Bone Metastasis Pain, from the Bench to the Bedside




{kind=link}
{kind=link}
{kind=link}
Abstract
1. The Healthy Bone Tissue
The Virtuous Cycle in the Physiology of Bone
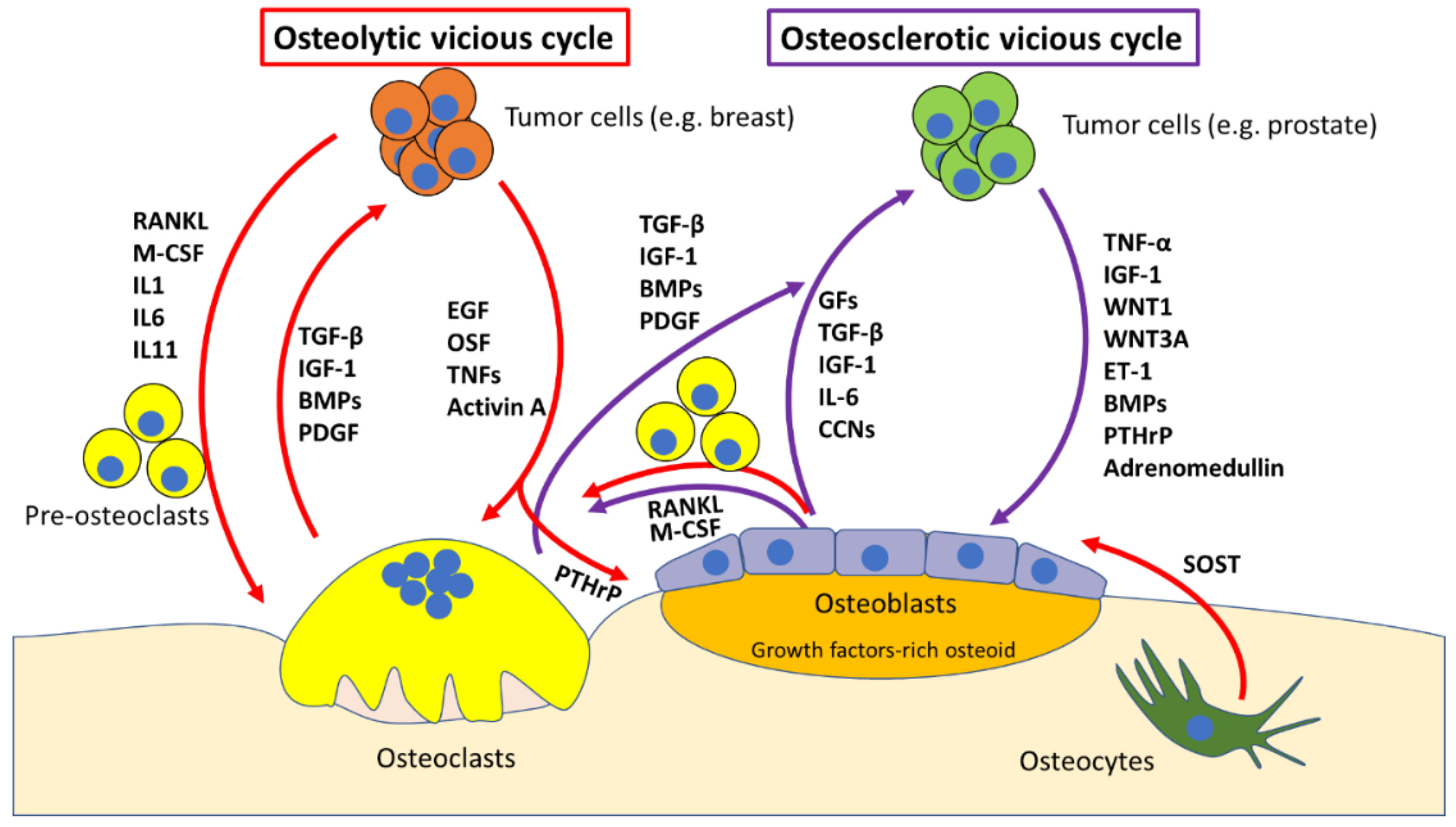
2. Molecular Mechanisms of Bone Metastases
The “Vicious Cycle”
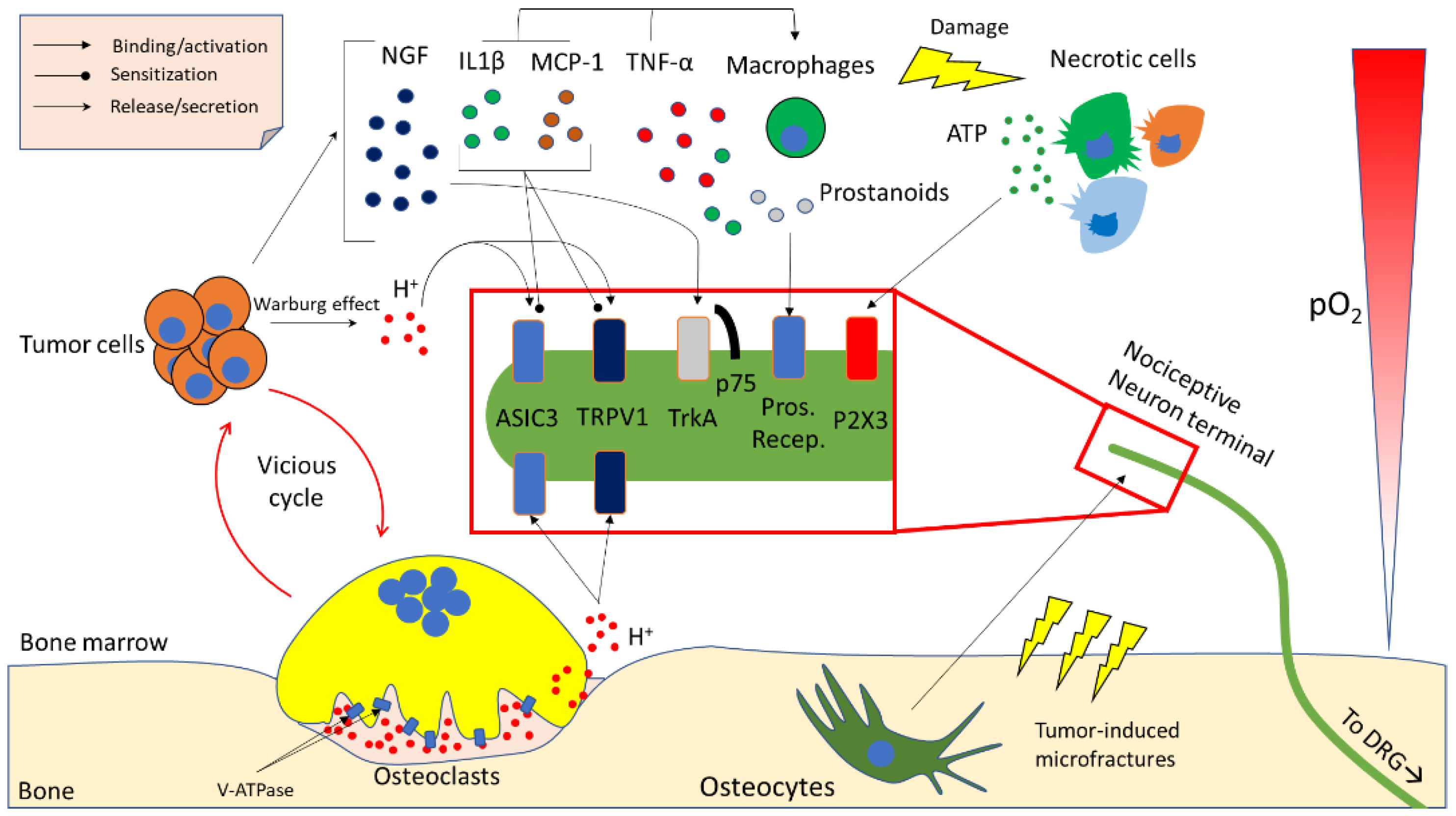
3. Molecular Determinants of Cancer-Induced Bone Pain (CIBP)
3.1. Acidity
3.2. Neurotrophins
3.3. Inflammatory Cytokines and Chemokines
3.4. Other Microenvironment- and Tumour-Derived Factors
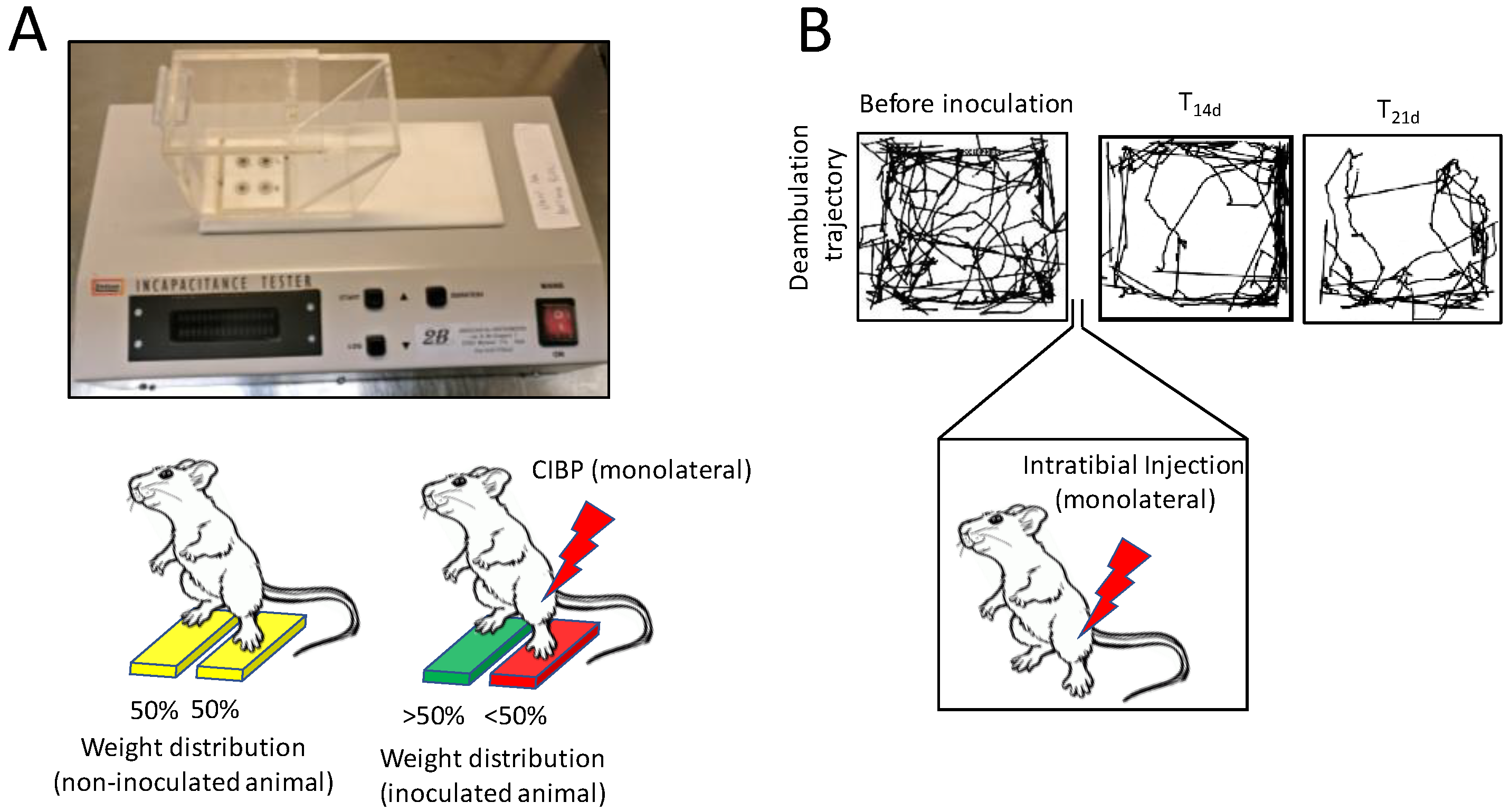
4. In Vivo Models of Bone Pain
5. Current Treatments for CIBP
5.1. Surgical Intervention
5.2. Radiotherapy
5.3. Drug Treatment
5.3.1. Nonsteroidal Anti-Inflammatory Drugs
5.3.2. Opioid Treatment
5.3.3. Anti-Resorptive Agents
5.3.4. Endothelin-A (ET-A) Receptor Antagonists: a New Avenue
Author Contributions
Funding
Conflicts of Interest
References
- Rucci, N.; Teti, A. The “love-hate” relationship between osteoclasts and bone matrix. Matrix Biol. 2016, 54, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Cappariello, A.; Ponzetti, M.; Rucci, N. The “soft” side of the bone: Unveiling its endocrine functions. Horm. Biol. Clin. Investig. 2016, 28, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Jähn, K.; Kelkar, S.; Zhao, H.; Xie, Y.; Tiede-Lewis, L.M.; Dusevich, V.; Dallas, S.L.; Bonewald, L.F. Osteocytes acidify their microenvironment in response to PTHrP in vitro and in lactating mice in vivo. J. Bone Miner. Res. 2017, 32, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Uda, Y.; Azab, E.; Sun, N.; Shi, C.; Pajevic, P.D. Osteocyte mechanobiology. Curr. Osteoporos. Rep. 2017, 15, 318–325. [Google Scholar] [CrossRef]
- Gong, Y.; Slee, R.B.; Fukai, N.; Rawadi, G.; Roman-Roman, S.; Reginato, A.M.; Wang, H.; Cundy, T.; Glorieux, F.H.; Lev, D.; et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001, 107, 513–523. [Google Scholar] [CrossRef]
- Boyden, L.M.; Mao, J.; Belsky, J.; Mitzner, L.; Farhi, A.; Mitnick, M.A.; Wu, D.; Insogna, K.; Lifton, R.P. High bone density due to a mutation in LDL-receptor-related protein 5. N. Engl. J. Med. 2002, 346, 1513–1521. [Google Scholar] [CrossRef]
- Little, R.D.; Carulli, J.P.; Del Mastro, R.G.; Dupuis, J.; Osborne, M.; Folz, C.; Manning, S.P.; Swain, P.M.; Zhao, S.C.; Eustace, B.; et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am. J. Hum. Genet. 2002, 70, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin signaling, disease, and emerging therapeutic modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Zhang, R.; Geoffry, V.; Ridall, A.I.; Karsenty, G. Osf/2Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef]
- Lee, B.; Thirunavukkarasu, K.; Zhou, L.; Pastore, L.; Baldini, A.; Hecht, J.; Geoffroy, V.; Ducy, P.; Karsenty, G. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat. Genet. 1997, 16, 307–310. [Google Scholar] [CrossRef]
- Paduano, F.; Marrelli, M.; Palmieri, F.; Tatullo, M. CD146 expression influences periapical cyst mesenchymal stem cell properties. Stem Cell Rev. 2016, 12, 592–603. [Google Scholar] [CrossRef]
- Aulino, P.; Costa, A.; Chiaravalloti, E.; Perniconi, B.; Adamo, S.; Coletti, D.; Marrelli, M.; Tatullo, M.; Teodori, L. Muscle extracellular matrix scaffold is a multipotent environment. Int. J. Med. Sci. 2015, 12, 336–340. [Google Scholar] [CrossRef]
- Anderson, H.C. Matrix vesicles and calcification. Curr. Rheumatol. Rep. 2003, 5, 222–226. [Google Scholar] [CrossRef]
- Brown, H.K.; Schiavone, K.; Gouin, F.; Heymann, M.F.; Heymann, D. Biology of bone sarcomas and new therapeutic developments. Calcif. Tissue Int. 2018, 102, 174–195. [Google Scholar] [CrossRef]
- Ottewell, P.D. The role of osteoblasts in bone metastasis. J. Bone Oncol. 2016, 5, 124–127. [Google Scholar] [CrossRef]
- Feng, X.; Teitelbaum, S. Osteoclasts: New insights. Bone Res. 2013, 1, 11–26. [Google Scholar] [CrossRef]
- Tondravi, M.M.; McKercher, S.R.; Anderson, K.; Erdmann, J.M.; Quiroz, M.; Maki, R.; Teitelbaum, S.L. Osteopetrosis in mice lacking hematopoietic transcription factor PU.1. Nature 1997, 386, 81–84. [Google Scholar] [CrossRef]
- Kawaguchi, N.; Noda, M. Mitf is expressed in osteoclast progenitors in vitro. Exp. Cell Res. 2000, 260, 284–291. [Google Scholar] [CrossRef]
- Biskobing, D.M.; Fan, X.; Rubin, J. Characterization of MCS-induced proliferation and subsequent osteoclast formation in murine marrow culture. J. Bone Miner. Res. 1995, 10, 1025–1032. [Google Scholar] [CrossRef]
