FcεRI-HDAC3-MCP1 Signaling Axis Promotes Passive Anaphylaxis Mediated by Cellular Interactions


Abstract
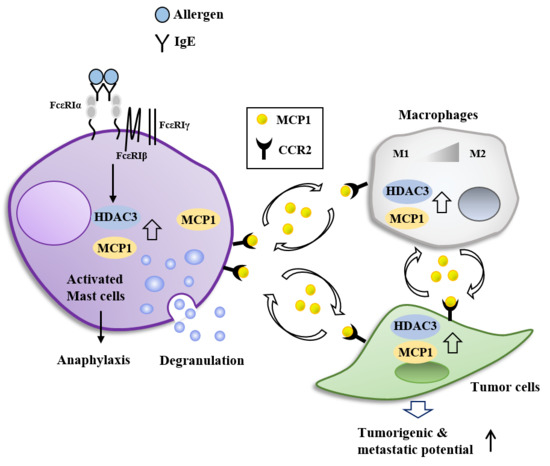
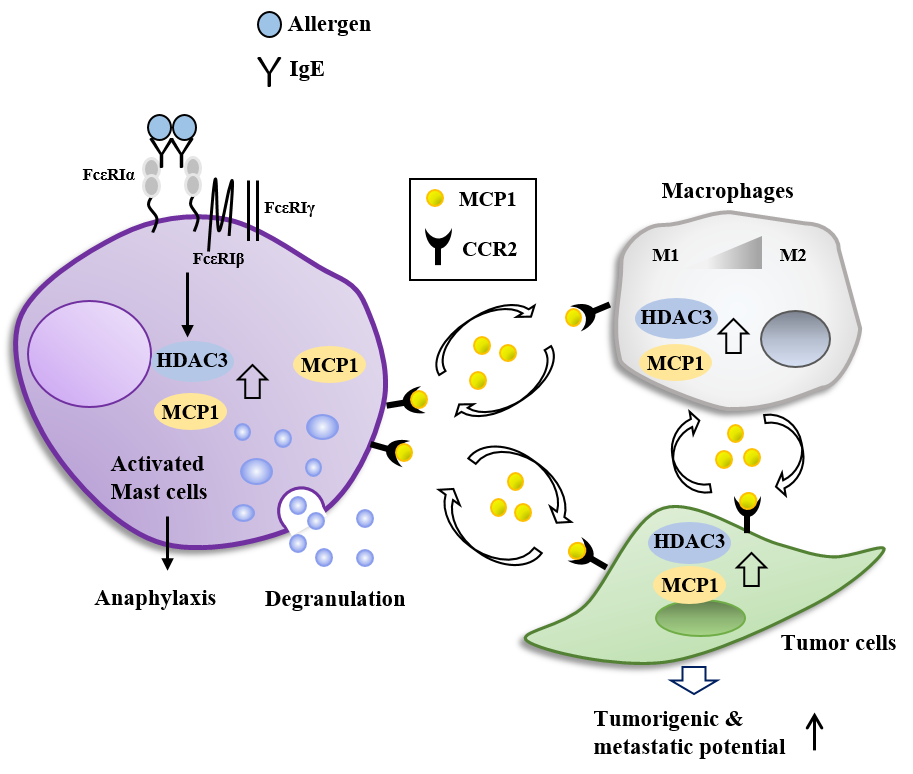
:
{kind=link}
{kind=link}
{kind=link}
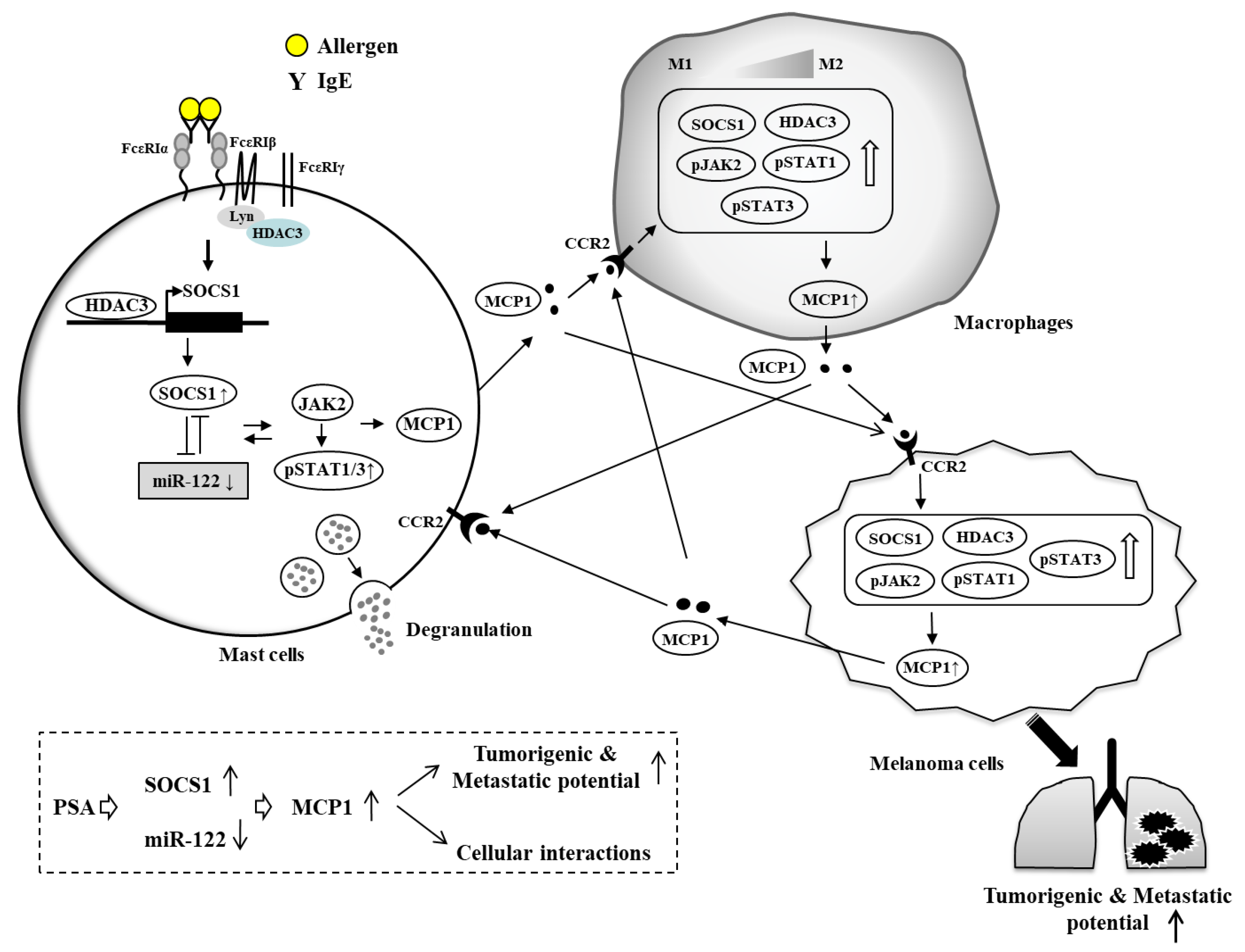{kind=link}
