Swim Training Modulates Mouse Skeletal Muscle Energy Metabolism and Ameliorates Reduction in Grip Strength in a Mouse Model of Amyotrophic Lateral Sclerosis

 , , , and
, , , and
Abstract
:1. Introduction
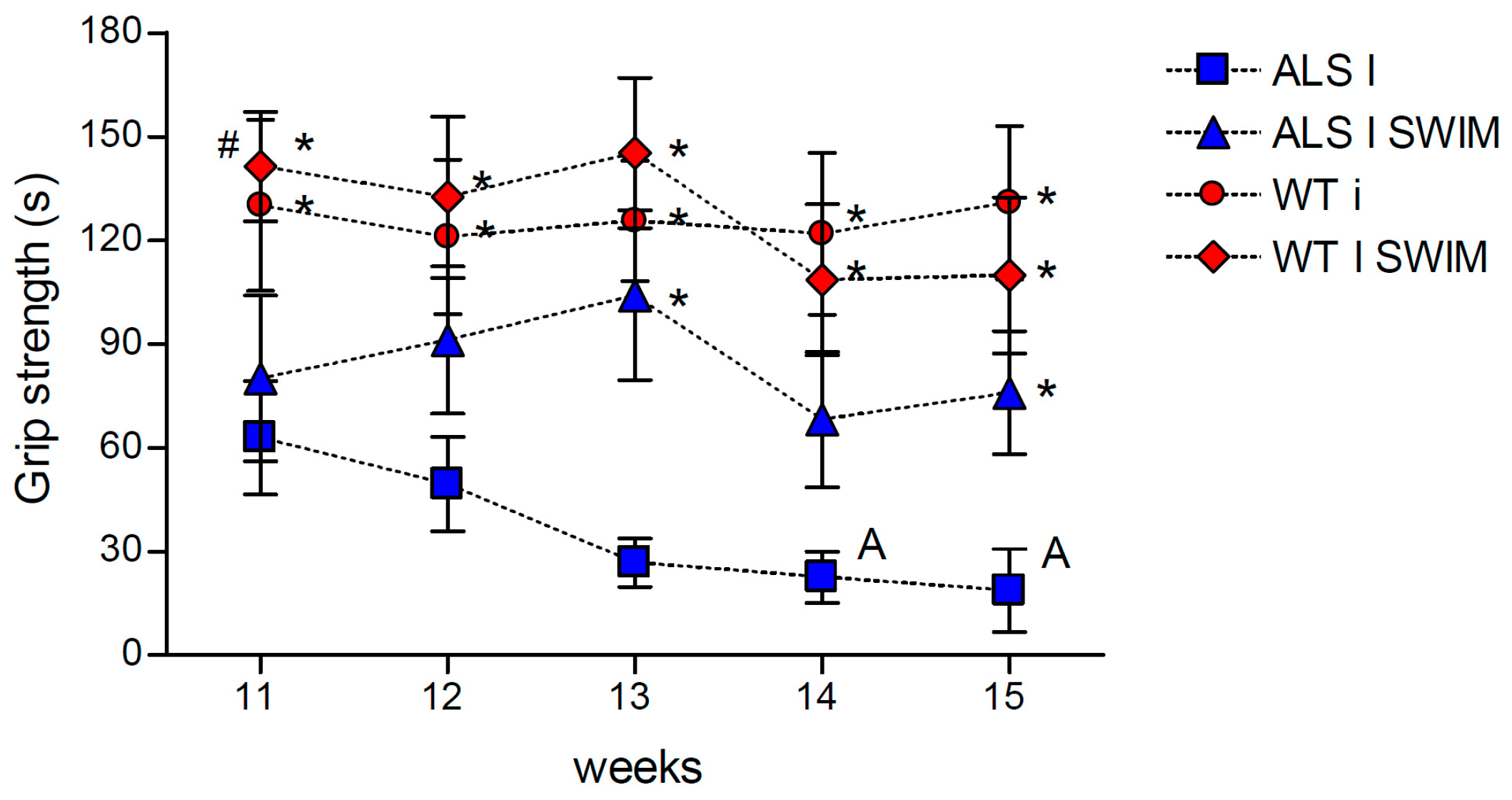
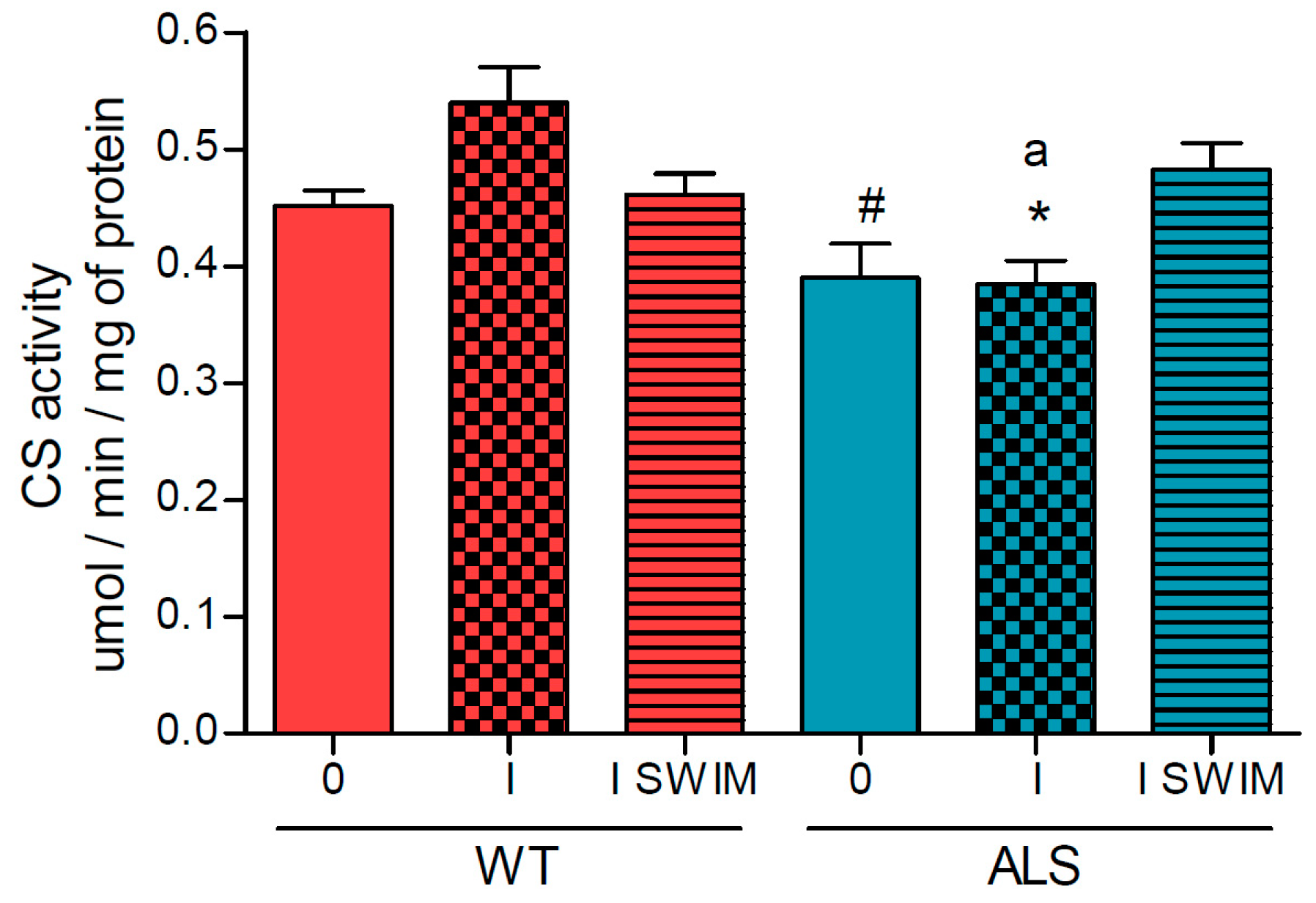
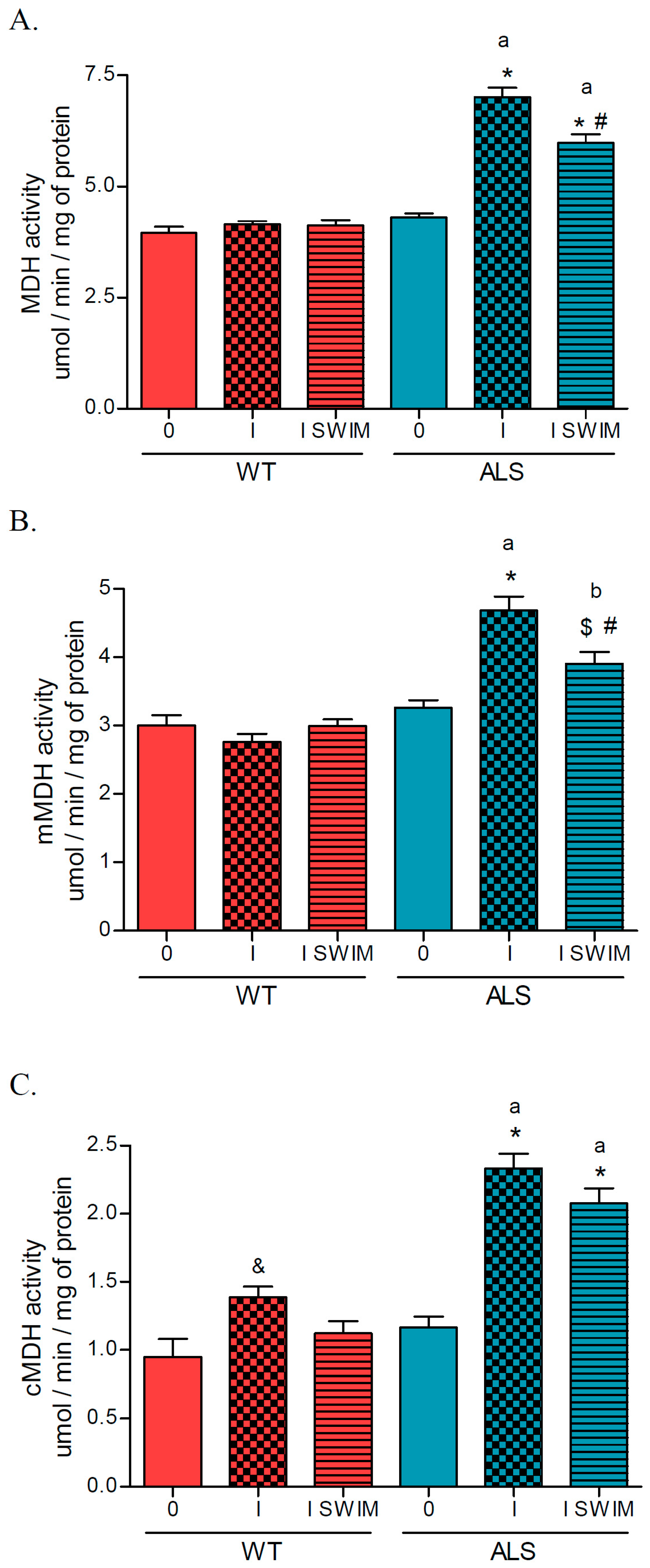
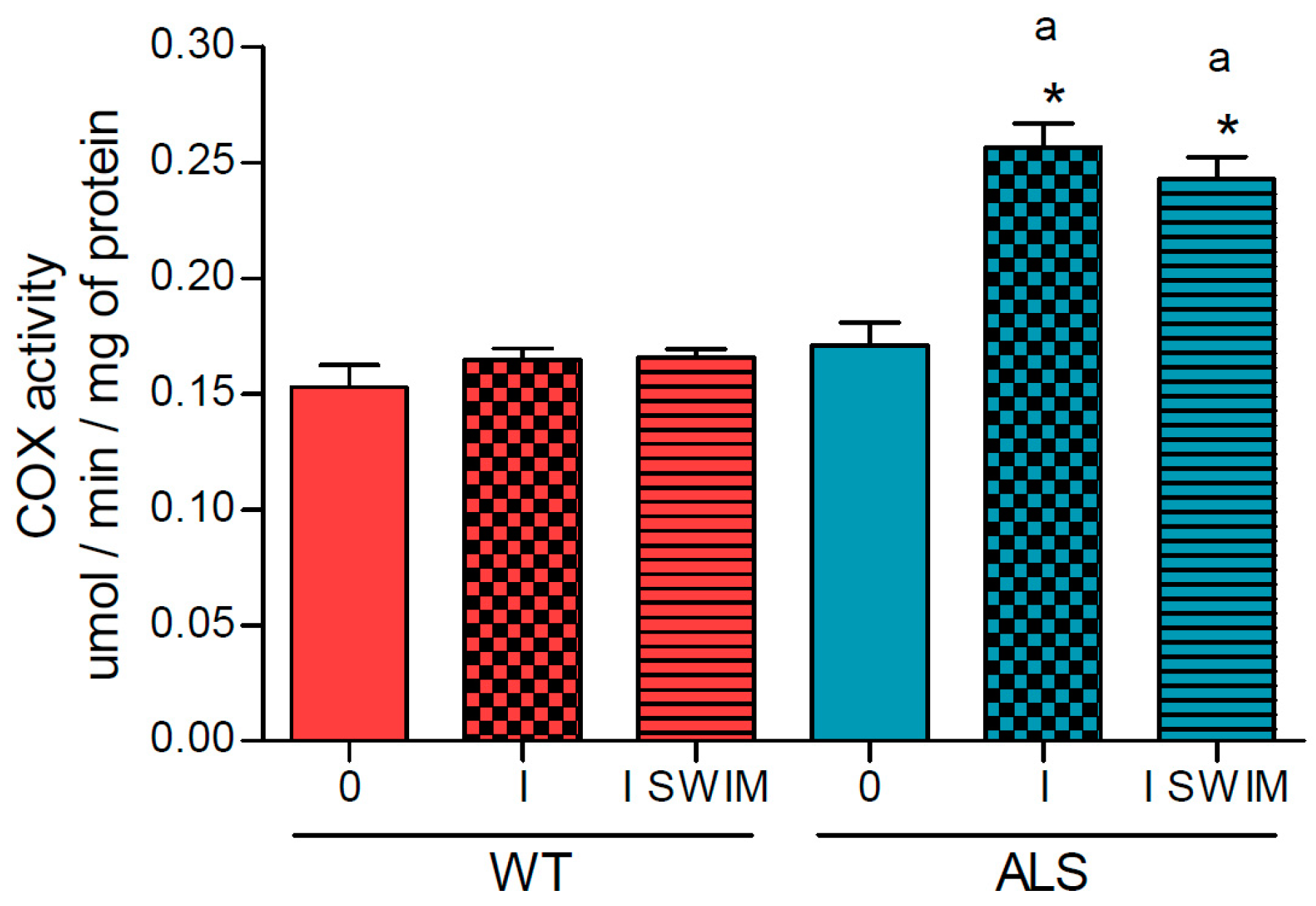
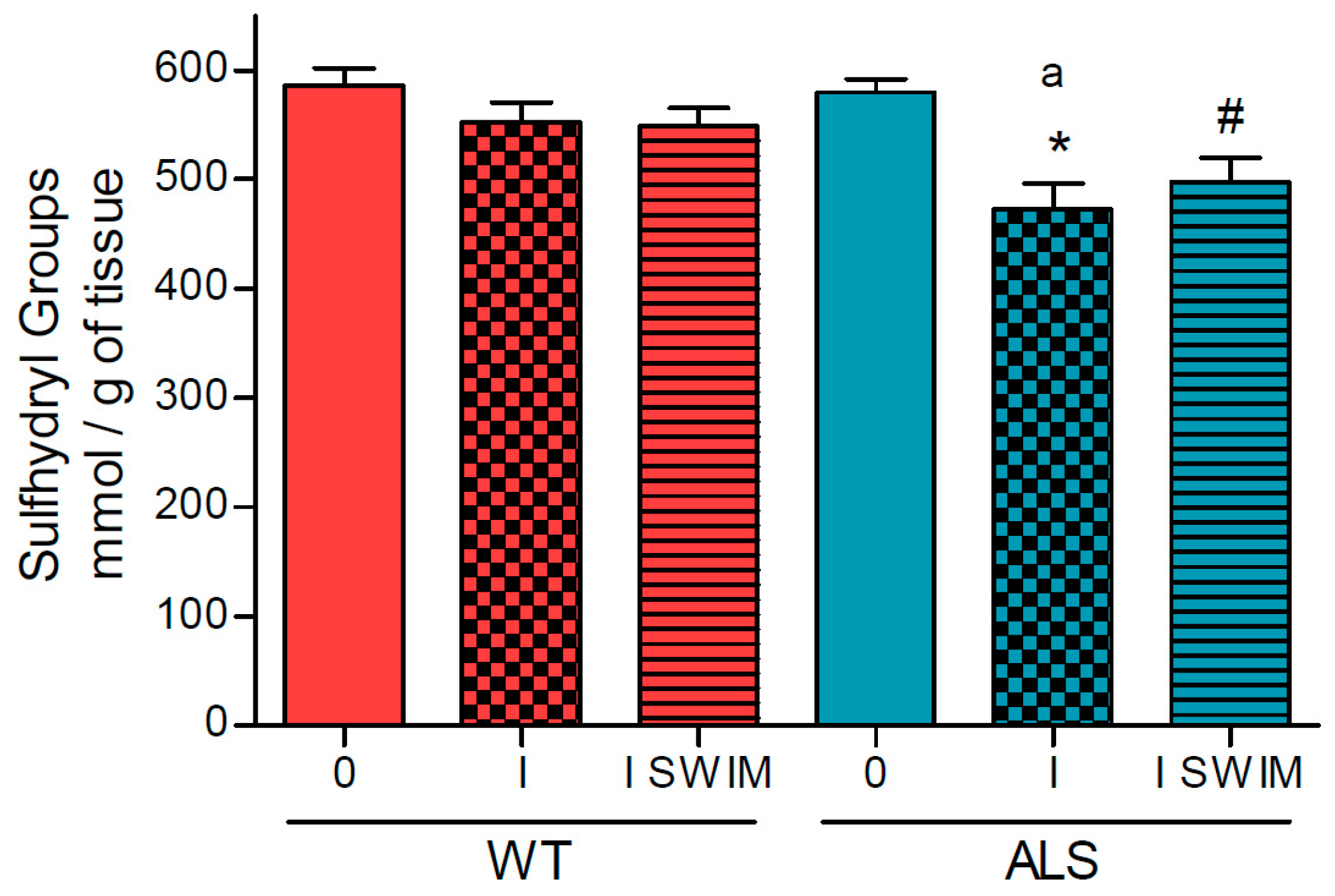
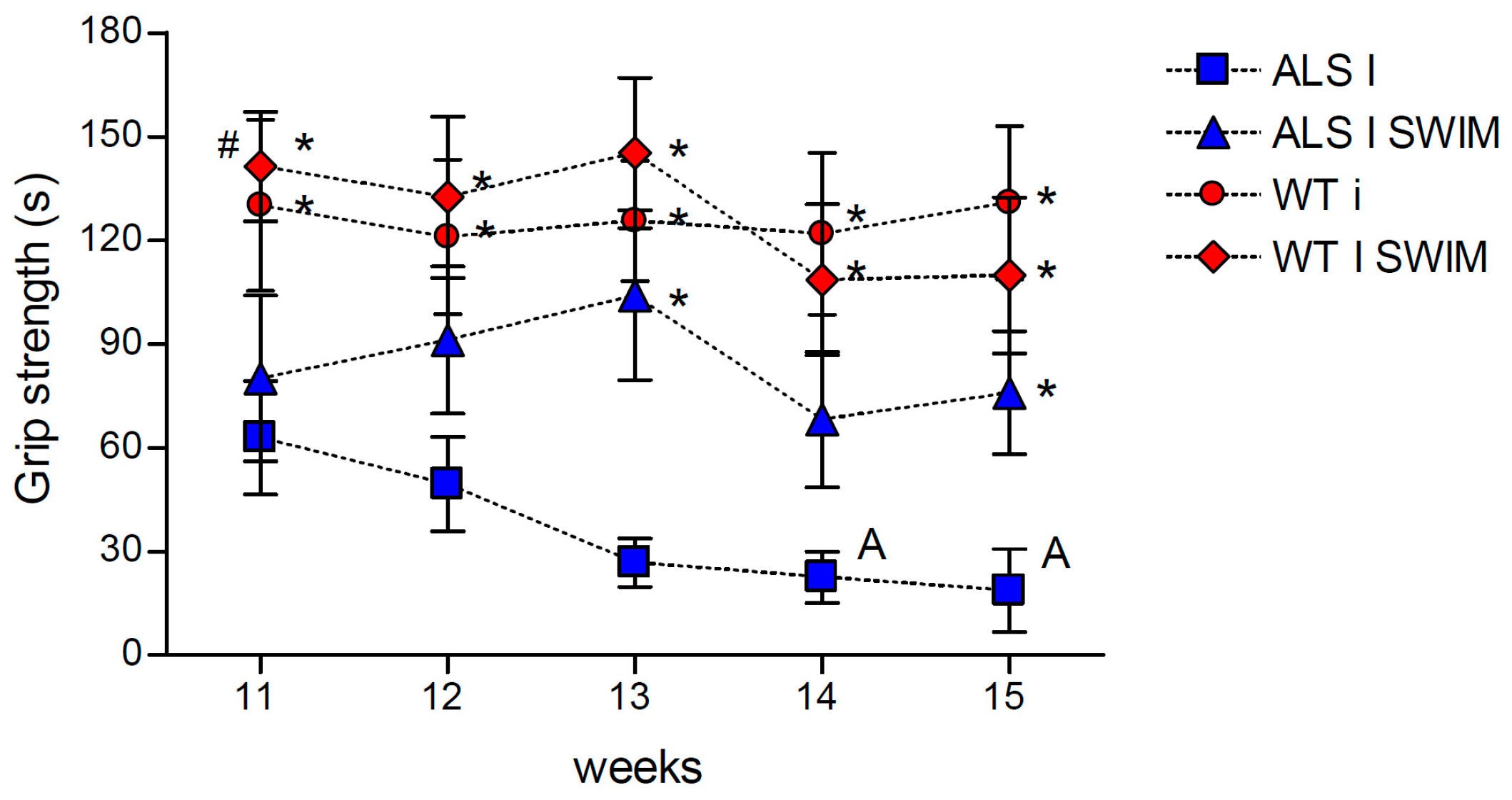
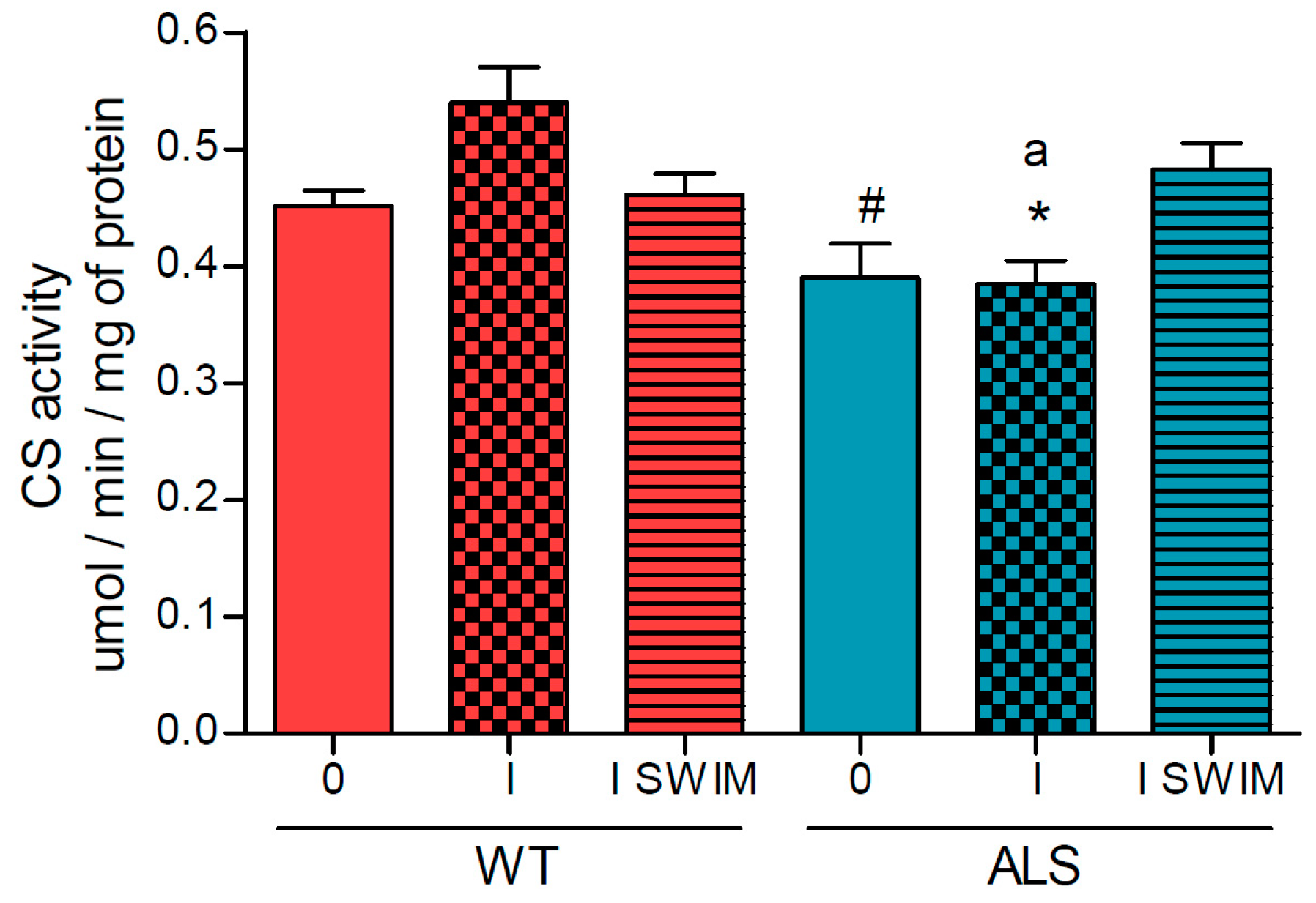
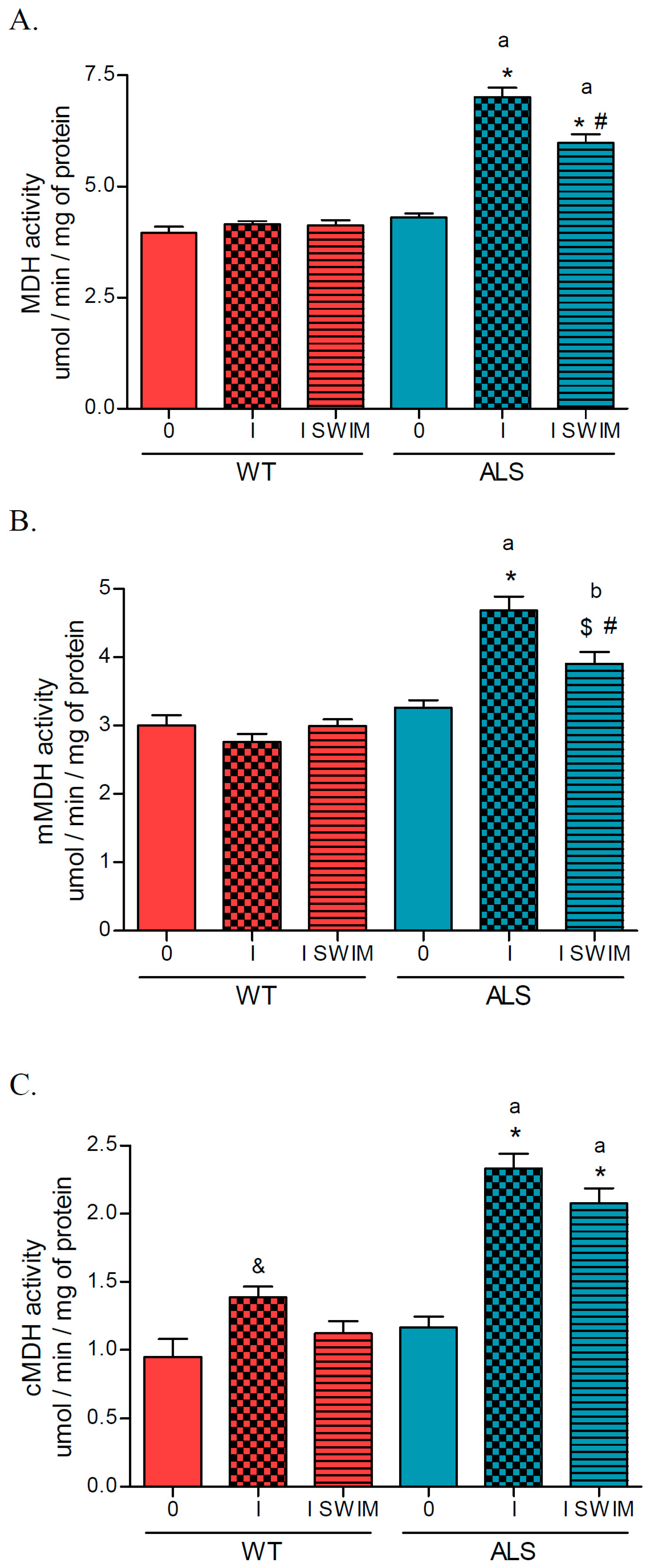
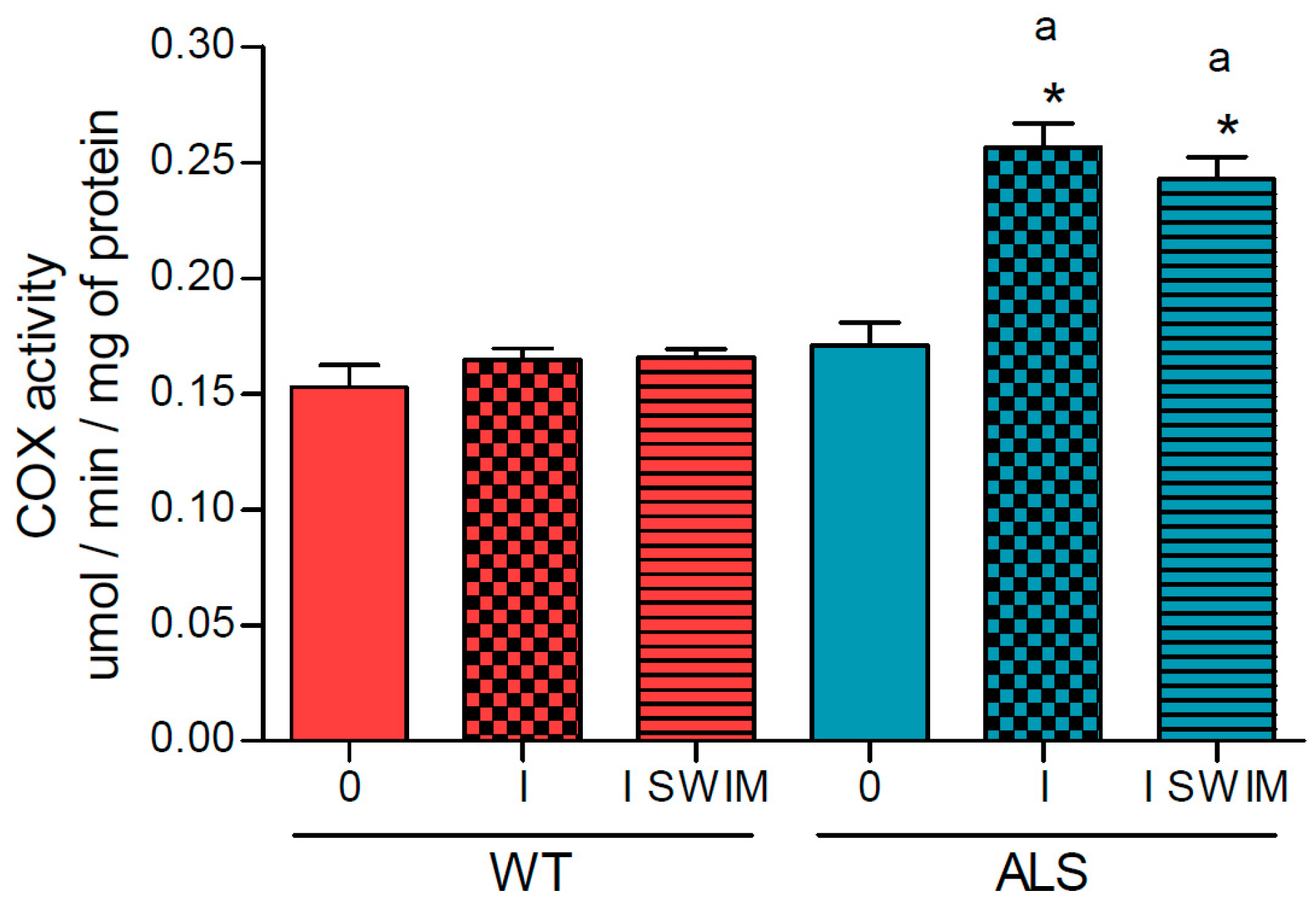
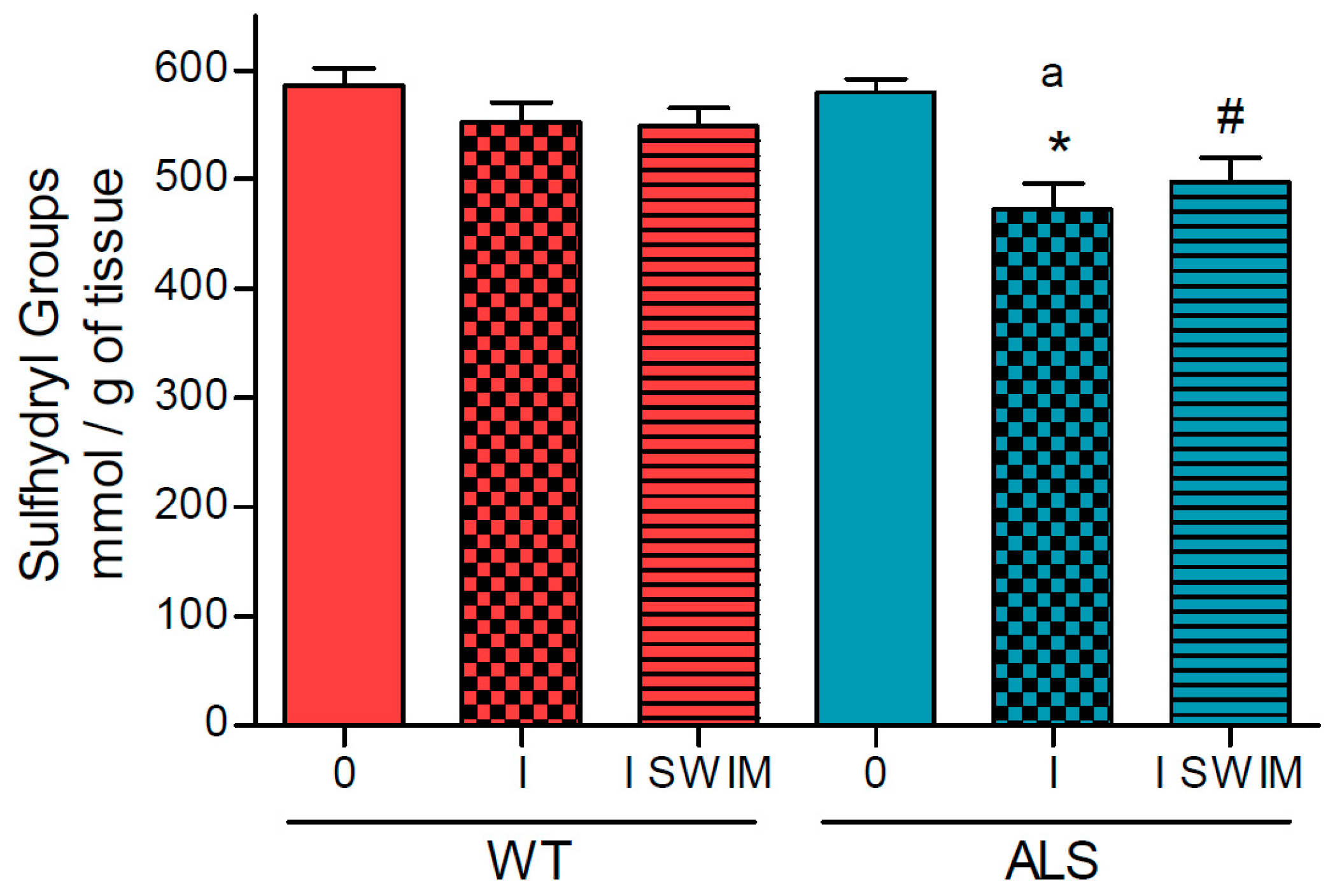
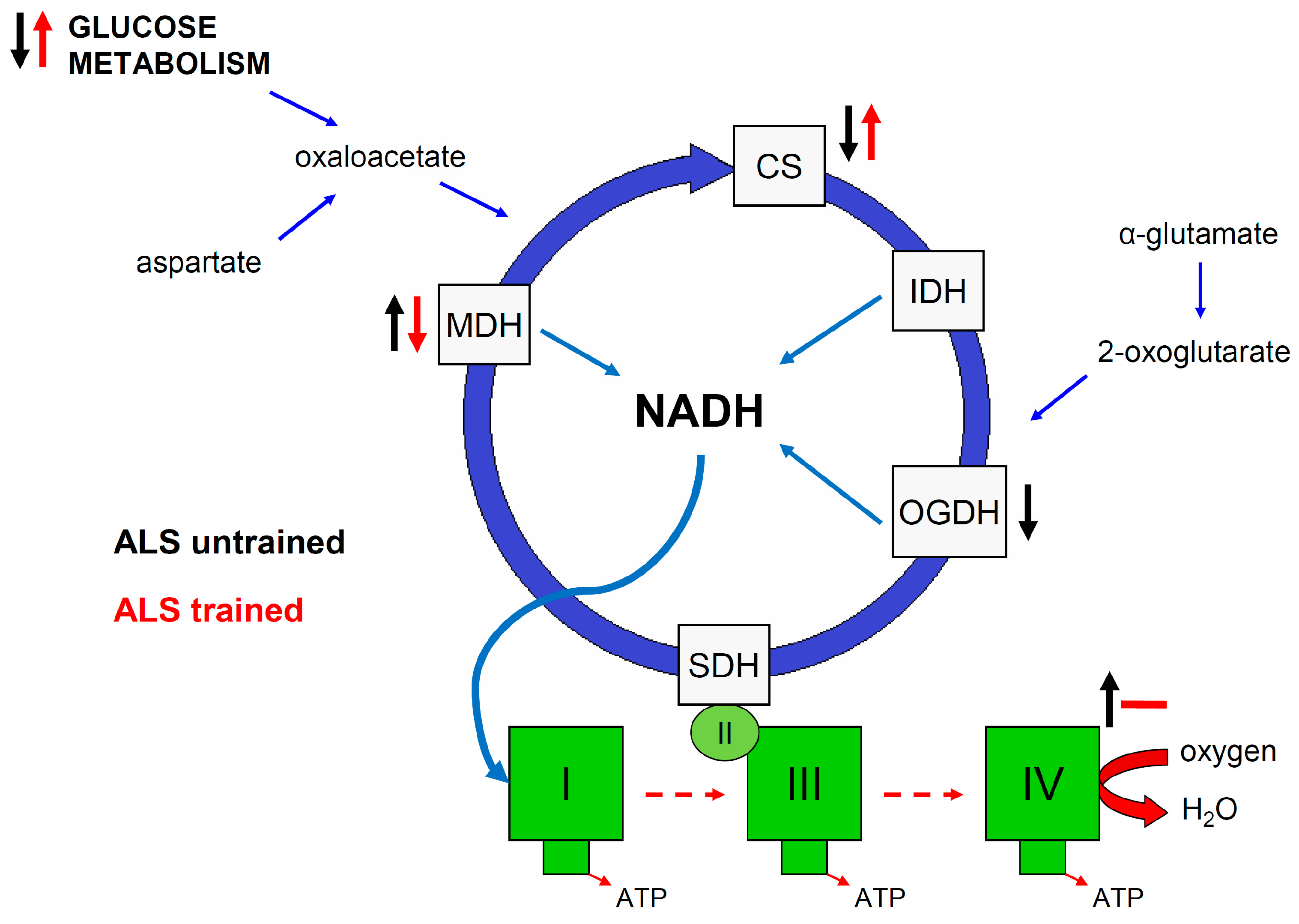
2. Results
3. Discussion
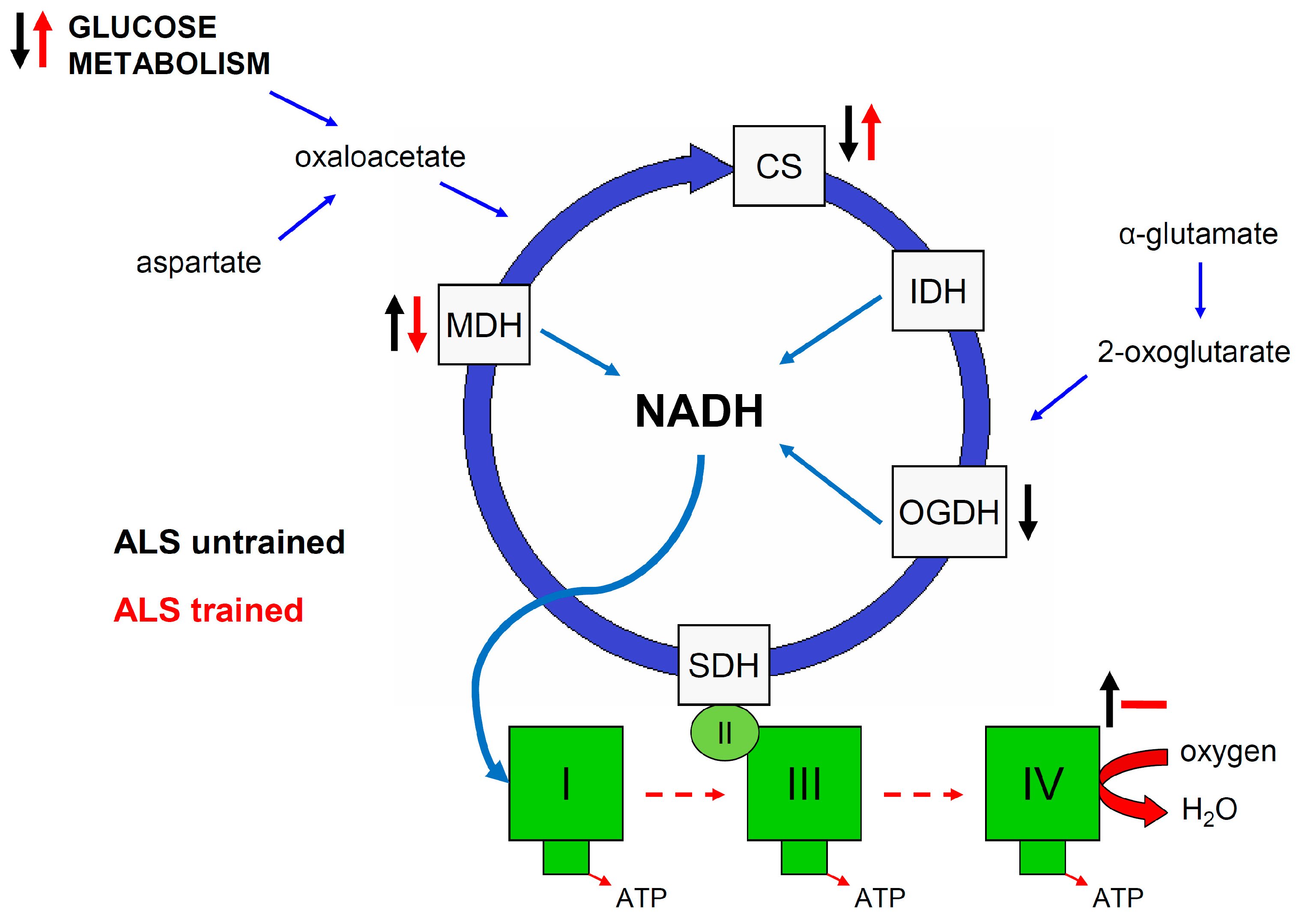
3.1. Effect of ALS on Energy Metabolism and Oxidative Stress in Symptomatic ALS Mice
3.2. Effect of Swim Training on Changes in Muscle Strength, Oxidative Stress, and Energy Metabolism in Symptomatic ALS Mice
4. Materials and Methods
4.1. Animals
4.2. Swim Training Protocol
4.3. Grip Strength
4.4. Isolation of Skeletal Muscle Mitochondria
4.5. High-Resolution Respirometry
4.6. Measurement of Cytochrome c Oxidase, Citrate Synthase and Malate Dehydrogenase Activities
4.7. Manifestation of Oxidative Stress
4.8. Data Analysis
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Andersen, P.M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr. Neurol. Neurosci. Rep. 2006, 6, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.P. Diagnosis of amyotrophic lateral sclerosis. J. Neurol. Sci. 1998, 160 (Suppl. 1), S6–S24. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in Amyotrophic Lateral Sclerosis: "Ambivalent" Behavior Connected to the Disease. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 2010, 19, 2284–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Boncompagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef]
