Natural Killer Cells and Current Applications of Chimeric Antigen Receptor-Modified NK-92 Cells in Tumor Immunotherapy

Abstract
:1. Introduction
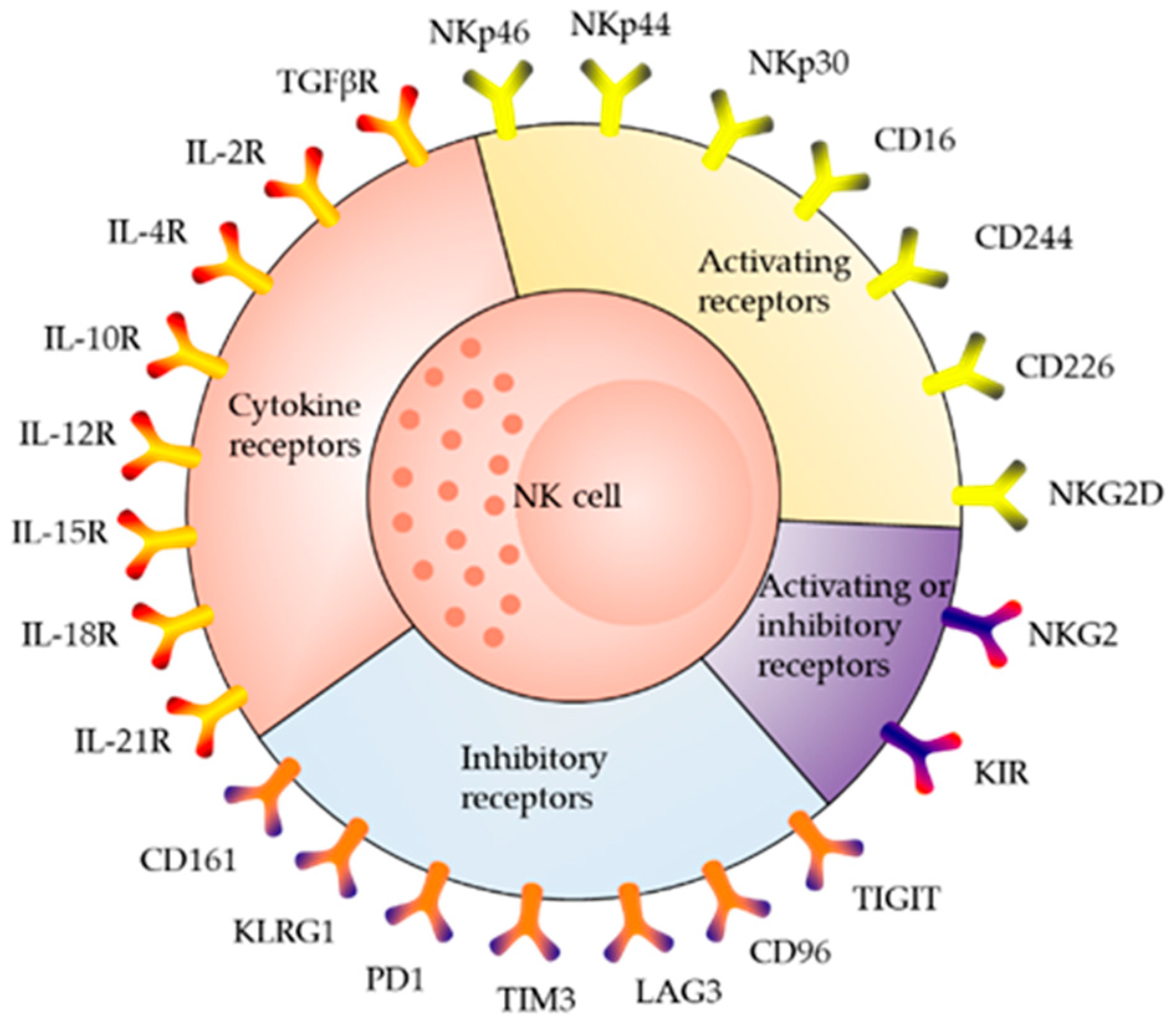
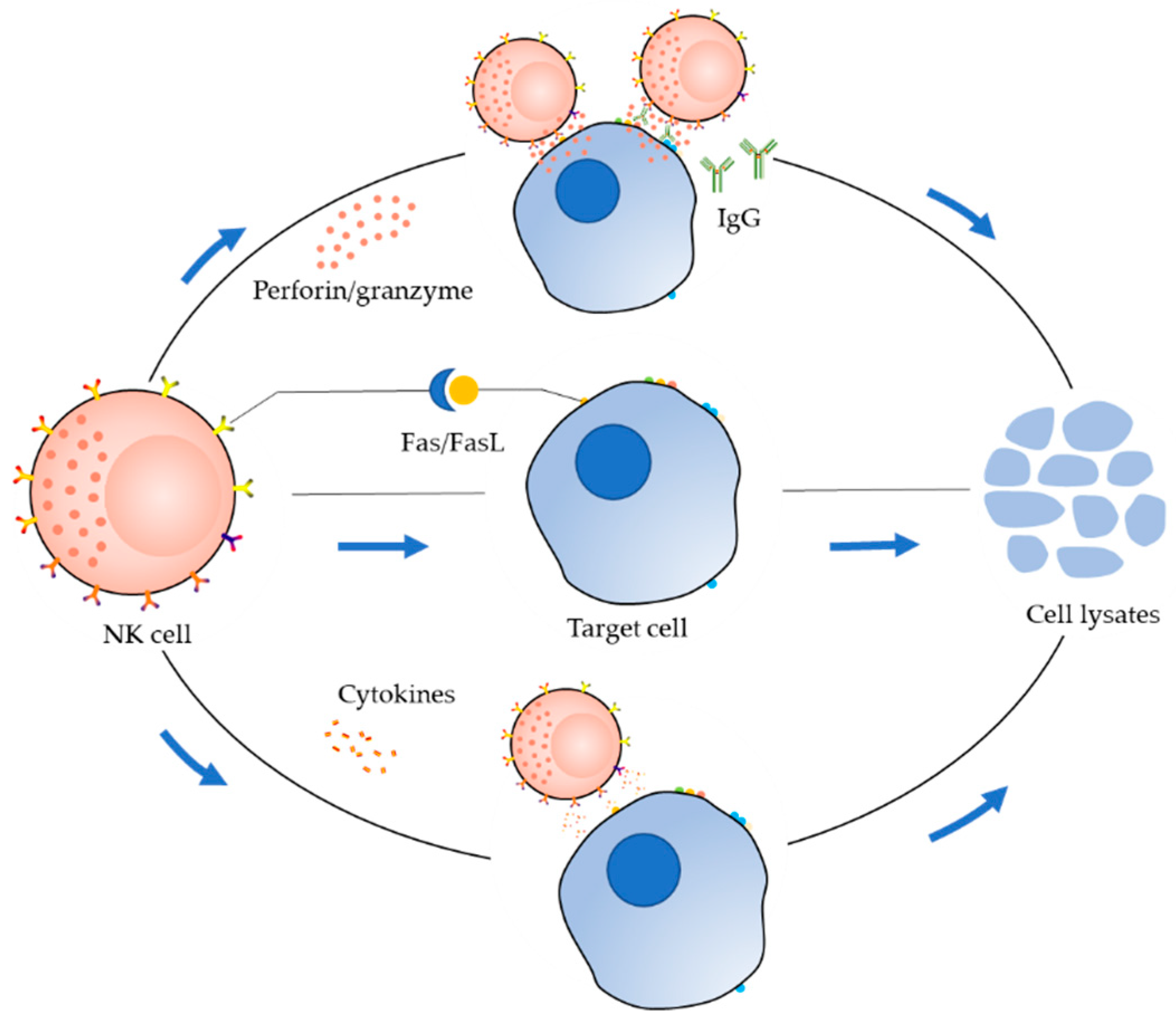
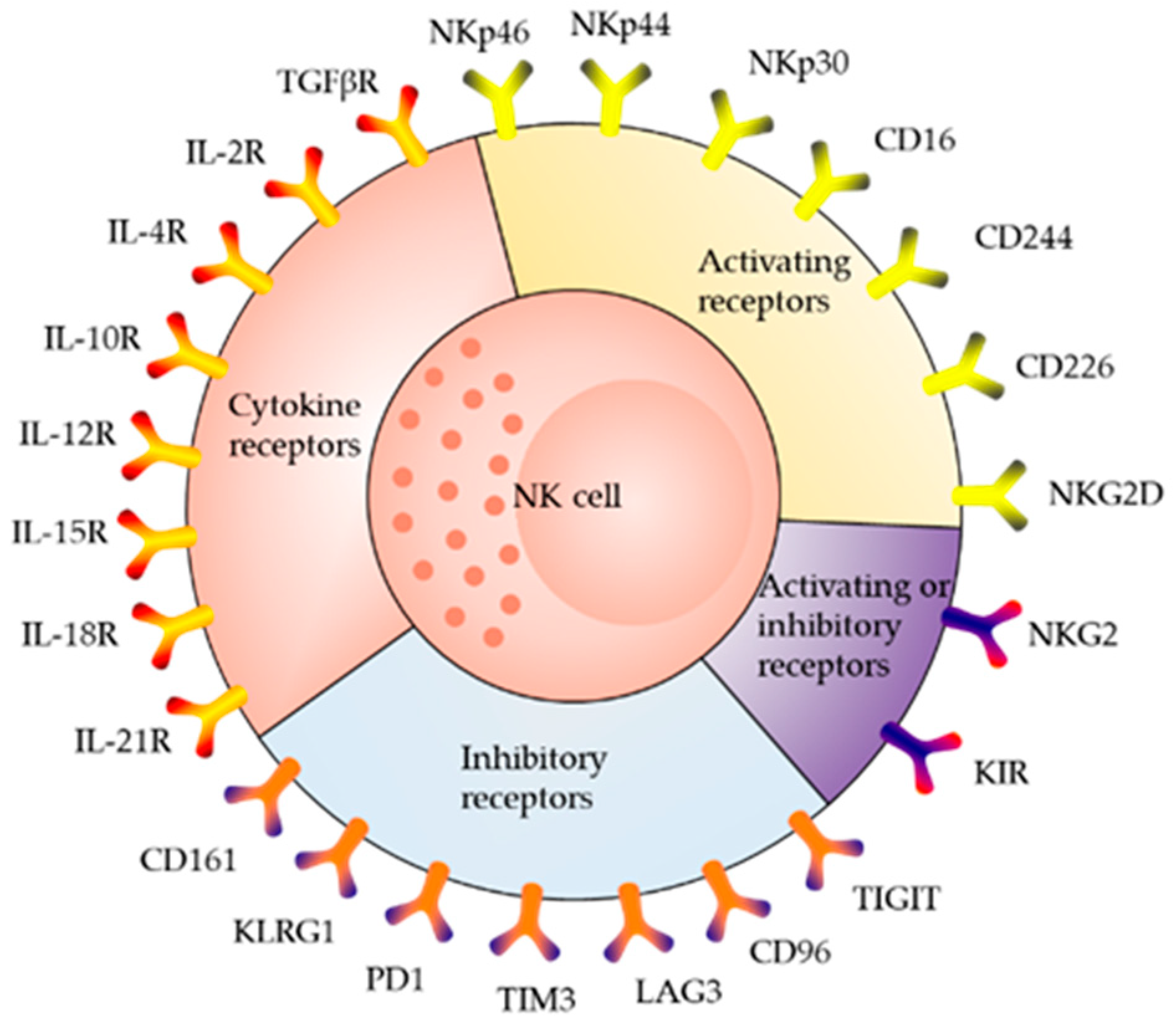
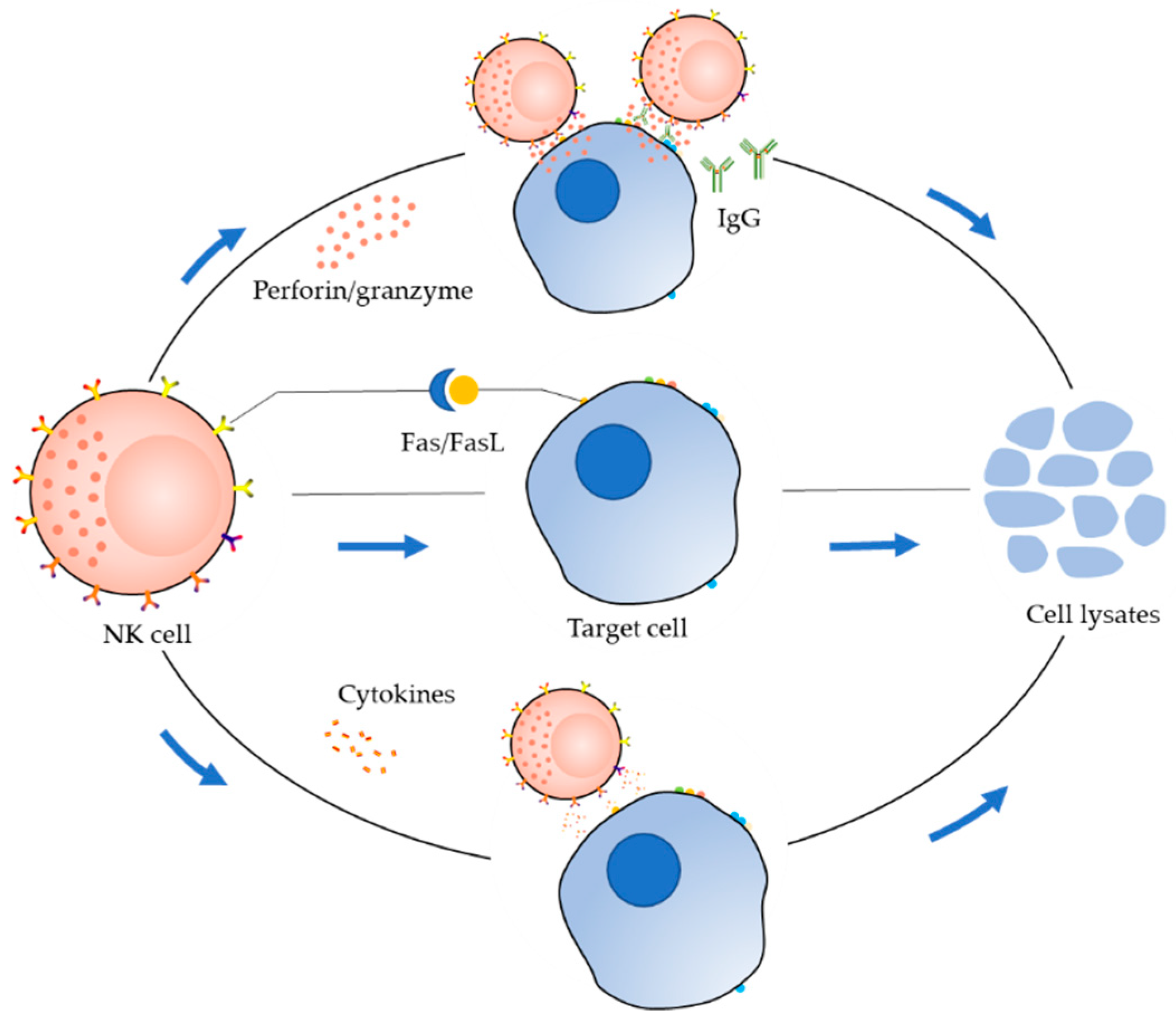
2. Receptor Distribution and Killing Mechanism of NK Cells
3. Currently Known NK Cell Lines
3.1. NK3.3 Cells
3.2. YT Cells
3.3. NKL Cells
3.4. HANK1 Cells
3.5. NK-YS Cells
3.6. KHYG-1 Cells
3.7. SNK-6 and SNT-8 Cells
3.8. IMC-1 Cells
3.9. NK-92 Cells
4. Progress in the Application of NK-92 Cells
5. Structure of CARs and Their Applications in NK-92 Cells
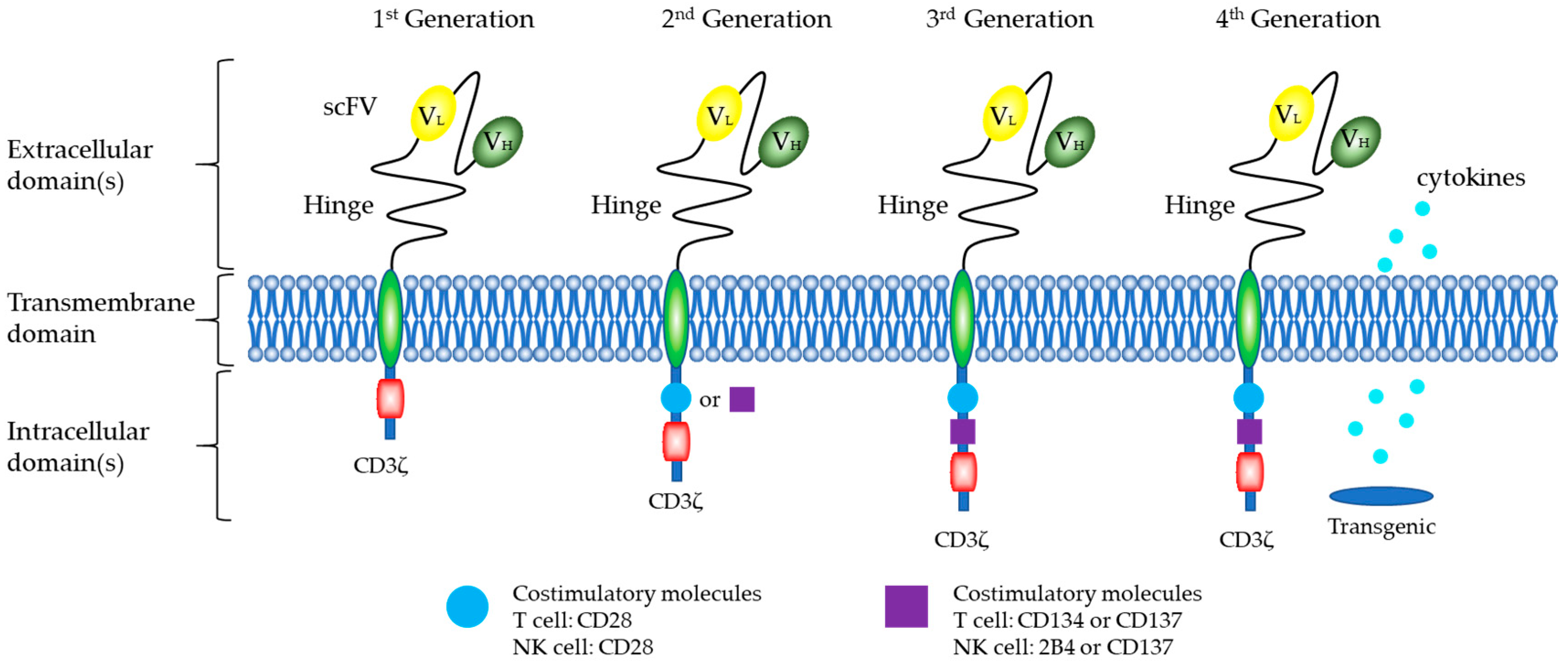
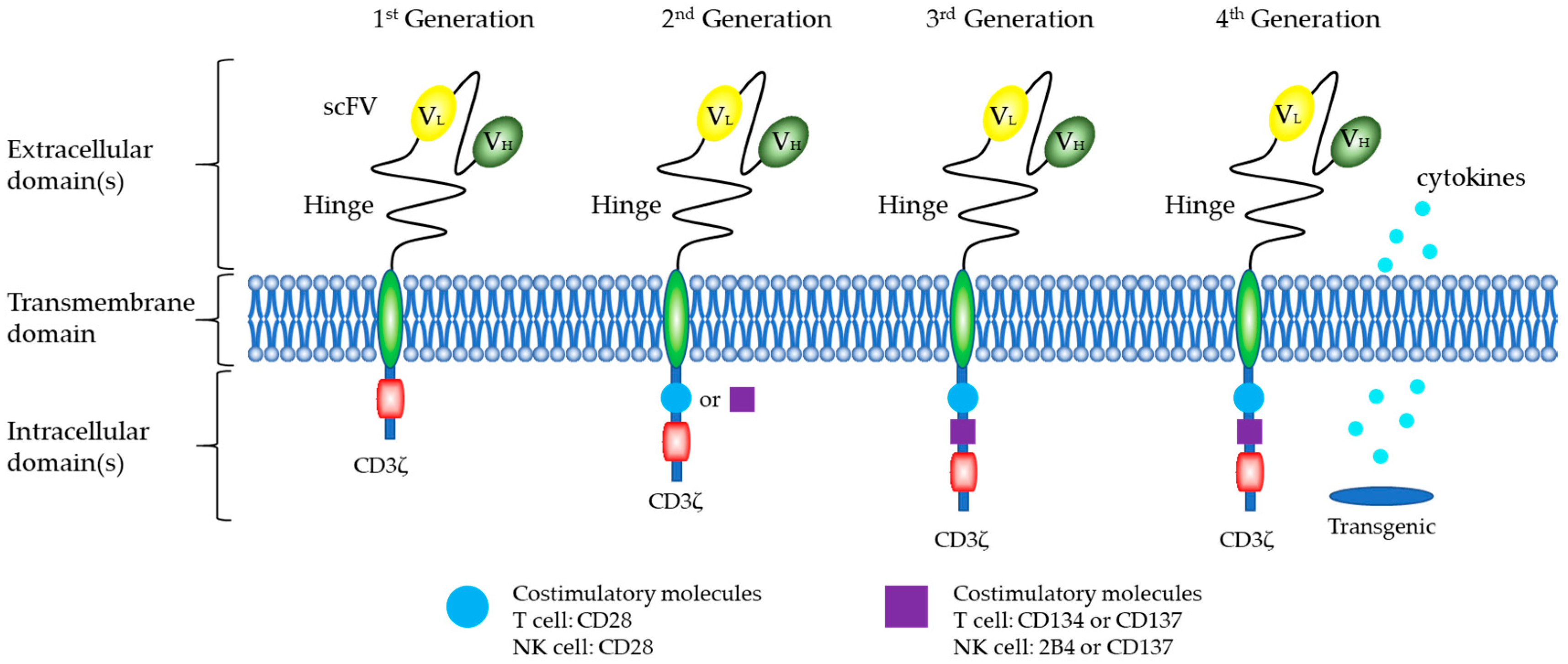
5.1. Structure of CARs
CARs of NK Cells
5.2. Preclinical Studies of CAR-NK-92 Cells
5.3. Ongoing Clinical Trials
5.4. Advantages of CAR-NK-92 Cells
5.5. Challenges and Coping Strategies
5.5.1. Tumor-Producing and Potential Epstein-Barr (EB) Virus Susceptibility
5.5.2. NK-92 Cells Have A Short Life Cycle after Irradiation
5.5.3. Defects in the Transfected Vector
5.5.4. Off-Target Effects
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADCC | Antibody dependent cell mediated cytotoxicity |
| CAR | Chimeric antigen receptor |
| cGMP | Current good manufacturing practice |
| EB | Epstein-Barr |
| FDA | Food and Drug Administration |
| GVHD | Graft-versus-host disease |
| HER2 | Human epidermal growth factor receptor 2 |
| IL-2 | Interleukin-2 |
| ITAMs | Immunoreceptor tyrosine-based activation motif |
| KIRs | Killer immunoglobulin-like receptor |
| LGLs | Large granular lymphocytes |
| MHC | Major histocompatibility complex |
| NK | Natural killer |
| PB | Peripheral blood |
| scFv | Single-chain variable fragment |
| TCR | T-cell receptor |
| TNF | Tumor necrosis factor |
References
- Fang, F.; Xiao, W.; Tian, Z. NK cell-based immunotherapy for cancer. Semin. Immunol. 2017, 31, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Hazenberg, M.D.; Spits, H. Human innate lymphoid cells. Blood 2014, 124, 700–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.S.; Hasegawa, J. Natural killer cell biology: An update and future directions. J. Allergy Clin. Immun. 2013, 132, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerwenka, A.; Lanier, L.L. Natural killer cell memory in infection, inflammation and cancer. Nat. Rev. Immunol. 2016, 16, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Hammer, Q.; Ruckert, T.; Romagnani, C. Natural killer cell specificity for viral infections. Nat. Immunol. 2018, 19, 800–808. [Google Scholar] [CrossRef]
- Mehta, R.S.; Rezvani, K. Chimeric Antigen Receptor Expressing Natural Killer Cells for the Immunotherapy of Cancer. Front. Immunol. 2018, 9, 283. [Google Scholar] [CrossRef]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef]
- Handgretinger, R.; Lang, P.; André, M.C. Exploitation of natural killer cells for the treatment of acute leukemia. Blood 2016, 127, 3341–3349. [Google Scholar] [CrossRef] [Green Version]
- Romagnani, C.; Juelke, K.; Falco, M.; Morandi, B.; D’Agostino, A.; Costa, R.; Ratto, G.; Forte, G.; Carrega, P.; Lui, G.; et al. CD56brightCD16- Killer Ig-Like Receptor- NK Cells Display Longer Telomeres and Acquire Features of CD56dim NK Cells upon Activation. J. Immunol. 2007, 178, 4947–4955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehniger, T.A.; Cooper, M.A.; Nuovo, G.J.; Cella, M.; Facchetti, F.; Colonna, M.; Caligiuri, M.A. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: A potential new link between adaptive and innate immunity. Blood 2003, 101, 3052–3057. [Google Scholar] [CrossRef] [PubMed]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Romain, G.; Senyukov, V.; Rey-Villamizar, N.; Merouane, A.; Kelton, W.; Liadi, I.; Mahendra, A.; Charab, W.; Georgiou, G.; Roysam, B.; et al. Antibody Fc engineering improves frequency and promotes kinetic boosting of serial killing mediated by NK cells. Blood 2014, 124, 3241–3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Zhang, J. Reformation in chimeric antigen receptor based cancer immunotherapy: Redirecting natural killer cell. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 200–215. [Google Scholar] [CrossRef] [PubMed]