- Arai, F.; Myamoto, T.; Ohneda, O.; Inada, T.; Sudo, T.; Brasel, K.; Miyata, T.; Anderson, D.M.; Susa, T. Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor kappaB (RANK) receptors. J. Exp. Med. 1999, 190, 1741–1754. [Google Scholar] [CrossRef] [PubMed]
- Lacey, D.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef]
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef]
- Franzoso, G.; Carlson, L.; Xing, L.; Poljak, L.; Shores, E.W.; Brown, K.D.; Leonardi, A.; Tran, T.; Boyce, B.F.; Siebenlist, U. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997, 11, 3482–3496. [Google Scholar] [CrossRef]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.R.; Besser, D.; Kim, N.; Arron, J.R.; Vologodskaia, M.; Hanafusa, H.; Choy, Y. TRANCE, a TNF family member, activates Akt/PKB through a signalling complex involving TRAF6 and c-Src. Mol. Cell 1999, 4, 1041–1049. [Google Scholar] [CrossRef]
- Kudo, O.; Sabokbar, A.; Pocock, A.; Itonaga, I.; Fujikawa, Y.; Athanasou, N.A. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone 2003, 32, 1–7. [Google Scholar] [CrossRef]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 2000, 106, 1481–1488. [Google Scholar] [CrossRef]
- Kim, J.H.; Jin, H.M.; Kim, K.; Song, I.; Youn, B.U.; Matsuo, K.; Kim, N. The mechanism of osteoclast differentiation induced by IL-1. J. Immunol. 2009, 183, 1862–1870. [Google Scholar] [CrossRef]
- Cappariello, A.; Maurizi, A.; Veeriah, V.; Teti, A. The great beauty of the osteoclast. Arch. Biochem. Biophys. 2014, 558, 70–78. [Google Scholar] [CrossRef]
- Harada, S.I.; Rodan, G.A. Control of osteoblast function and regulation of bone mass. Nature 2003, 423, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Woodward, E.; Jagdev, S.; McPartland, I.; Clark, K.; Gregory, W.; Newsham, A.; Rogerson, S.; Hayward, K.; Selby, P.; Brown, J. Skeletal complications and survival in renal cancer patients with bone metastases. Bone 2011, 48, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin. Cancer Res. 2006, 12, 6243s–6249s. [Google Scholar] [CrossRef] [PubMed]
- Van Moos, R.; Body, J.J.; Egerdie, B.; Stopeck, A.; Brown, J.; Fallowfield, I.; Patrick, D.L.; Cleeland, C.; Damyanov, D.; Palazzo, F.S.; et al. Pain and analgesic use associated with skeletal-related events in patients with advanced cancer and bone metastases. Support. Care Cancer 2016, 24, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Brodowicz, T.; Hadji, P.; Niepel, D.; Diel, I. Early identification and intervention matters: A comprehensive review of current evidence and recommendations for the monitoring of bone health in patients with cancer. Cancer Treat. Rev. 2017, 61, 23–34. [Google Scholar] [CrossRef]
- Coleman, R.E. Bone cancer 2011: Prevention and treatment of bone metastases. Nat. Rev. Clin. Oncol. 2011, 9, 76–78. [Google Scholar] [CrossRef]
- Rucci, N.; Teti, A. Osteomimicry: How the seed growth in the soil. Calcif. Tissue Int. 2018, 102, 131–140. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metast. Rev. 1889, 8, 98–101. [Google Scholar] [CrossRef]
- Maurizi, A.; Rucci, N. The osteoclast in bone metastasis: Player and target. Cancers 2018, 10, 218. [Google Scholar] [CrossRef]
- Ibrahim, T.; Flamini, E.; Mercatali, L.; Sacanna, E.; Serra, P.; Amadori, D. Pathogenesis of osteoblastic bone metastases from prostate cancer. Cancer 2010, 116, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Guise, T.A.; Mohammad, K.S.; Clines, G.; Stebbins, E.G.; Wong, D.H.; Higgins, L.S.; Vessella, R.; Corey, E.; Padalecki, S.; Suva, L.; et al. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin. Cancer Res. 2006, 12 (Suppl. 20), 6213s–6246s. [Google Scholar] [CrossRef]
- Keller, E.T.; Brown, J. Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J. Cell Biochem. 2004, 91, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Roudier, M.P.; Morrissey, C.; True, L.D.; Higano, C.S.; Vessella, R.L.; Ott, S.M. Histopathologic assessment of prostate cancer bone “osteoblastic” metastases. J. Urol. 2008, 180, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Carducci, M.; Smith, M.; Damião, R.; Brown, J.; Karsh, L.; Milecki, P.; Shore, N.; Rader, M.; Wang, H.; et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: A randomised, double-blind study. Lancet 2011, 377, 813–822. [Google Scholar] [CrossRef]
- Saad, F.; Gleason, D.M.; Murray, R.; Tchekmedyian, S.; Venner, P.; Lacombe, L.; Chin, J.L.; Vinholes, J.J.; Goas, J.A.; Zheng, M.; et al. Long-term efficacy of zoledronic acid for the prevention of skeletal complications in patients with metastatic hormone-refractory prostate cancer. J. Natl. Cancer Inst. 2004, 96, 879–882. [Google Scholar] [CrossRef]