- Dupuis, L.; Gonzalez de Aguilar, J.L.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE 2009, 4, e5390. [Google Scholar] [CrossRef]
- Halon, M.; Sielicka-Dudzin, A.; Wozniak, M.; Ziolkowski, W.; Nyka, W.; Herbik, M.; Grieb, P.; Figarski, A.; Antosiewicz, J. Up-regulation of ferritin ubiquitination in skeletal muscle of transgenic rats bearing the G93A hmSOD1 gene mutation. Neuromuscul. Disord. NMD 2010, 20, 29–33. [Google Scholar] [CrossRef]
- Halon, M.; Kaczor, J.J.; Ziolkowski, W.; Flis, D.J.; Borkowska, A.; Popowska, U.; Nyka, W.; Wozniak, M.; Antosiewicz, J. Changes in skeletal muscle iron metabolism outpace amyotrophic lateral sclerosis onset in transgenic rats bearing the G93A hmSOD1 gene mutation. Free Radic. Res. 2014, 48, 1363–1370. [Google Scholar] [CrossRef]
- Halon-Golabek, M.; Borkowska, A.; Kaczor, J.J.; Ziolkowski, W.; Flis, D.J.; Knap, N.; Kasperuk, K.; Antosiewicz, J. hmSOD1 gene mutation-induced disturbance in iron metabolism is mediated by impairment of Akt signalling pathway. J. Cachexia Sarcopenia Muscle 2018. [Google Scholar] [CrossRef] [PubMed]
- Olivan, S.; Calvo, A.C.; Gasco, S.; Munoz, M.J.; Zaragoza, P.; Osta, R. Time-Point Dependent Activation of Autophagy and the UPS in SOD1G93A Mice Skeletal Muscle. PLoS ONE 2015, 10, e0134830. [Google Scholar] [CrossRef] [PubMed]
- Dibaj, P.; Schomburg, E.D.; Steffens, H. Contractile characteristics of gastrocnemius-soleus muscle in the SOD1G93A ALS mouse model. Neurol. Res. 2015, 37, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Beqollari, D.; Romberg, C.F.; Dobrowolny, G.; Martini, M.; Voss, A.A.; Musaro, A.; Bannister, R.A. Progressive