- De Maria, A.; Bozzano, F.; Cantoni, C.; Moretta, L. Revisiting human natural killer cell subset function revealed cytolytic CD56(dim)CD16+ NK cells as rapid producers of abundant IFN-gamma on activation. Proc. Natl. Acad. Sci. USA 2011, 108, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, P.V.; Gunturi, A.; Salcedo, M.; Schatzle, J.D.; Lai, W.C.; Kurepa, Z.; Pitcher, L.; Seaman, M.S.; Lemonnier, F.A.; Bennett, M.; et al. Cutting edge: Expression of functional CD94/NKG2A inhibitory receptors on fetal NK1.1+Ly-49- cells: A possible mechanism of tolerance during NK cell development. J. Immunol. 1999, 162, 6976–6980. [Google Scholar]
- Kumar, S. Natural killer cell cytotoxicity and its regulation by inhibitory receptors. Immunology 2018, 154, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H. The Fas Signaling Pathway: More Than a Paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef] [PubMed]
- Waring, P.; Mullbacher, A. Cell death induced by the Fas/Fas ligand pathway and its role in pathology. Immunol. Cell Biol. 1999, 77, 312–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornbluth, J.; Flomenberg, N.; Dupont, B. Cell surface phenotype of a cloned line of human natural killer cells. J. Immunol. 1982, 129, 2831–2837. [Google Scholar] [PubMed]
- Le Bouteiller, P.; Barakonyi, A.; Giustiniani, J.; Lenfant, F.; Marie-Cardine, A.; Aguerre-Girr, M.; Rabot, M.; Hilgert, I.; Mami-Chouaib, F.; Tabiasco, J.; et al. Engagement of CD160 receptor by HLA-C is a triggering mechanism used by circulating natural killer (NK) cells to mediate cytotoxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 16963–16968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umehara, H.; Huang, J.Y.; Kono, T.; Tabassam, F.H.; Okazaki, T.; Bloom, E.T.; Domae, N. Involvement of protein tyrosine kinase p72syk and phosphatidylinositol 3-kinase in CD2-mediated granular exocytosis in the natural killer cell line, NK3.3. J. Immunol. 1997, 159, 1200–1207. [Google Scholar] [PubMed]
- Mahle, N.H.; Radcliff, G.; Sevilla, C.L.; Kornbluth, J.; Callewaert, D.M. Kinetics of cellular cytotoxicity mediated by a cloned human natural killer cell line. Immunobiology 1989, 179, 230–243. [Google Scholar] [CrossRef]
- Yodoi, J.; Teshigawara, K.; Nikaido, T.; Fukui, K.; Noma, T.; Honjo, T.; Takigawa, M.; Sasaki, M.; Minato, N.; Tsudo, M.; et al. TCGF (IL 2)-receptor inducing factor(s). I. Regulation of IL 2 receptor on a natural killer-like cell line (YT cells). J. Immunol. 1985, 134, 1623–1630. [Google Scholar]
- Yoneda, N.; Tatsumi, E.; Kawano, S.; Teshigawara, K.; Oka, T.; Fukuda, M.; Yamaguchi, N. Detection of Epstein-Barr virus genome in natural-killer-like cell line, YT. Leukemia 1992, 6, 136–141. [Google Scholar]
- Chen, X.; Allan, D.; Krzewski, K.; Ge, B.; Kopcow, H.; Strominger, J.L. CD28-stimulated ERK2 phosphorylation is required for polarization of the microtubule organizing center and granules in YTS NK cells. Proc. Natl. Acad. Sci. USA 2006, 103, 10346–10351. [Google Scholar] [CrossRef] [Green Version]
- Robertson, M.J.; Cochran, K.J.; Cameron, C.; Le, J.M.; Tantravahi, R.; Ritz, J. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. Exp. Hematol. 1996, 24, 406–415. [Google Scholar]