- Deftosa, L.J.; Barkend, I.; Burtona, D.W.; Hoffmanb, R.M.; Gellere, J. Direct evidence that PTHrP expression promotes prostate cancer progression in bone. Biochem. Biophys. Res. Commun. 2005, 327, 468–472. [Google Scholar] [CrossRef]
- Iwamura, M.; di Santagnese, P.A.; Wu, G.; Benning, C.M.; Cockett, A.T.; Deftosa, L.J.; Abrahamsson, P.A. Immunohistochemical localization of parathyroid hormone-related protein in human prostate cancer. Cancer Res. 2001, 61, 2572–2578. [Google Scholar]
- Sohail, A.; Sherin, L.; Butt, S.I.; Javed, S.; Li, Z.; Iqbal, S.; Be’g, O.A. Role of key players in paradigm shifts of prostate cancer bone metastasis. Cancer Manag. Res. 2018, 10, 1619–1626. [Google Scholar] [CrossRef]
- Nandana, S.; Tripathi, M.; Duan, P.; Chu, C.Y.; Mishra, R.; Liu, C.; Jin, R.; Yamashita, H.; Zayzafoon, M.; Bhowmick, N.A.; et al. Bone metastasis of prostate cancer can be therapeutically targeted at the TBX2–WNT signaling axis. Cancer Res. 2017, 77, 1331–1334. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.J.; Mohammad, K.S.; Käkönen, S.M.; Harris, S.; Wu-Wong, J.R.; Wessale, J.L.; Padley, R.J.; Garrett, I.R.; Chirgwin, J.M.; Guise, T.A. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc. Natl. Acad. Sci. USA 2003, 100, 10954–10959. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.M.; Shay, G.; Aruajo, A.; Lynch, C.C. Integrating new discoveries into the “vicious cycle” paradigm of prostate to bone metastases. Cancer Metast. Rev. 2014, 33, 511–525. [Google Scholar] [CrossRef]
- Hernandez, R.K.; Wade, S.W.; Reich, A.; Pirolli, M.; Liede, A.; Lyman, G.H. Incidence of bone metastases in patients with solid tumors: Analysis of oncology electronic medical records in the United States. BMC Cancer 2018, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- Guise, T.A.; Yin, J.J.; Taylor, S.D.; Kumagai, Y.; Dallas, M.; Boyce, B.F.; Yoneda, T.; Mundy, G.R. Evidence for a causal role of parathyroid hormone–related protein in the pathogenesis of human breast cancer–mediated osteolysis. J. Clin. Investig. 1996, 98, 1544–1549. [Google Scholar] [CrossRef] [PubMed]
- Wein, M.N. Parathyroid hormone signaling in osteocytes. JBMR Plus 2018, 2, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Rana, T.; Chakrabarti, A.; Freeman, M.; Biswasbirra, S. Doxorubicin-mediated bone loss in breast cancer bone metastases is driven by an interplay between oxidative stress and induction of TGFβ. PLoS ONE 2013, 8, e78043. [Google Scholar] [CrossRef]
- Tatullo, M.; Simone, G.M.; Tarullo, F.; Irlandese, G.; De Vito, D.; Marrelli, M.; Santacroce, L.; Cocco, T.; Ballini, A.; Scacco, S. Antioxidant and Antitumor Activity of a Bioactive Polyphenolic Fraction Isolated from the Brewing Process. Sci. Rep. 2016, 6, 36042. [Google Scholar] [CrossRef]
- Van den Beuken-van Everdingen, M.H.; Hochstenbach, L.M.; Joosten, E.A.; Tjan-Heijnen, V.C.; Janssen, D.J. Update on prevalence of pain in patients with cancer: Systematic review and meta-analysis. J. Pain Symptom Manag. 2016, 51, 1070–1090. [Google Scholar] [CrossRef]
- Middlemiss, T.; Laird, B.J.A.; Fallon, M.T. Mechanisms of cancer-induced bone pain. Clin. Oncol. 2011, 23, 387–392. [Google Scholar] [CrossRef]
- Lozano-Ondoua, A.N.; Symons-Liguori, A.M.; Vanderah, T.W. Cancer-induced bone pain: Mechanisms and models. Neurosci. Lett. 2013, 557, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Eber, M.R.; Widner, D.B.; Shiozawa, Y. Role of the bone microenvironment in the development of painful complications of skeletal metastases. Cancers 2018, 10, 141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Levy, D. The sensory innervation of the calvarial periosteum is nociceptive and contributes to headache-like behavior. Pain 2014, 155, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Mahns, D.A.; Ivanusic, J.J.; Sahai, V.; Rowe, M.J. An intact peripheral nerve preparation for monitoring the activity of single, periosteal afferent nerve fibres. J. Neurosci. Methods 2006, 156, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Ivanusic, J. The size, neurochemistry and segmental distribution of sensory neurons innervating the rat tibia. J. Comp. Neurol. 2009, 517, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Tanaka, S.; Sekiguchi, T.; Sugiyama, D.; Kawamata, M. Spinal nociceptive transmission by mechanical stimulation of bone marrow. Mol. Pain 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Fock, S.; Mense, S. Excitatory effects of 5-hydroxytryptamine, histamine and potassium ions on muscular group IV afferent units: A comparison with bradykinin. Brain Res. 1976, 105, 459–469. [Google Scholar] [CrossRef]