impairment of CaV1.1 function in the skeletal muscle of mice expressing a mutant type 1 Cu/Zn superoxide dismutase (G93A) linked to amyotrophic lateral sclerosis. Skelet Muscle 2016, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Deforges, S.; Branchu, J.; Biondi, O.; Grondard, C.; Pariset, C.; Lecolle, S.; Lopes, P.; Vidal, P.P.; Chanoine, C.; Charbonnier, F. Motoneuron survival is promoted by specific exercise in a mouse model of amyotrophic lateral sclerosis. J. Physiol. 2009, 587, 3561–3572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PLoS ONE 2013, 8, e82112. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Gonzalez de Aguilar, J.L.; Oudart, H.; de Tapia, M.; Barbeito, L.; Loeffler, J.P. Mitochondria in amyotrophic lateral sclerosis: A trigger and a target. Neurodegener. Dis. 2004, 1, 245–254. [Google Scholar] [CrossRef]
- Chen, D.; Wang, Y.; Chin, E.R. Activation of the endoplasmic reticulum stress response in skeletal muscle of G93A*SOD1 amyotrophic lateral sclerosis mice. Front. Cell. Neurosci. 2015, 9, 170. [Google Scholar] [CrossRef]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Kaczor, J.J.; Bourgeois, J.; Yasuda, N.; Tarnopolsky, M.A. Oxidative stress and antioxidant enzyme upregulation in SOD1-G93A mouse skeletal muscle. Muscle Nerve 2006, 33, 809–816. [Google Scholar] [CrossRef]
- Martin, L.J.; Fancelli, D.; Wong, M.; Niedzwiecki, M.; Ballarini, M.; Plyte, S.; Chang, Q. GNX-4728, a novel small molecule drug inhibitor of mitochondrial permeability transition, is therapeutic in a mouse model of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 433. [Google Scholar] [CrossRef]