- Kagami, Y.; Nakamura, S.; Suzuki, R.; Iida, S.; Yatabe, Y.; Okada, Y.; Kobayashi, T.; Tsurumi, T.; Seto, M.; Ogura, M.; et al. Establishment of an IL-2-dependent cell line derived from ‘nasal-type’ NK/T-cell lymphoma of CD2+, sCD3-, CD3epsilon+, CD56+ phenotype and associated with the Epstein-Barr virus. Br. J. Haematol. 1998, 103, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiyama, J.; Yoshino, T.; Mori, M.; Kondoh, E.; Oka, T.; Akagi, T.; Hiraki, A.; Nakayama, H.; Shibuya, A.; Ma, Y.; et al. Characterization of a novel human natural killer-cell line (NK-YS) established from natural killer cell lymphoma/leukemia associated with Epstein-Barr virus infection. Blood 1998, 92, 1374–1383. [Google Scholar]
- Yagita, M.; Huang, C.L.; Umehara, H.; Matsuo, Y.; Tabata, R.; Miyake, M.; Konaka, Y.; Takatsuki, K. A novel natural killer cell line (KHYG-1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia 2000, 14, 922–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suck, G.; Branch, D.R.; Smyth, M.J.; Miller, R.G.; Vergidis, J.; Fahim, S.; Keating, A. KHYG-1, a model for the study of enhanced natural killer cell cytotoxicity. Exp. Hematol. 2005, 33, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Nagata, H.; Konno, A.; Kimura, N.; Zhang, Y.; Kimura, M.; Demachi, A.; Sekine, T.; Yamamoto, K.; Shimizu, N. Characterization of novel natural killer (NK)-cell and gammadelta T-cell lines established from primary lesions of nasal T/NK-cell lymphomas associated with the Epstein-Barr virus. Blood 2001, 97, 708–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, I.; Whalen, M.; Bankhurst, A.; Sever, C.E.; Doshi, R.; Hardekopf, D.; Montgomery, K.; Willman, C.L. A new human natural killer leukemia cell line, IMC-1. A complex chromosomal rearrangement defined by spectral karyotyping: Functional and cytogenetic characterization. Leukemia Res. 2004, 28, 275–284. [Google Scholar] [CrossRef]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994, 8, 652–658. [Google Scholar] [PubMed]
- Tam, Y.K.; Maki, G.; Miyagawa, B.; Hennemann, B.; Tonn, T.; Klingemann, H.G. Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy. Hum. Gene Ther. 1999, 10, 1359–1373. [Google Scholar] [CrossRef] [PubMed]
- Tam, Y.K.; Miyagawa, B.; Ho, V.C.; Klingemann, H.G. Immunotherapy of malignant melanoma in a SCID mouse model using the highly cytotoxic natural killer cell line NK-92. J. Hematother. 1999, 8, 281. [Google Scholar] [CrossRef] [PubMed]
- Klingemann, H.G.; Miyagawa, B. Purging of malignant cells from blood after short ex vivo incubation with NK-92 cells. Blood 1996, 87, 4913–4914. [Google Scholar]
- Isobe, Y.; Sugimoto, K.; Yang, L.; Tamayose, K.; Egashira, M.; Kaneko, T.; Takada, K.; Oshimi, K. Epstein-Barr virus infection of human natural killer cell lines and peripheral blood natural killer cells. Cancer Res. 2004, 64, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Maki, G.; Klingemann, H.G.; Martinson, J.A.; Tam, Y.K. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J. Hematother. Stem Cell. Res. 2001, 10, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Boyiadzis, M.; Agha, M.; Redner, R.L.; Sehgal, A.; Im, A.; Hou, J.; Farah, R.; Dorritie, K.A.; Raptis, A.; Lim, S.H.; et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy 2017, 19, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef]
- Williams, B.A.; Law, A.D.; Routy, B.; DenHollander, N.; Gupta, V.; Wang, X.H.; Chaboureau, A.; Viswanathan, S.; Keating, A. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget 2017, 8, 89256–89268. [Google Scholar] [CrossRef]
- Tonn, T.; Becker, S.; Esser, R.; Schwabe, D.; Seifried, E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J. Hematother. Stem Cell. Res. 2001, 10, 535–544. [Google Scholar] [CrossRef]
- Yan, Y.; Steinherz, P.; Klingemann, H.G.; Dennig, D.; Childs, B.H.; McGuirk, J.; O’Reilly, R.J. Antileukemia activity of a natural killer cell line against human leukemias. Clin. Cancer Res. 1998, 4, 2859–2868. [Google Scholar]
