belangero, S.I.; Ejnisman, B.; et al. Identification of suitable reference genes for gene expression studies of shoulder instability. PLoS ONE 2014, 9, e105002. [Google Scholar] [CrossRef] [PubMed]
- Koppelkamm, A.; Vennemann, B.; Fracasso, T.; Lutz-Bonengel, S.; Schmidt, U.; Heinrich, M. Validation of adequate endogenous reference genes for the normalisation of qPCR gene expression data in human post mortem tissue. Int. J. Leg. Med. 2010, 124, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Stern-Straeter, J.; Bonaterra, G.A.; Hormann, K.; Kinscherf, R.; Goessler, U.R. Identification of valid reference genes during the differentiation of human myoblasts. BMC Mol. Boil. 2009, 10, 66. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession # | Protein Name | Unique Peptides | Fold of Regulation (Log2) | p-ANOVA | Localization | Function |
|---|---|---|---|---|---|---|
| Q8BGY2 | Eukaryotic translation initiation factor 5A-2 (IF5A2_MOUSE) | 2 | −2.45 | 0.001 | Nucleus/ ER | Mediates effects of polyamines on neuronal process extension and survival |
| Q62264 | Thyroid hormone-inducible hepatic protein (THRSP_MOUSE) | 1 | −2.36 | 0.048 | Nucleus | Mediates biosynthesis of triglycerides with medium-length fatty acid chains and modulates transcription factor activity of THRB |
| Q8K0E8 | Fibrinogen beta chain (FIBB_MOUSE) | 24 | 2.06 | 0.006 | ECM | Polymerizes to form an insoluble fibrin matrix |
| P03995 | Glial fibrillary acidic protein (GFAP_MOUSE) | 5 | 2.08 | 0.009 | Cytoplasm | Class-III intermediate filament; cell-specific marker that, during the development of the central nervous system, distinguishes astrocytes from other glial cells |
| Q8BK62 | Olfactomedin-like protein 3 (OLFL3_MOUSE) | 4 | 2.14 | <0.0005 | ECM | Critical for early development and functional organization of the nervous system as well as hematopoiesis |
| Q60847 | Collagen alpha-1(XII) chain (COCA1_MOUSE) | 51 | 2.16 | <0.0005 | ECM | In general, collagens have been described to be important for the structural and functional integrity of the neuromuscular junction |
| P21981 | Protein-glutamine gamma-glutamyltransferase 2 (TGM2_MOUSE) | 12 | 2.17 | <0.0005 | Multiple | Catalyzes the cross-linking of proteins and the conjugation of polyamines to proteins |
| P14106 | Complement C1q subcomponent subunit B (C1QB_MOUSE) | 1 | 2.21 | 0.047 | ECM | First component of the serum complement system |
| P08121 | Collagen alpha-1(III) chain (CO3A1_MOUSE) | 3 | 2.42 | 0.042 | ECM | In general, collagens have been described to be important for the structural and functional integrity of the neuromuscular junction |
| Q8VCM7 | Fibrinogen gamma chain (FIBG_MOUSE) | 20 | 2.47 | 0.004 | ECM | Polymerizes to form an insoluble fibrin matrix |
| Q61001 | Laminin subunit alpha-5 (LAMA5_MOUSE) | 24 | 2.50 | 0.020 | ECM | Mediate the attachment, migration and organization of cells into tissues during embryonic development by interacting with other extracellular matrix components |
| P55002 | Microfibrillar-associated protein 2 (MFAP2_MOUSE) | 3 | 2.78 | 0.050 | ECM | Fibrinogen binding; component of the elastin-associated microfibrils |
| P51910 | Apolipoprotein D (APOD_MOUSE) | 9 | 2.92 | <0.0005 | ECM | Glia-derived apolipoprotein required for peripheral nerve functional integrity and a timely response to injury |
| Q8CC35 | Synaptopodin (SYNPO_MOUSE) | 1 | 3.02 | 0.005 | Cytoskeleton | modulating actin-based shape and motility of dendritic spines; involved in synaptic plasticity |
| P11087 | Collagen alpha-1(I) chain (CO1A1_MOUSE) | 8 | 3.08 | 0.036 | ECM | In general, collagens have been described to be important for the structural and functional integrity of the neuromuscular junction |
| P22935 | Cellular retinoic acid-binding protein 2 (RABP2_MOUSE | 2 | 3.14 | <0.0005 | Nucleus/ ER | Participates in retinoic acid-modulated nerve regeneration |
| Q01149 | Collagen alpha-2(I) chain (CO1A2_MOUSE) | 7 | 3.32 | 0.033 | ECM | In general, collagens have been described to be important for the structural and functional integrity of the neuromuscular junction |
| O08746 | Matrilin-2 (MATN2_MOUSE) | 11 | 3.40 | 0.003 | ECM | Involved in matrix assembly and axon guidance |
| P08226 | Apolipoprotein E (APOE_MOUSE) | 18 | 3.52 | <0.0005 | ECM | Regulation of regeneration in the peripheral nervous system |
| Q2UY11 | Collagen alpha-1(XXVIII) chain (COSA1_MOUSE | 19 | 4.10 | 0.006 | ECM | In general, collagens have been described to be important for the structural and functional integrity of the neuromuscular junction |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cipriani, S.; Phan, V.; Médard, J.-J.; Horvath, R.; Lochmüller, H.; Chrast, R.; Roos, A.; Spendiff, S. Neuromuscular Junction Changes in a Mouse Model of Charcot-Marie-Tooth Disease Type 4C. Int. J. Mol. Sci. 2018, 19, 4072. https://doi.org/10.3390/ijms19124072
Cipriani S, Phan V, Médard J-J, Horvath R, Lochmüller H, Chrast R, Roos A, Spendiff S. Neuromuscular Junction Changes in a Mouse Model of Charcot-Marie-Tooth Disease Type 4C. International Journal of Molecular Sciences. 2018; 19(12):4072. https://doi.org/10.3390/ijms19124072
Chicago/Turabian StyleCipriani, Silvia, Vietxuan Phan, Jean-Jacques Médard, Rita Horvath, Hanns Lochmüller, Roman Chrast, Andreas Roos, and Sally Spendiff. 2018. "Neuromuscular Junction Changes in a Mouse Model of Charcot-Marie-Tooth Disease Type 4C" International Journal of Molecular Sciences 19, no. 12: 4072. https://doi.org/10.3390/ijms19124072
APA StyleCipriani, S., Phan, V., Médard, J.-J., Horvath, R., Lochmüller, H., Chrast, R., Roos, A., & Spendiff, S. (2018). Neuromuscular Junction Changes in a Mouse Model of Charcot-Marie-Tooth Disease Type 4C. International Journal of Molecular Sciences, 19(12), 4072. https://doi.org/10.3390/ijms19124072