- Martin, L.J. The mitochondrial permeability transition pore: A molecular target for amyotrophic lateral sclerosis therapy. Biochim. Biophys. Acta 2010, 1802, 186–197. [Google Scholar] [CrossRef] [Green Version]
- Szymanski, J.; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M.; Ziolkowski, W.; Duszynski, J.; Pinton, P.; Dobrzyn, A.; Wieckowski, M.R. Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Derave, W.; Van Den Bosch, L.; Lemmens, G.; Eijnde, B.O.; Robberecht, W.; Hespel, P. Skeletal muscle properties in a transgenic mouse model for amyotrophic lateral sclerosis: Effects of creatine treatment. Neurobiol. Dis. 2003, 13, 264–272. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; Baik, M.J.; Gurney, M.; Brown, R.H., Jr.; Beal, M.F. Metabolic dysfunction in familial, but not sporadic, amyotrophic lateral sclerosis. J. Neurochem. 1998, 71, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Desseille, C.; Deforges, S.; Biondi, O.; Houdebine, L.; D’Amico, D.; Lamaziere, A.; Caradeuc, C.; Bertho, G.; Bruneteau, G.; Weill, L.; et al. Specific Physical Exercise Improves Energetic Metabolism in the Skeletal Muscle of Amyotrophic-Lateral- Sclerosis Mice. Front. Mol. Neurosci. 2017, 10, 332. [Google Scholar] [CrossRef] [PubMed]
- Bowling, A.C.; Schulz, J.B.; Brown, R.H., Jr.; Beal, M.F. Superoxide dismutase activity, oxidative damage, and mitochondrial energy metabolism in familial and sporadic amyotrophic lateral sclerosis. J. Neurochem. 1993, 61, 2322–2325. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, C.; Scapagini, G.; Curro, D.; Giuffrida Stella, A.M.; De Marco, C.; Butterfield, D.A.; Calabrese, V. Mitochondrial dysfunction, free radical generation and cellular stress response in neurodegenerative disorders. Front. Biosci. 