- Swift, B.E.; Williams, B.A.; Kosaka, Y.; Wang, X.H.; Medin, J.A.; Viswanathan, S.; Martinez-Lopez, J.; Keating, A. Natural killer cell lines preferentially kill clonogenic multiple myeloma cells and decrease myeloma engraftment in a bioluminescent xenograft mouse model. Haematologica 2012, 97, 1020–1028. [Google Scholar] [CrossRef] [Green Version]
- Konstantinidis, K.V.; Alici, E.; Aints, A.; Christensson, B.; Ljunggren, H.G.; Dilber, M.S. Targeting IL-2 to the endoplasmic reticulum confines autocrine growth stimulation to NK-92 cells. Exp. Hematol. 2005, 33, 159–164. [Google Scholar] [CrossRef]
- Tam, Y.K.; Martinson, J.A.; Doligosa, K.; Klingemann, H.G. Ex vivo expansion of the highly cytotoxic human natural killer-92 cell-line under current good manufacturing practice conditions for clinical adoptive cellular immunotherapy. Cytotherapy 2003, 5, 259–272. [Google Scholar] [PubMed]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy—Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Pegram, H.J.; Smith, E.L.; Rafiq, S.; Brentjens, R.J. CAR therapy for hematological cancers: Can success seen in the treatment of B-cell acute lymphoblastic leukemia be applied to other hematological malignancies? Immunotherapy 2015, 7, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 139r–303r. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224r–225r. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Sredni, B.; Longo, D.L. Cancer immunotherapy: Are we there yet? Semin. Cancer Biol. 2012, 22, 1–2. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Wilson, W.H. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef] [Green Version]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 138r–177r. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Second CAR T-cell Therapy. Cancer Discov. 2018, 8, 5–6. [CrossRef] [PubMed]
- First-ever CAR T-cell therapy approved in U.S. Cancer Discov. 2017. [CrossRef]
- Wang, Z.; Wu, Z.; Liu, Y.; Han, W. New development in CAR-T cell therapy. J. Hematol. Oncol. 2017, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, E.; Yang, Y.; Qin, H.; Chien, C.D.; Kochenderfer, J.N.; Fry, T.J. Murine allogeneic CD19 CAR T cells harbor potent antileukemic activity but have the potential to mediate lethal GVHD. Blood 2016, 127, 1361–1370. [Google Scholar] [CrossRef]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef]
- Fan, M.; Li, M.; Gao, L.; Geng, S.; Wang, J.; Wang, Y.; Yan, Z.; Yu, L. Chimeric antigen receptors for adoptive T cell therapy in acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 151. [Google Scholar] [CrossRef]
- Jensen, M.C.; Riddell, S.R. Design and implementation of adoptive therapy with chimeric antigen receptor-modified T cells. Immunol. Rev. 2014, 257, 127–144. [Google Scholar] [CrossRef]
- Wang, J.; Jensen, M.; Lin, Y.; Sui, X.; Chen, E.; Lindgren, C.G.; Till, B.; Raubitschek, A.; Forman, S.J.; Qian, X.; et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum. Gene Ther. 2007, 18, 712–725. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [PubMed]
- Love, P.E.; Hayes, S.M. ITAM-mediated signaling by the T-cell antigen receptor. Cold Spring Harb. Perspect. Biol. 2010, 2, a2485. [Google Scholar] [CrossRef] [PubMed]
- Arnon, T.I.; Markel, G.; Mandelboim, O. Tumor and viral recognition by natural killer cells receptors. Semin. Cancer Biol. 2006, 16, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Tassev, D.V.; Cheng, M.; Cheung, N.K. Retargeting NK92 cells using an HLA-A2-restricted, EBNA3C-specific chimeric antigen receptor. Cancer Gene Ther. 2012, 19, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Gilfillan, S.; Ho, E.L.; Cella, M.; Yokoyama, W.M.; Colonna, M. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat. Immunol. 2002, 3, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. The choices of a natural killer. Nat. Immunol. 2003, 4, 509–510. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.H.; Connolly, J.; Shimasaki, N.; Mimura, K.; Kono, K.; Campana, D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013, 73, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- McNerney, M.E.; Lee, K.; Kumar, V. 2B4 (CD244) is a non-MHC binding receptor with multiple functions on natural killer cells and CD8+ T cells. Mol. Immunol. 2005, 42, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Altvater, B.; Landmeier, S.; Pscherer, S.; Temme, J.; Schweer, K.; Kailayangiri, S.; Campana, D.; Juergens, H.; Pule, M.; Rossig, C. 2B4 (CD244) Signaling by Recombinant Antigen-specific Chimeric Receptors Costimulates Natural Killer Cell Activation to Leukemia and Neuroblastoma Cells. Clin. Cancer Res. 2009, 15, 4857–4866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uherek, C.; Tonn, T.; Uherek, B.; Becker, S.; Schnierle, B.; Klingemann, H.G.; Wels, W. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood 2002, 100, 1265–1273. [Google Scholar] [PubMed]
- Romanski, A.; Uherek, C.; Bug, G.; Seifried, E.; Klingemann, H.; Wels, W.S.; Ottmann, O.G.; Tonn, T. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J. Cell. Mol. Med. 2016, 20, 1287–1294. [Google Scholar] [CrossRef] [Green Version]
- Oelsner, S.; Friede, M.E.; Zhang, C.; Wagner, J.; Badura, S.; Bader, P.; Ullrich, E.; Ottmann, O.G.; Klingemann, H.; Tonn, T.; et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017, 19, 235–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boissel, L.; Betancur, M.; Wels, W.S.; Tuncer, H.; Klingemann, H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leukemia Res. 2009, 33, 1255–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boissel, L.; Betancur, M.; Lu, W.; Krause, D.; Van Etten, R.; Wels, W.; Klingemann, H. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology 2014, 2, e26527. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhang, W.; Shang, P.; Zhang, H.; Fu, W.; Ye, F.; Zeng, T.; Huang, H.; Zhang, X.; Sun, W.; et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol. Oncol. 2014, 8, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Han, J.; Chu, J.; Zhang, L.; Zhang, J.; Chen, C.; Chen, L.; Wang, Y.; Wang, H.; Yi, L.; et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 2016, 7, 27764. [Google Scholar] [CrossRef]
- Alkins, R.; Burgess, A.; Kerbel, R.; Wels, W.S.; Hynynen, K. Early treatment of HER2-amplified brain tumors with targeted NK-92 cells and focused ultrasound improves survival. Neuro-Oncology 2016, 18, 974–981. [Google Scholar] [CrossRef] [Green Version]
- Alkins, R.; Burgess, A.; Ganguly, M.; Francia, G.; Kerbel, R.; Wels, W.S.; Hynynen, K. Focused Ultrasound Delivers Targeted Immune Cells to Metastatic Brain Tumors. Cancer Res. 2013, 73, 1892–1899. [Google Scholar] [CrossRef] [Green Version]
- Sahm, C.; Schonfeld, K.; Wels, W.S. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol. Immunother. 2012, 61, 1451–1461. [Google Scholar] [CrossRef]
- Daldrup-Link, H.E.; Meier, R.; Rudelius, M.; Piontek, G.; Piert, M.; Metz, S.; Settles, M.; Uherek, C.; Wels, W.; Schlegel, J.R.; et al. In vivo tracking of genetically engineered, anti-HER2/neu directed natural killer cells to HER2/neu positive mammary tumors with magnetic resonance imaging. Eur. Radiol. 2005, 15, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Meier, R.; Piert, M.; Piontek, G.; Rudelius, M.; Oostendorp, R.A.; Senekowitsch-Schmidtke, R.; Henning, T.D.; Wels, W.S.; Uherek, C.; Rummeny, E.J.; et al. Tracking of [18F]FDG-labeled natural killer cells to HER2/neu-positive tumors. Nucl. Med. Biol. 2008, 35, 579–588. [Google Scholar] [CrossRef]
- Schonfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bonig, H.; Kohl, U.; Kloess, S.; et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, B.; Sun, T.; Lin, L.; Hu, Y.; Deng, M.; Yang, J.; Liu, T.; Li, J.; Sun, S.; et al. Specific growth inhibition of ErbB2expressing human breast cancer cells by genetically modified NK92 cells. Oncol. Rep. 2015, 33, 95–102. [Google Scholar] [PubMed]