- Inman, V.; Saunders, J. Referred pain from skeletal structures. J. Nerv. Ment. Dis. 1944, 99, 660–667. [Google Scholar] [CrossRef]
- Brjussowa, S.S.; Lebedenko, W.W. Zur Schmerzleitungsfähigkeit der Gefäße. Z. Gesamte Exp. Med. 1930, 69, 29–40. [Google Scholar] [CrossRef]
- Lemperg, R.K.; Arnoldi, C.C. The significance of intraosseous pressure in normal and diseased states with special reference to the intraosseous engorgement-pain syndrome. Clin. Orthop. Relat. Res. 1978, 136, 143–156. [Google Scholar]
- Arnoldi, C.C.; Djurhuus, J.C.; Heerfordt, J.; Karle, A. Intraosseous phlebography, intraosseous pressure measurements and 99mTC-polyphosphate scintigraphy in patients with various painful conditions in the hip and knee. Acta Orthop. Scand. 1980, 51, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Sottnik, J.L.; Dai, J.; Zhang, H.; Campbell, B.; Keller, E.T. Tumor-induced pressure in the bone microenvironment causes osteocytes to promote the growth of prostate cancer bone metastases. Cancer Res. 2015, 75, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Teti, A.; Zallone, A. Do osteocytes contribute to bone mineral homeostasis? Osteocytic osteolysis revisited. Bone 2009, 44, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, P.; Jonsson, J.I. The hematopoietic stem cell niche: Low in oxygen but a nice place to be. J. Cell. Physiol. 2010, 222, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, J.R.; Röhrich, H.; Lindsay, T.H.; Sevcik, M.A.; Schwei, M.J.; Kubota, K.; Halvorson, K.G.; Poblete, J.; Chaplan, S.R.; Dubin, A.E.; et al. Selective blockade of the capsaicin receptor TRPV1 attenuates bone cancer pain. J. Neurosci. 2005, 25, 3126–3131. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, H.; Wakisaka, S.; Hiraga, T.; Sakurai, T.; Tominaga, M.; Yoneda, T. Role of acid-sensing TRPV1 in bone pain associated with cancer colonization in bone. J. Bone Miner. Res. 2005, 20 (Suppl. 1), S32. [Google Scholar]
- Nagae, M.; Hiraga, T.; Yoneda, T. Acidic microenvironment created by osteoclasts causes bone pain associated with tumor colonization. J. Bone Miner. Metab. 2007, 25, 99–104. [Google Scholar] [CrossRef]
- Pan, H.L.; Zhang, Y.Q.; Zhao, Z.Q. Involvement of lysophosphatidic acid in bone cancer pain by potentiation of TRPV1 via PKCepsilon pathway in dorsal root ganglion neurons. Mol. Pain 2010, 6, 85. [Google Scholar] [CrossRef]
- Lautner, M.A.; Ruparel, S.B.; Patil, M.J.; Hargreaves, K.M. In vitro sarcoma cells release a lipophilic substance that activates the pain transduction system via TRPV1. Ann. Surg. Oncol. 2011, 18, 866–871. [Google Scholar] [CrossRef]
- Qiu, F.; Wei, X.L.; Zhang, S.Z.; Yuan, W.X.; Mi, W.D. Increased expression of acid sensing ion channel 3 within dorsal root ganglia in a rat model of bone cancer pain. Neuroreport 2014, 25, 887–893. [Google Scholar] [CrossRef]
- Caterina, M.J.; Leffler, A.; Malmberg, A.B.; Martin, W.J.; Trafton, J.; Petersen-Zeitz, K.R.; Koltzenburg, M.; Basbaum, A.I.; Julius, D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 2000, 288, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Hata, K.; Nagayama, T.; Sakurai, T.; Nishisho, T.; Wakabayashi, H.; Hiraga, T.; Ebisu, S.; Yoneda, T. Acid activation of Trpv1 leads to an up-regulation of calcitonin gene-related peptide expression in dorsal root ganglion neurons via the CaMK-CREB cascade: A potential mechanism of inflammatory pain. Mol. Biol. Cell 2010, 21, 2568–2577. [Google Scholar] [CrossRef] [PubMed]
- Niiyama, Y.; Kawamata, T.; Yamamoto, J.; Furuse, S.; Namiki, A. SB366791, a TRPV1 antagonist, potentiates analgesic effects of systemic morphine in a murine model of bone cancer pain. Br. J. Anaesth. 2009, 102, 251–258. [Google Scholar] [CrossRef]
- Deval, E.; Gasull, X.; Noël, J.; Salinas, M.; Baron, A.; Diochot, S.; Lingueglia, E. Acid-sensing ion channels (ASICs): Pharmacology and implication in pain. Pharmacol. Ther. 2010, 128, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-G.; Xu, T.-L. ASIC3 channels in multimodal sensory perception. ACS Chem. Neurosci. 2011, 2, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Hiasa, M.; Okui, T.; Allette, Y.M.; Ripsch, M.S.; Sun-Wada, G.H.; Wakabayashi, H.; Roodman, G.D.; White, F.A.; Yoneda, T. Bone pain induced by multiple myeloma is reduced by targeting V.-ATPase and ASIC3. Cancer Res. 2017, 77, 1283–1295. [Google Scholar] [CrossRef]