2007, 12, 1107–1123. [Google Scholar] [CrossRef]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Xiao, Y.; Karam, C.; Yi, J.; Zhang, L.; Li, X.; Yoon, D.; Wang, H.; Dhakal, K.; Ramlow, P.; Yu, T.; et al. ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharmacol. Res. 2018, 138, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 2006, 1762, 1051–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weishaupt, J.H.; Bartels, C.; Polking, E.; Dietrich, J.; Rohde, G.; Poeggeler, B.; Mertens, N.; Sperling, S.; Bohn, M.; Huther, G.; et al. Reduced oxidative damage in ALS by high-dose enteral melatonin treatment. J. Pineal Res. 2006, 41, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012, 2012, 428010. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, E.; Furukawa, Y. Copper Homeostasis as a Therapeutic Target in Amyotrophic Lateral Sclerosis with SOD1 Mutations. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Flis, D.J.; Dzik, K.; Kaczor, J.J.; Halon-Golabek, M.; Antosiewicz, J.; Wieckowski, M.R.; Ziolkowski, W. Swim Training Modulates Skeletal Muscle Energy Metabolism, Oxidative Stress, and Mitochondrial Cholesterol Content in Amyotrophic Lateral Sclerosis Mice. Oxid. Med. Cell. Longev. 2018, 2018, 5940748. [Google Scholar] [CrossRef] [PubMed]
- Grondard, C.; Biondi, O.; Pariset, C.; Lopes, P.; Deforges, S.; Lecolle, S.; Gaspera, B.D.; Gallien, C.L.; Chanoine, C.; Charbonnier, F. Exercise-induced modulation of calcineurin activity parallels the time course of myofibre transitions. J. Cell. Physiol. 2008, 214, 126–135. [Google Scholar] [CrossRef]