- Seidel, D.; Shibina, A.; Siebert, N.; Wels, W.S.; Reynolds, C.P.; Huebener, N.; Lode, H.N. Disialoganglioside-specific human natural killer cells are effective against drug-resistant neuroblastoma. Cancer Immunol. Immunother. 2015, 64, 621–634. [Google Scholar] [CrossRef]
- Esser, R.; Müller, T.; Stefes, D.; Kloess, S.; Seidel, D.; Gillies, S.D.; Aperlo-Iffland, C.; Huston, J.S.; Uherek, C.; Schönfeld, K.; et al. NK cells engineered to express a GD2-specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J. Cell. Mol. Med. 2012, 16, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Binyamin, L.; Alpaugh, R.K.; Hughes, T.L.; Lutz, C.T.; Campbell, K.S.; Weiner, L.M. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J. Immunol. 2008, 180, 6392–6401. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192. [Google Scholar] [CrossRef]
- Martín-Antonio, B.; Suñe, G.; Perez-Amill, L.; Castella, M.; Urbano-Ispizua, A. Natural Killer Cells: Angels and Devils for Immunotherapy. Int J. Mol. Sci. 2017, 18, 1868. [Google Scholar] [CrossRef]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083. [Google Scholar]
- Müller, T.; Uherek, C.; Maki, G.; Chow, K.U.; Schimpf, A.; Klingemann, H.; Tonn, T.; Wels, W.S. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol. Immunother. 2008, 57, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Tavri, S.; Jha, P.; Meier, R.; Henning, T.D.; Muller, T.; Hostetter, D.; Knopp, C.; Johansson, M.; Reinhart, V.; Boddington, S.; et al. Optical imaging of cellular immunotherapy against prostate cancer. Mol. Imaging 2009, 8, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Meier, R.; Golovko, D.; Tavri, S.; Henning, T.D.; Knopp, C.; Piontek, G.; Rudelius, M.; Heinrich, P.; Wels, W.S.; Daldrup-Link, H. Depicting adoptive immunotherapy for prostate cancer in an animal model with magnetic resonance imaging. Magn. Reson. Med. 2011, 65, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, R.; Zhu, X.; Wang, L.; Ma, J.; Han, H.; Wang, X.; Zhang, G.; He, W.; Wang, W.; et al. Retargeting NK-92 for anti-melanoma activity by a TCR-like single-domain antibody. Immunol. Cell Biol. 2013, 91, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Deng, Y.; Benson, D.M.; He, S.; Hughes, T.; Zhang, J.; Peng, Y.; Mao, H.; Yi, L.; Ghoshal, K.; et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014, 28, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chu, J.; Keung, C.W.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.H.; Grandi, P.; et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genssler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 2016, 5, e1119354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108, djv375. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Wada, M.; Firor, A.E.; Pinz, K.G.; Jares, A.; Liu, H.; Salman, H.; Golightly, M.; Lan, F.; Jiang, X.; et al. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget 2016, 7, 56219. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Wada, M.; Pinz, K.G.; Liu, H.; Lin, K.W.; Jares, A.; Firor, A.E.; Shuai, X.; Salman, H.; Golightly, M.; et al. Preclinical targeting of aggressive T-cell malignancies using anti-CD5 chimeric antigen receptor. Leukemia 2017, 31, 2151–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domogala, A.; Madrigal, J.A.; Saudemont, A. Natural Killer Cell Immunotherapy: From Bench to Bedside. Front. Immunol. 2015, 6, 264. [Google Scholar] [CrossRef] [PubMed]
- Glienke, W.; Esser, R.; Priesner, C.; Suerth, J.D.; Schambach, A.; Wels, W.S.; Grez, M.; Kloess, S.; Arseniev, L.; Koehl, U. Advantages and applications of CAR-expressing natural killer cells. Front. Pharmacol. 2015, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Yu, Y.P.; Zuo, Z.H.; Nelson, J.B.; Michalopoulos, G.K.; Monga, S.; Liu, S.; Tseng, G.; Luo, J.H. Targeting genomic rearrangements in tumor cells through Cas9-mediated insertion of a suicide gene. Nat. Biotechnol. 2017, 35, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Cell Line | Year | Disease Diagnosis | Patient | Doubling Time | Viral Status | Cytokine | Primary Reference |