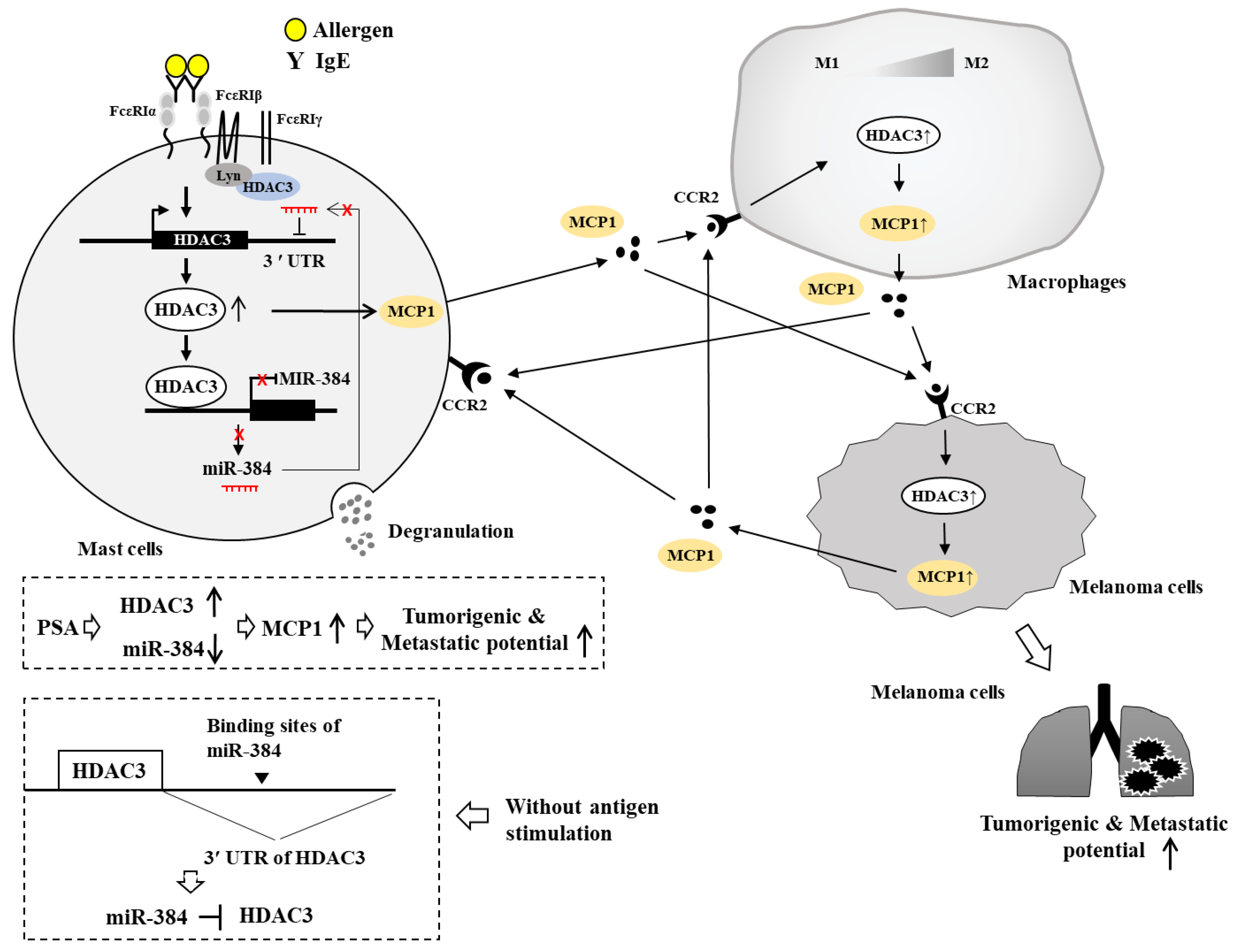{kind=link}
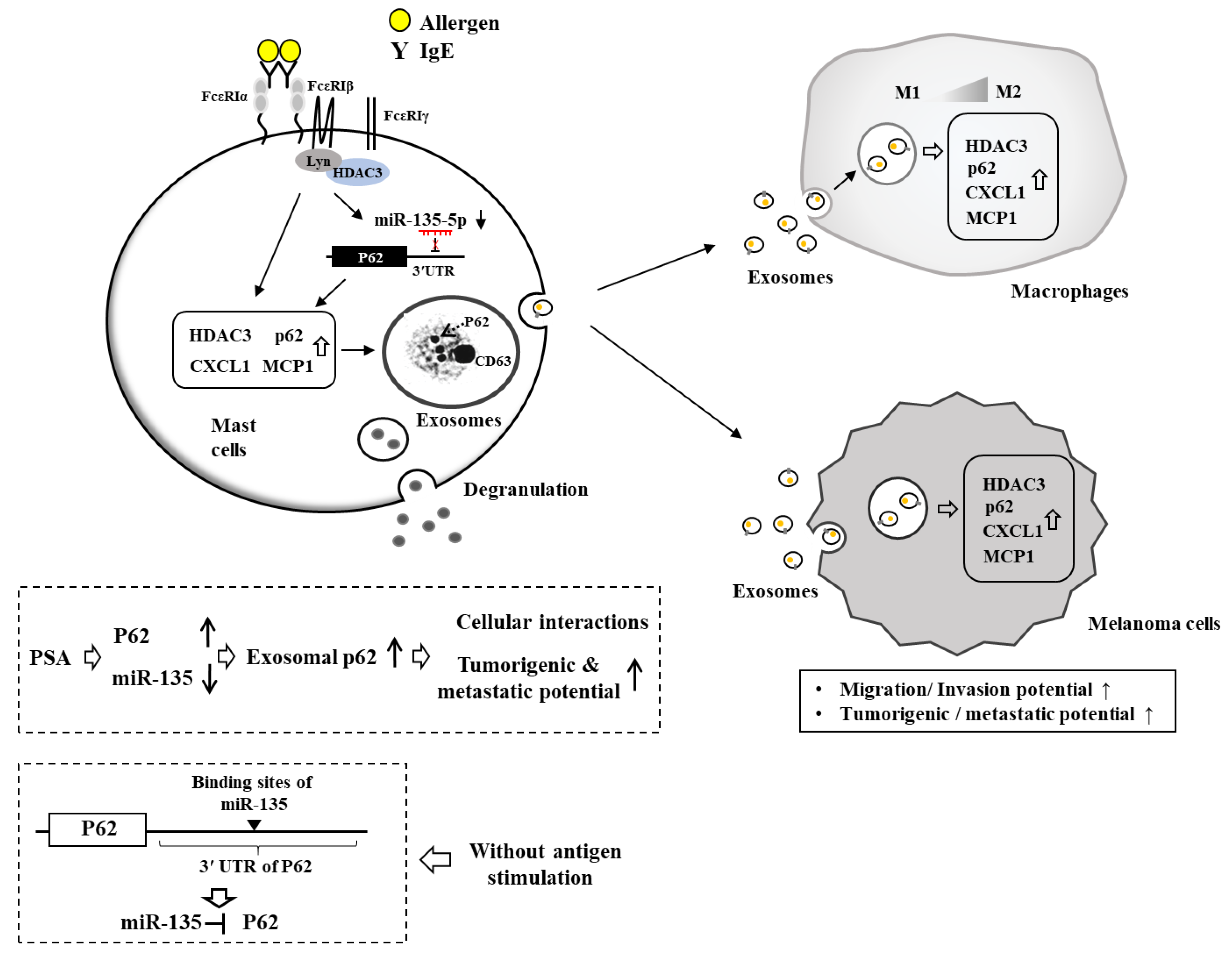{kind=link}
1. Introduction
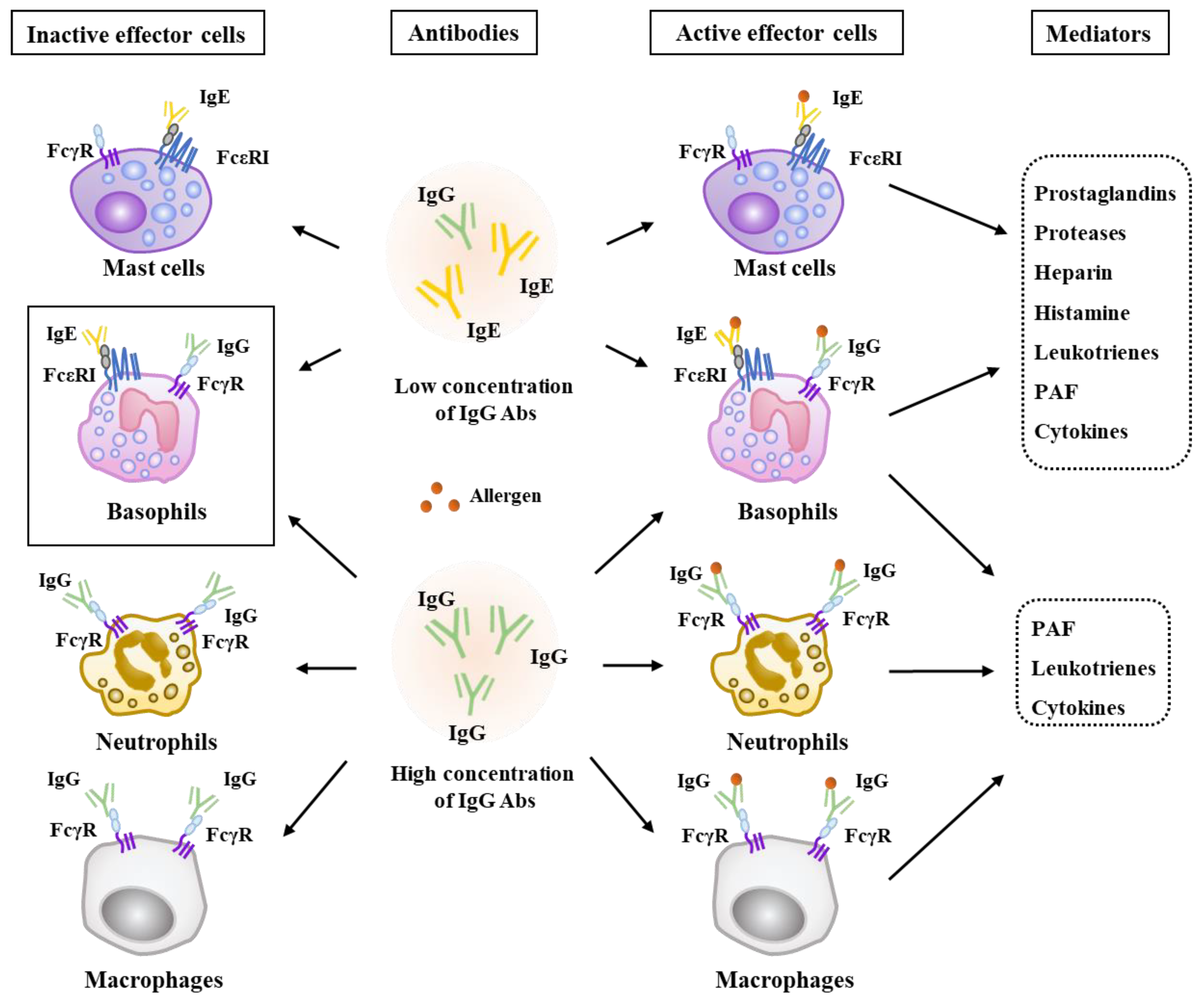