- Izumi, M.; Ikeuchi, M.; Ji, Q.; Tani, T. Local ASIC3 modulates pain and disease progression in a rat model of osteoarthritis. J. Biomed. Sci. 2012, 19, 77. [Google Scholar] [CrossRef]
- Yoneda, T.; Hiasa, M.; Nagata, Y.; Okui, T.; White, F.A. Acidic microenvironment and bone pain in cancer-colonized bone. Bonekey Rep. 2015, 4, 690. [Google Scholar] [CrossRef]
- Di Pompo, G.; Lemma, S.; Canti, L.; Rucci, N.; Ponzetti, M.; Errani, C.; Donati, D.M.; Russell, S.; Gillies, R.; Chano, T.; et al. Intratumoral acidosis fosters cancer-induced bone pain through the activation of the mesenchymal tumor-associated stroma in bone metastasis from breast carcinoma. Oncotarget 2017, 8, 54478–54496. [Google Scholar] [CrossRef]
- Skaper, S.D. The neurotrophin family of neurotrophic factors: An overview. Methods Mol. Biol. 2012, 846, 1–12. [Google Scholar] [CrossRef]
- Halvorson, K.G.; Kubota, K.; Sevcik, M.A.; Lindsay, T.H.; Sotillo, J.E.; Ghilardi, J.R.; Rosol, T.J.; Boustany, L.; Shelton, D.L.; Mantyh, P.W. A blocking antibody to nerve growth factor attenuates skeletal pain induced by prostate tumor cells growing in bone. Cancer Res. 2005, 65, 9426–9435. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Andrade, J.M.; Ghilardi, J.R.; Castañeda-Corral, G.; Kuskowski, M.A.; Mantyh, P.W. Preventive or late administration of anti-NGF therapy attenuates tumor-induced nerve sprouting, neuroma formation, and cancer pain. Pain 2011, 152, 2564–2574. [Google Scholar] [CrossRef] [PubMed]
- Buehlmann, D.; Ielacqua, G.D.; Xandry, J.; Rudin, M. Prospective administration of anti-nerve growth factor treatment effectively suppresses functional connectivity alterations after cancer-induced bone pain in mice. Pain 2019, 160, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.N.; Yang, J.P.; Ji, F.H.; Zhan, Y.; Jin, X.H.; Xu, Q.N.; Wang, X.Y.; Zuo, J.L. Brain-derived neurotrophic factor modulates N-methyl-D-aspartate receptor activation in a rat model of cancer-induced bone pain. J. Neurosci. Res. 2012, 90, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, J.R.; Freeman, K.T.; Jimenez-Andrade, J.M.; Mantyh, W.G.; Bloom, A.P.; Kuskowski, M.A.; Mantyh, P.W. Administration of a tropomyosin receptor kinase inhibitor attenuates sarcoma-induced nerve sprouting, neuroma formation and bone cancer pain. Mol. Pain 2010, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Hondermarck, H. Neurotrophins and their receptors in breast cancer. Cytokine Growth Factor Rev. 2012, 23, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Molloy, N.H.; Read, D.E.; Gorman, A.E. Nerve growth factor in cancer cell death and survival. Cancers 2011, 3, 510–530. [Google Scholar] [CrossRef]
- Williams, K.S.; Killebrew, D.A.; Clary, G.P.; Seawell, J.A.; Meeker, R.B. Differential regulation of macrophage phenotype by mature and pro-nerve growth factor. J. Neuroimmunol. 2015, 285, 76–93. [Google Scholar] [CrossRef]
- Zhang, X.C.; Kainz, V.; Burstein, R.; Levy, D. Tumor necrosis factor-alpha induces sensitization of meningeal nociceptors mediated via local COX and P38 map kinase actions. Pain 2011, 152, 140–149. [Google Scholar] [CrossRef]
- Binshtok, A.M.; Wang, H.; Zimmermann, K.; Amaya, F.; Vardeh, D.; Shi, L.; Brenner, G.J.; Ji, R.R.; Bean, B.P.; Woolf, C.J.; et al. Nociceptors are interleukin-1beta sensors. J. Neurosci. 2008, 28, 14062–14073. [Google Scholar] [CrossRef]
- Mamet, J.; Baron, A.; Lazdunski, M.; Voilley, N. Proinflammatory mediators, stimulators of sensory neuron excitability via the expression of acid-sensing ion channels. J. Neurosci. 2002, 22, 10662–10670. [Google Scholar] [CrossRef] [PubMed]
- Barrios-Rodiles, M.; Chadee, K. Novel regulation of cyclooxygenase-2 expression and prostaglandin e2 production by IFN-gamma in human macrophages. J. Immunol. 1998, 161, 2441–2448. [Google Scholar] [PubMed]
- Sabino, M.A.; Ghilardi, J.R.; Jongen, J.L.; Keyser, C.P.; Luger, N.M.; Mach, D.B.; Peters, C.M.; Rogers, S.D.; Schwei, M.J.; de Felipe, C.; et al. Simultaneous reduction in cancer pain, bone destruction, and tumor growth by selective inhibition of cyclooxygenase-2. Cancer Res. 2002, 62, 7343–7349. [Google Scholar]