|---|---|---|---|---|---|---|---|
| NK3.3 | 1982 | NR | NR | NR | EBV− | IL-2-dependent | [23] |
| YT | 1983 | Acute lymphoblastic lymphoma (with thymoma) | 15-year-old male | 40–50 h | EBV+ | Independent of IL-2 | [27] |
| NKL | 1996 | NK-LGLL | 63-year-old male | 24–48 h | NR | IL-2-dependent | [30,41] |
| HANK1 | 1998 | Nasal-like NK/T-cell lymphoma | 46-year-old female | 3 day | EBV+ | IL-2-dependent | [31] |
| NK-YS | 1996 | NK cell lymphoma, Nasal angiocentric, Leukemic state with systemic skin infiltration | 19-year-old female | 48 h | EBV+ | IL-2-dependent | [32] |
| KHYG-1 | 1997 | Aggressive NK leukemia | 45-year-old female | 24–48 h | EBV− | IL-2-dependent | [33] |
| SNK-6 | 1998 | Nasal NK/T-cell lymphoma | 62-year-old male | NR | EBV+ | IL-2-dependent | [35] |
| SNT-8 | 1998 | Nasal NK/T-cell lymphoma | 48-year-old female | NR | EBV+ | IL-2-dependent | [35] |
| IMC-1 | 2004 | Aggressive NK cell leukemia | 42-year-old male | 24–36 h | EBV− | IL-2-dependent | [36] |
| NK-92 | 1992 | LGL-NHL | 50-year-old male | 24 h | EBV− | IL-2-dependent; Growth stimulation:IL-7 | [37] |
| Cancer Type | Antigen Targeted | Hinge | TM | Intracellular Signal Domain | Genetic Modification Method | Effector Cell | Year | References |
|---|---|---|---|---|---|---|---|---|
| Multiple myeloma | CD138 | CD8 | CD3ζ | CD3ζ | lentiviral vector | NK-92MI | 2014 | [86] |
| B-cell malignancies | CD19 | CD8 | NR | CD3ζ | Retrovirus | NK-92 | 2016 | [82] |
| B-cell malignancies | CD19 | CD8 | CD28 | CD3ζ | Lentiviral | NK-92 | 2017 | [83] |
| CLL | CD19 | CD8 | CD3ζ | CD3ζ | Electroporation | NK-92 | 2009 | [84] |
| ALL CLL | CD19 CD20 | NR | NR | CD3ζ | Lentivirus | NK-92 | 2014 | [85] |
| B-cell malignancies | CD20 | CD8 | CD3ζ | CD3ζ | Retroviral | NK-92 | 2008 | [101] |
| Prostate cancer | EpCAM | CD8 | CD3ζ | CD3ζ | Retrovirus | NK-92 | 2009 | [102] |
| Prostate cancer | EpCAM | CD8 | CD3ζ | CD3ζ | Retrovirus | NK-92 | 2011 | [103] |
| Neuroblastoma | GD2 | CD8 | CD3ζ | CD3ζ | Retrovirus | NK-92 | 2012 | [96] |
| Neuroblastoma | GD2 | CD8 | CD3ζ | CD3ζ | Retrovirus | NK-92 | 2015 | [95] |
| Melanoma | GPA7 | NR | HLA-A2 | CD3ζ | Electroporation | NK-92MI | 2013 | [104] |
| Brain metastasis | HER2 | CD8α | CD3ζ | CD3ζ | Retrovirus | NK-92 | 2016 | [88] |
| Brain metastasis | HER2 | CD8 | CD3ζ | CD3ζ | Retrovirus | NK-92 | 2013 | [89] |
| Breast cancer | HER2 | CD8 | CD3ζ | CD3ζ | Retrovirus | NK-92 | 2005 | [91] |
| Breast cancer | HER2 | CD8 | CD3ζ | CD3ζ | Retrovirus | NK-92 | 2008 | [92] |
| Breast/ovarian cancer | HER2 | CD8 | CD3ζ | CD3ζ | Retrovirus | NK-92 | 2002 | [81] |
| Breast cancer, Ovarian cancer, Melanoma Renal cell carcinoma | HER2 | CD8 | CD3ζ | CD3ζ | Lentiviral | NK-92 | 2015 | [93] |
| Ovarian cancer Mesothelin-expressing tumors | Mesothelin | CD8 | NKG2D | CD3ζ | Transposon plasmids | NK-92 | 2018 | [98] |
| Cancer Type | Antigen Targeted | Hinge | TM | Intracellular Signal Domain | Genetic Modification Method | Effector Cell | Year | References |
|---|---|---|---|---|---|---|---|---|
| B-cell malignancies | CD19 | CD8 | CD28 | CD28-CD3ζ CD137-CD3ζ | Lentiviral | NK-92 | 2017 | [83] |
| Multiple myeloma | CS1 | NR | NR | CD28-CD3ζ | Lentivirus | NK-92 | 2014 | [105] |
| EBV+ cells | EBNA3C | NR | NR | CD137-CD3ζ | Retrovirus | NK-92MI | 2012 | [75] |
| Glioblastoma | EGFR EGFRvIII | NR | CD28 | CD28-CD3ζ | Lentivirus | NK-92 and NKL | 2015 | [106] |
| Brain metastasis | EGFR | NR | NR | CD28-CD3ζ | Lentivirus | NK-92 | 2016 | [87] |
| Glioblastoma | EGFR EGFRvIII | CD8 | CD28 | CD28-CD3ζ | Lentivirus | NK-92 | 2015 | [107] |