- Flis, D.J.; Olek, R.A.; Kaczor, J.J.; Rodziewicz, E.; Halon, M.; Antosiewicz, J.; Wozniak, M.; Gabbianelli, R.; Ziolkowski, W. Exercise-Induced Changes in Caveolin-1, Depletion of Mitochondrial Cholesterol, and the Inhibition of Mitochondrial Swelling in Rat Skeletal Muscle but Not in the Liver. Oxid. Med. Cell. Longev. 2016, 2016, 3620929. [Google Scholar] [CrossRef]
- Ziolkowski, W.; Vadhana, M.S.D.; Kaczor, J.J.; Olek, R.A.; Flis, D.J.; Halon, M.; Wozniak, M.; Fedeli, D.; Carloni, M.; Antosiewicz, J.; et al. Exercise-induced heart mitochondrial cholesterol depletion influences the inhibition of mitochondrial swelling. Exp. Physiol. 2013, 98, 1457–1468. [Google Scholar] [CrossRef] [Green Version]
- Tefera, T.W.; Wong, Y.; Barkl-Luke, M.E.; Ngo, S.T.; Thomas, N.K.; McDonald, T.S.; Borges, K. Triheptanoin Protects Motor Neurons and Delays the Onset of Motor Symptoms in a Mouse Model of Amyotrophic Lateral Sclerosis. PLoS ONE 2016, 11, e0161816. [Google Scholar] [CrossRef]
- Russell, A.P.; Wada, S.; Vergani, L.; Hock, M.B.; Lamon, S.; Leger, B.; Ushida, T.; Cartoni, R.; Wadley, G.D.; Hespel, P.; et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 49, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Crugnola, V.; Lamperti, C.; Lucchini, V.; Ronchi, D.; Peverelli, L.; Prelle, A.; Sciacco, M.; Bordoni, A.; Fassone, E.; Fortunato, F.; et al. Mitochondrial respiratory chain dysfunction in muscle from patients with amyotrophic lateral sclerosis. Arch. Neurol. 2010, 67, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Schaardenburgh, M.; Wohlwend, M.; Rognmo, O.; Mattsson, E.J.R. Exercise in claudicants increase or decrease walking ability and the response relates to mitochondrial function. J. Transl. Med. 2017, 15, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, B.P.; Safdar, A.; Raha, S.; Tarnopolsky, M.A.; Hamadeh, M.J. Caloric restriction shortens lifespan through an increase in lipid peroxidation, inflammation and apoptosis in the G93A mouse, an animal model of ALS. PLoS ONE 2010, 5, e9386. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; Di Meo, S. Effect of training on antioxidant capacity, tissue damage, and endurance of adult male rats. Int. J. Sports Med. 1997, 18, 497–502. [Google Scholar] [CrossRef]
- Makinen, M.W.; Lee, C.P. Biochemical studies of skeletal muscle mitochondria. I. Microanalysis of cytochrome content, oxidative and phosphorylative activities of mammalian skeletal muscle mitochondria. Arch. Biochem. Biophys. 1968, 126, 75–82. [Google Scholar] [CrossRef]
- Wharton, D.C.; Tzagoloff, A. Cytochrome C oxidase from beef heart mitochondria. In Methods in Enzymol; Estabrook, R.W., Pullman, M.E., Eds.; Academic Press: New York, NY, USA, 1967; Volume 10, pp. 245–250. [Google Scholar]
- De Lisio, M.; Kaczor, J.J.; Phan, N.; Tarnopolsky, M.A.; Boreham, D.R.; Parise, G. Exercise training enhances the skeletal muscle response to radiation-induced oxidative stress. Muscle Nerve 2011, 43, 58–64. [Google Scholar] [CrossRef]
- Schantz, P.G. Plasticity of human skeletal muscle with special reference to effects of physical training on enzyme levels of the NADH shuttles and phenotypic expression of slow and fast myofibrillar proteins. Acta Physiol. Scand. Suppl. 1986, 558, 1–62. [Google Scholar] [PubMed]
- Rice-Evans, C.A.; Diplock, A.T.; Symons, M.C.R. Techniques in Free Radical Research; Elsevier: Amsterdam, The Netherlands, 1991; Volume 22. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | ALS | |||||
|---|---|---|---|---|---|---|
| 0 | I | I SWIM | 0 | I | I SWIM | |
| state NL | 27.86 ± 2.06 | 29.43 ± 1.18 | 30.06 ± 1.96 | 23.87 ± 1.78 | 28.96 ± 1.12 | 29.84 ± 1.16 |