- Baamonde, A.; Curto-Reyes, V.; Juarez, L.; Meana, A.; Hidalgo, A.; Menendez, L. Antihyperalgesic effects induced by the IL-1 receptor antagonist anakinra and increased IL-1beta levels in inflamed and osteosarcoma-bearing mice. Life Sci. 2007, 81, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Constantin, C.E.; Mair, N.; Sailer, C.A.; Andratsch, M.; Xu, Z.Z.; Blumer, M.J.; Scherbakov, N.; Davis, J.B.; Bluethmann, H.; Ji, R.R.; et al. Endogenous tumor necrosis factor alpha (TNFalpha) requires TNF receptor type 2 to generate heat hyperalgesia in a mouse cancer model. J. Neurosci. 2008, 28, 5072–5081. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Zheng, X.Y.; Yang, J.P.; Wang, L.N.; Ji, F.H. Involvement of spinal monocyte chemoattractant protein-1 (MCP-1) in cancer-induced bone pain in rats. Neurosci. Lett. 2012, 517, 60–63. [Google Scholar] [CrossRef]
- Giuliani, A.L.; Sarti, A.C.; Di Virgilio, F. Extracellular nucleotides and nucleosides as signalling molecules. Immunol. Lett. 2018. [Google Scholar] [CrossRef]
- Kaan, T.K.; Yip, P.K.; Patel, S.; Davies, M.; Marchand, F.; Cockayne, D.A.; Nunn, P.A.; Dickenson, A.H.; Ford, A.P.; Zhong, Y.; et al. Systemic blockade of P2X3 and P2X2/3 receptors attenuates bone cancer pain behaviour in rats. Brain 2010, 133, 2549–2564. [Google Scholar] [CrossRef]
- Hansen, R.R.; Nasser, A.; Falk, S.; Baldvinsson, S.B.; Ohlsson, P.H.; Bahl, J.M.; Jarvis, M.F.; Ding, M.; Heegaard, A.M. Chronic administration of the selective P2X3, P2X2/3 receptor antagonist, A-317491, transiently attenuates cancer-induced bone pain in mice. Eur. J. Pharmacol. 2012, 688, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Baron, R. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism; Favus, M.J., Ed.; ASBMR: Washington, DC, USA, 2003; pp. 1–8. [Google Scholar]
- Breuksch, I.; Weinert, M.; Brenner, W. The role of extracellular calcium in bone metastasis. J. Bone Oncol. 2016, 5, 143–145. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, X.M.; Duan, K.Z.; Gu, X.Y.; Han, M.; Liu, B.L.; Zhao, Z.Q.; Zhang, Y.Q. Peripheral TGF-β1 signaling is a critical event in bone cancer-induced hyperalgesia in rodents. J. Neurosci. 2013, 33, 19099–19111. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cai, J.; Han, Y.; Xiao, X.; Meng, X.L.; Su, L.; Liu, F.Y.; Xing, G.G.; Wan, Y. Enhanced function of TRPV1 via up-regulation by insulin-like growth factor-1 in a rat model of bone cancer pain. Eur. J. Pain 2014, 18, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Condon, K.W.; Kuhstoss, S.A.; Plotkin, L.I.; Bellido, T.; Roodman, G.D. Genetic deletion of Sost or pharmacological inhibition of sclerostin prevent multiple myeloma-induced bone disease without affecting tumor growth. Leukemia 2017, 31, 2686–2694. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, W.G.; Yu, Y.; Xiao, X.; Cheng, J.; Zeng, W.Z.; Peng, Z.; Xi Zhu, M.; Xu, T.L. Serotonin facilitates peripheral pain sensitivity in a manner that depends on the nonproton ligand sensing domain of ASIC3 channel. J. Neurosci. 2013, 33, 4265–4279. [Google Scholar] [CrossRef] [PubMed]
- Holzer, P. The pharmacological challenge to tame the transient receptor potential vanilloid-1 (TRPV1) nocisensor. Br. J. Pharmacol. 2008, 155, 1145–1162. [Google Scholar] [CrossRef] [PubMed]
- Studer, M.; McNaughton, P.A. Modulation of single-channel properties of TRPV1 by phosphorylation. J. Physiol. 2010, 588, 3743–3756. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, K.K. Glial fibrillary acidic protein: From intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef]
- Jia, R.; Bertaa, T.; Nedergaardb, M. Glia and pain: Is chronic pain a gliopathy? Pain 2013, 154, S10–S28. [Google Scholar] [CrossRef]
- De Ciantis, P.D.; Yashpal, K.; Henry, J.; Singh, G. Characterization of a rat model of metastatic prostate cancer bone pain. J. Pain Res. 2010, 3, 213–221. [Google Scholar] [CrossRef]
- Hald, A.; Hansen, R.R.; Thomsen, M.W.; Ding, M.; Croucher, P.I.; Gallagher, O.; Ebetino, F.H.; Kassem, M.; Heegaard, A.M. Cancer-induced bone loss and associated pain-related behavior is reduced by risedronate but not its phosphonocarboxylate analog NE-10790. Int. J. Cancer 2009, 125, 1177–1185. [Google Scholar] [CrossRef]
- Zhu, X.C.; Ge, C.T.; Wang, P.; Zhang, J.L.; Yu, Y.Y.; Fu, C.Y. Analgesic effects of lappaconitine in leukemia bone pain in a mouse model. Peer J. 2015, 3, e936. [Google Scholar] [CrossRef] [PubMed]
- Majutaa, L.A.; Guedona, J.G.; Mitchella, S.A.T.; Kuskowskib, M.A.; Mantyh, P.W. Mice with cancer-induced bone pain show a marked decline in day/night activity. Pain Rep. 2017, 2, e614. [Google Scholar] [CrossRef] [PubMed]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]
- Randall, L.O.; Selitto, J.J. A method for measurement of analgesic activity on inflamed tissue. Arch. Int. Pharmacodyn. Ther. 1957, 111, 409–419. [Google Scholar] [PubMed]