| Breast cancer | EpCAM | CD8 | CD28 | CD28-CD3ζ | Lentivirus | NK-92 | 2012 | [90] |
| Breast cancer Renal cell carcinoma Ovarian carcinoma Melanoma | HER2 | CD8 | CD28 CD137 | CD28-CD3ζ CD137-CD3ζ | Lentiviral | NK-92 | 2015 | [93] |
| Glioblastoma | HER2 | CD8 | CD28 | CD28-CD3ζ | Lentiviral | NK-92 | 2016 | [108] |
| Breast cancer | HER2 | CD8 | CD28 | CD28-CD3ζ | Electroporation | NK-92 | 2015 | [94] |
| Ovarian cancer Mesothelin-expressing tumors | Mesothelin | CD8 | CD16 | 2B4-CD3ζ | Transposon plasmids | NK-92 | 2018 | [98] |
| Ovarian cancer mesothelin-expressing tumors | Mesothelin | CD8 | NKp44 | DAP10-CD3ζ 2B4-CD3ζ | Transposon plasmids | NK-92 | 2018 | [98] |
| Ovarian cancer Mesothelin-expressing tumors | Mesothelin | CD8 | NKG2D | 2B4-CD3ζ CD137-CD3ζ | Transposon plasmids | NK-92 | 2018 | [98] |
| Ovarian cancer Mesothelin-expressing tumors | Mesothelin | CD8 | CD28 | CD28-CD137-CD3ζ | Transposon plasmids | NK-92 | 2018 | [98] |
| Ovarian cancer Mesothelin-expressing tumors | Mesothelin | CD8 | NKG2D | 2B4-DAP12-CD3ζ 2B4-DAP10-CD3ζ CD137-2B4-CD3ζ | Transposon plasmids | NK-92 | 2018 | [98] |
| Aggressive T cell malignancies | CD3 | CD8 | CD8 | CD28-CD137-CD3ζ | Lentivirus | NK-92 | 2016 | [109] |
| Aggressive T-cell malignancies | CD5 | CD8 | CD8 | CD28-CD137-CD3ζ | Lentivirus | NK-92 | 2017 | [110] |
| NCT Number | NK Cell Source | Target Antigen | Disease | Phase | Estimated Enrollment | Age | Location | References |
|---|---|---|---|---|---|---|---|---|
| NCT02742727 | NK-92 | CD7 | Acute Myeloid Leukemia;Precursor T-Cell Lymphoblastic Leukemia-Lymphoma; T-cell Prolymphocytic Leukemia; T-cell Large Granular Lymphocytic Leukemia; Peripheral T-cell Lymphoma, NOS; Angioimmunoblastic T-cell Lymphoma Extranodal NK/T-cell Lymphoma, Nasal Type; Enteropathy-type Intestinal T-cell Lymphoma; Hepatosplenic T-cell Lymphoma | Phase 1 Phase 2 | 10 participants | 18 Years and older (Adult, Older Adult) | China | NR |
| NCT02892695 | NK-92 | CD19 | Acute Lymphocytic Leukemia; Chronic Lymphocytic Leukemia; Follicular Lymphoma; Mantle Cell Lymphoma; B-cell Prolymphocytic Leukemia; Diffuse Large Cell Lymphoma; | Phase 1 Phase 2 | 10 participants | 3 Years to 80 Years (Child, Adult, Older Adult) | China | NR |
| NCT02944162 | NK-92 | CD33 | Acute Myelogenous Leukemia; Acute Myeloid Leukemia; Acute Myeloid Leukemia with Maturation; Acute Myeloid Leukemia Without Maturation; ANLL | Phase 1 Phase 2 | 10 participants | 3 Years to 80 Years (Child, Adult, Older Adult) | China | [100] |
| NCT03383978 | NK-92 | HER2 | Glioblastoma | Phase 1 | 30 participants | 18 Years and older (Adult, Older Adult) | Germany | NR |
| NCT03656705 | NK-92 | NR | Non-small Cell Lung Cancer | Phase 1 | 5 participants | 18 Years to 75 Years (Adult, Older Adult) | China | NR |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Zheng, H.; Diao, Y. Natural Killer Cells and Current Applications of Chimeric Antigen Receptor-Modified NK-92 Cells in Tumor Immunotherapy. Int. J. Mol. Sci. 2019, 20, 317. https://doi.org/10.3390/ijms20020317
Zhang J, Zheng H, Diao Y. Natural Killer Cells and Current Applications of Chimeric Antigen Receptor-Modified NK-92 Cells in Tumor Immunotherapy. International Journal of Molecular Sciences. 2019; 20(2):317. https://doi.org/10.3390/ijms20020317
Chicago/Turabian StyleZhang, Jianguang, Huifang Zheng, and Yong Diao. 2019. "Natural Killer Cells and Current Applications of Chimeric Antigen Receptor-Modified NK-92 Cells in Tumor Immunotherapy" International Journal of Molecular Sciences 20, no. 2: 317. https://doi.org/10.3390/ijms20020317
APA StyleZhang, J., Zheng, H., & Diao, Y. (2019). Natural Killer Cells and Current Applications of Chimeric Antigen Receptor-Modified NK-92 Cells in Tumor Immunotherapy. International Journal of Molecular Sciences, 20(2), 317. https://doi.org/10.3390/ijms20020317