| state NP | 410.1 ± 38.53 | 492.5 ± 19.78 | 434.6 ± 47.36 | 371.4 ± 41.26 | 424.2 ± 38.54 | 379.7 ± 29.95 |
| OCE | 0.930 ± 0.006 | 0.931 ± 0.007 | 0.933 ± 0.008 | 0.920 ± 0.013 | 0.920 ± 0.010 | 0.928 ± 0.005 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flis, D.J.; Dzik, K.; Kaczor, J.J.; Cieminski, K.; Halon-Golabek, M.; Antosiewicz, J.; Wieckowski, M.R.; Ziolkowski, W. Swim Training Modulates Mouse Skeletal Muscle Energy Metabolism and Ameliorates Reduction in Grip Strength in a Mouse Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2019, 20, 233. https://doi.org/10.3390/ijms20020233
Flis DJ, Dzik K, Kaczor JJ, Cieminski K, Halon-Golabek M, Antosiewicz J, Wieckowski MR, Ziolkowski W. Swim Training Modulates Mouse Skeletal Muscle Energy Metabolism and Ameliorates Reduction in Grip Strength in a Mouse Model of Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2019; 20(2):233. https://doi.org/10.3390/ijms20020233
Chicago/Turabian StyleFlis, Damian Jozef, Katarzyna Dzik, Jan Jacek Kaczor, Karol Cieminski, Malgorzata Halon-Golabek, Jedrzej Antosiewicz, Mariusz Roman Wieckowski, and Wieslaw Ziolkowski. 2019. "Swim Training Modulates Mouse Skeletal Muscle Energy Metabolism and Ameliorates Reduction in Grip Strength in a Mouse Model of Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 20, no. 2: 233. https://doi.org/10.3390/ijms20020233
APA StyleFlis, D. J., Dzik, K., Kaczor, J. J., Cieminski, K., Halon-Golabek, M., Antosiewicz, J., Wieckowski, M. R., & Ziolkowski, W. (2019). Swim Training Modulates Mouse Skeletal Muscle Energy Metabolism and Ameliorates Reduction in Grip Strength in a Mouse Model of Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 20(2), 233. https://doi.org/10.3390/ijms20020233