- Hargreaves, K.; Dubner, R.; Brown, F.; Flores, C.; Joris, J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988, 32, 77–88. [Google Scholar] [CrossRef]
- Nakamura, A.; Ono, H.; Ando, A.; Hinata, M.; Niidome, K.; Omachi, S.; Sakaguchi, G.; Shinohara, S. Suppression of the acute upregulation of phosphorylated-extracellular regulated kinase in ventral tegmental area by a mu-opioid receptor agonist is related to resistance to rewarding effects in a mouse model of bone cancer. J. Pharmacol. Sci. 2017, 133, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, G.; Nagakura, Y.; Takeshita, N.; Shimizu, Y. Efficacy of drugs with different mechanisms of action in relieving spontaneous pain at rest and during movement in a rat model of osteoarthritis. Eur. J. Pharmacol. 2014, 738, 111–117. [Google Scholar] [CrossRef] [PubMed]
- DeBoer, E.M.; Azevedo, R.; Vega, T.A.; Brodkin, J.; Akamatsu, W.; Okano, H.; Wagner, G.C.; Rasin, M.L. Prenatal deletion of the RNA-binding protein HuD disrupts postnatal cortical circuit maturation and behavior. J. Neurosci. 2014, 34, 3674–3686. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G. Frequency of pain in patients with cancer. Recent Res. Cancer Res. 1984, 89, 64–71. [Google Scholar]
- Coleman, R.E. Monitoring of bone metastases. Eur. J. Cancer 1998, 34, 252–259. [Google Scholar] [CrossRef]
- Zhu, X.C.; Zhang, J.L.; Ge, C.T.; Yun, Y.Y.; Wang, P.; Yuan, T.F.; Fu, C.Y. Advances in cancer pain from bone metastasis. Drug Des. Dev. Ther. 2015, 9, 4239–4245. [Google Scholar] [CrossRef]
- Jehn, C.F.; Diel, I.J.; Overkamp, F.; Kurth, A.; Schaefer, R.; Miller, K.; Luftner, D. Management of metastatic bone disease algorithms for diagnostics and treatment. Anticancer Res. 2016, 36, 2631–2638. [Google Scholar]
- Suva, L.J.; Washam, C.; Nicholas, R.W.; Griffin, R.J. Bone metastasis: Mechanisms and therapeutic opportunities. Nat. Rev. Endocrinol. 2011, 7, 208–218. [Google Scholar] [CrossRef]
- Ahmad, I.; Ahmed, M.M.; Ahsraf, M.F.; Naeem, A.; Tasleem, A.; Ahmed, M.; Farooqi, M.S. Pain management in metastatic bone disease: A literature review. Cureus 2018, 10, e3286. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.; Finlay, I.; Ray, A.; Simpson, B. Is there still a role for open cordotomy in cancer pain management? J. Pain Symptom Manag. 2003, 25, 179–184. [Google Scholar] [CrossRef]
- Chow, E. Update on radiation treatment for cancer pain. Curr. Opin. Support. Palliat. Care 2007, 1, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.; Gillick, L.; Hendrickson, F.R. The palliation of symptomatic osseous metastases: Final results of the study by the Radiation Therapy Oncology Group. Cancer 1982, 50, 893–899. [Google Scholar] [CrossRef]
- McQuay, H.J.; Collins, S.L.; Carroll, D.; Moore, R.A. Radiotherapy for the palliation of painful bone metastases. Cochrane Database Syst. Rev. 2000. [Google Scholar] [CrossRef]
- Nomiya, T.; Teruyama, K.; Wada, H.; Nemoto, K. Time course of pain relief in atients treated with radiotherapy for cancer pain: A prospective study. Clin. J. Pain 2010, 26, 38–42. [Google Scholar] [CrossRef]
- Liepe, K.; Kotzerke, J. A comparative study of 188Re-HEDP, 186Re-HEDP, 153Sm-EDTMP and 89Sr in the treatment of painful skeletal metastases. Nucl. Med. Commun. 2007, 28, 623–630. [Google Scholar] [CrossRef]
- Porter, A.T.; Davis, L.P. Systemic radionuclide therapy of bone metastases with strontium-89. Oncology 1994, 8, 93–96. [Google Scholar] [PubMed]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fossa, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alsympca Investigators: Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef]
- Ma, X.; Yang, Q.; Wilson, K.T.; Kundu, N.; Meltzer, S.J.; Fulton, A.M. Promoter methylation regulates cyclooxygenase expression in breast cancer. Breast Cancer Res. 2004, 6, R316–R321. [Google Scholar] [CrossRef]
- Sabino, M.C.; Ghilardi, J.R.; Feia, K.J.; Jongen, J.L.; Keyser, C.P.; Luger, N.M.; Mach, D.B.; Peters, C.M.; Rogers, S.D.; Schwei, M.J.; et al. The involvement of prostaglandins in tumorigenesis, tumor-induced osteolysis and bone cancer pain. J. Musculoskelet. Neuronal Interact. 2002, 2, 561–562. [Google Scholar] [PubMed]
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.B.; Hawkey, C.J.; Hochberg, M.C.; et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. Vigor study group. N. Engl. J. Med. 2000, 343, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Otis, V.; Sarret, P.; Gendron, L. Spinal activation of delta opioid receptors alleviates cancerrelated bone pain. Neuroscience 2011, 183, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Ferrari, S.; Russel, R.G. Denosumab and bisphosphonates: Different mechanisms of action and effects. Bone 2011, 48, 677–692. [Google Scholar] [CrossRef]
- Hadji, P.; Ziller, M.; Maurer, T.; Autenrieth, M.; Muth, M.; Ruebel, M.; May, C.; Birkholz, K.; Diebel, E.; Gleissner, J.; et al. The ZOTEC study: Effect of zoledronic acid on bone metabolism in patients with bone metastases from prostate or breast cancer. J. Bone Oncol. 2012, 1, 88–94. [Google Scholar] [CrossRef]
- Coleman, R.E. Adjuvant bone-targeted therapy to prevent metastasis: Lessons from the AZURE study. Curr. Opin. Support. Palliat. Care 2012, 6, 322–329. [Google Scholar] [CrossRef]
- Nigro, C.; Donadio, M.; Ardine, M.; Beano, A.; Mistrangelo, M.; Coccorullo, Z.; Bertetto, O. Pain control with zoledronic acid in patients with breast cancer and metastatic bone disease. Am. J. Cancer 2004, 3, 257–263. [Google Scholar] [CrossRef]
- Clemons, M.; Dranitsaris, G.; Ooi, W.; Cole, D.E.C. A phase II trial evaluating the palliative benefit of secondline oral ibandronate in breast cancer patients with breast cancer: A systematic review and meta-analysis. PLoS ONE 2008, 108, 79–85. [Google Scholar] [CrossRef]
- Eleutherakis-Papaiakovou, E.; Barmias, A. Antiresorptive treatment-associated ONJ. Eur. J. Cancer Care (Engl.) 2017, 26. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, K.H.; Wanyan, P.; Tian, J.H. Comparison of the efficacy and safety of denosumab versus bisphosphonates in breast cancer and metastases treatment: A meta-analysis of randomized controlled trials. Oncol. Lett. 2014, 7, 1997–2002. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.S.; Gordon, D.; Kaminski, M.; Howell, A.; Belch, A.; Mackey, J.; Apffelstaedt, J.; Hussein, M.A.; Coleman, R.E.; Reitsma, D.J.; et al. Long-term efficacy and safety of zoledronic acid compared with pamidronate disodium in the treatment of skeletal complications in patients with advanced multiple myeloma or breast carcinoma: A randomized, double-blind, multicentre, comparative trial. Cancer 2003, 98, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Coleman, R.E.; Klotz, L.; Pittman, K.; Milecki, P.; Ng, S.; Chi, K.N.; Balakumaran, A.; Wei, R.; Wang, H.; et al. Denosumab for the prevention of skeletal complications in metastatic castration-resistant prostate cancer: Comparison of skeletal-related events and symptomatic skeletal events. Ann. Oncol. 2015, 26, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Posta-Sales, J.; Garzon-Rodriguez, C.; Llorens-Torromé, S.; Brunelli, C.; Pigni, A.; Caraceni, A. Evidence of the analgesic role of bisphosphonates and denosumab in the treatment of pain due to bone metastases: A systematic review within the European Association for the Palliative Care guidelines project. Palliat. Med. 2017, 31, 5–25. [Google Scholar] [CrossRef]
- Khodorova, A.; Montmayeur, J.P.; Strichartz, G. Endothelin receptors and pain. J. Pain 2009, 10, 4–28. [Google Scholar] [CrossRef]
- Wacnik, P.W.; Eikmeier, L.J.; Ruggles, T.R.; Ramnaraine, M.L.; Walcheck, B.K.; Beitz, A.J.; Wilcox, G.L. Functional interactions between tumor and peripheral nerve: Morphology, algogen identification, and behavioural characterization of a new murine model of cancer pain. J. Neurosci. 2001, 21, 9355–9366. [Google Scholar] [CrossRef]
- Rove, K.O.; Crawford, E.D. Evolution of treatment options for patients with CRPC and bone metastases: Bone-target agents that go beyond palliation of symptoms to improve overall survival. Oncology 2011, 25, 1362–1370. [Google Scholar]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aielli, F.; Ponzetti, M.; Rucci, N. Bone Metastasis Pain, from the Bench to the Bedside. Int. J. Mol. Sci. 2019, 20, 280. https://doi.org/10.3390/ijms20020280
Aielli F, Ponzetti M, Rucci N. Bone Metastasis Pain, from the Bench to the Bedside. International Journal of Molecular Sciences. 2019; 20(2):280. https://doi.org/10.3390/ijms20020280
Chicago/Turabian StyleAielli, Federica, Marco Ponzetti, and Nadia Rucci. 2019. "Bone Metastasis Pain, from the Bench to the Bedside" International Journal of Molecular Sciences 20, no. 2: 280. https://doi.org/10.3390/ijms20020280
APA StyleAielli, F., Ponzetti, M., & Rucci, N. (2019). Bone Metastasis Pain, from the Bench to the Bedside. International Journal of Molecular Sciences, 20(2), 280. https://doi.org/10.3390/ijms20020280