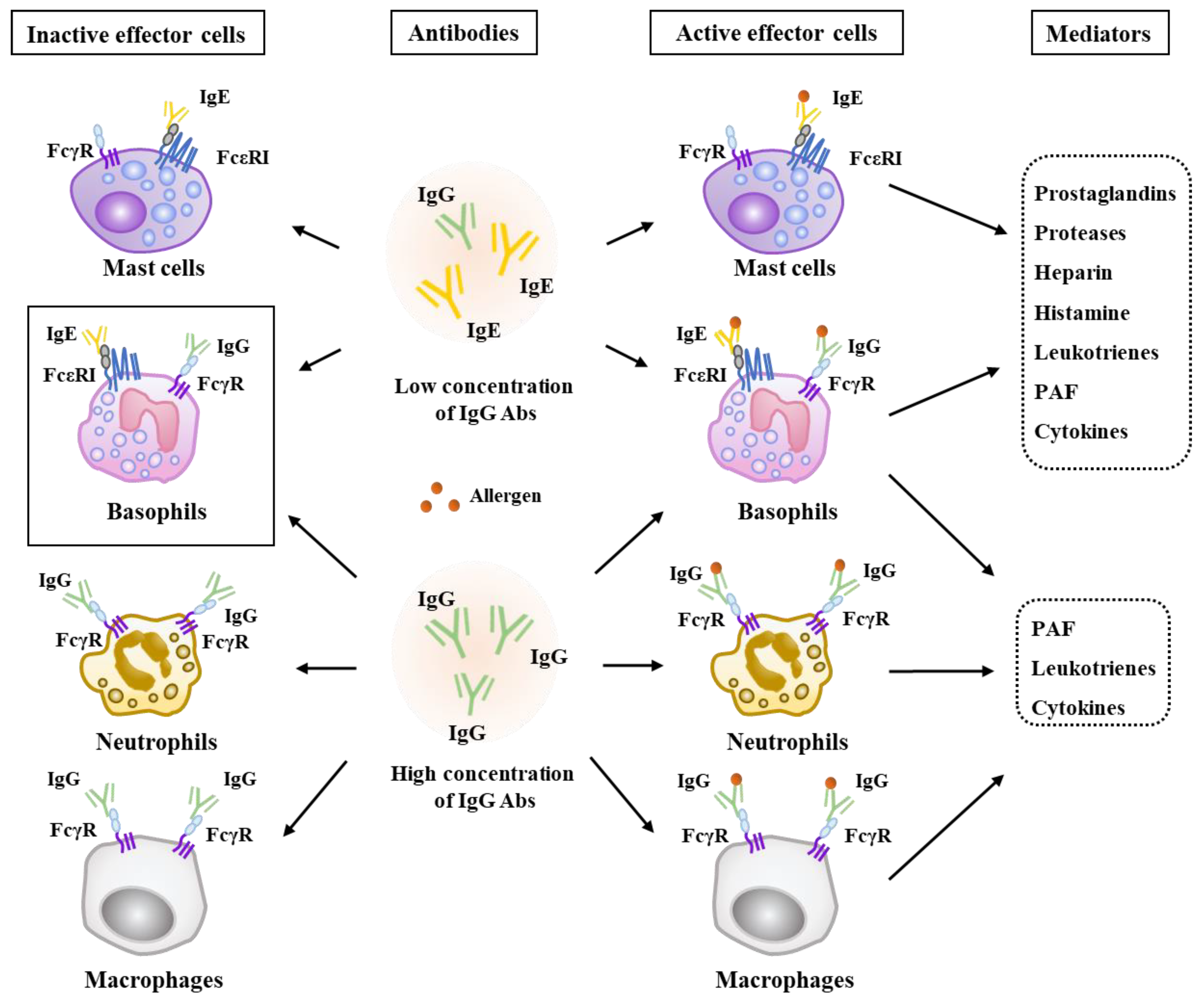
2. Effector Cells, Effector Molecules, and Cellular Interactions in Anaphylaxis
3. HDACs (Histone Deacetylases)
4. HDACs Inhibitors and Allergic Inflammatory Diseases
5. A Negative Regulatory Role of HDAC2 in Allergic Inflammation
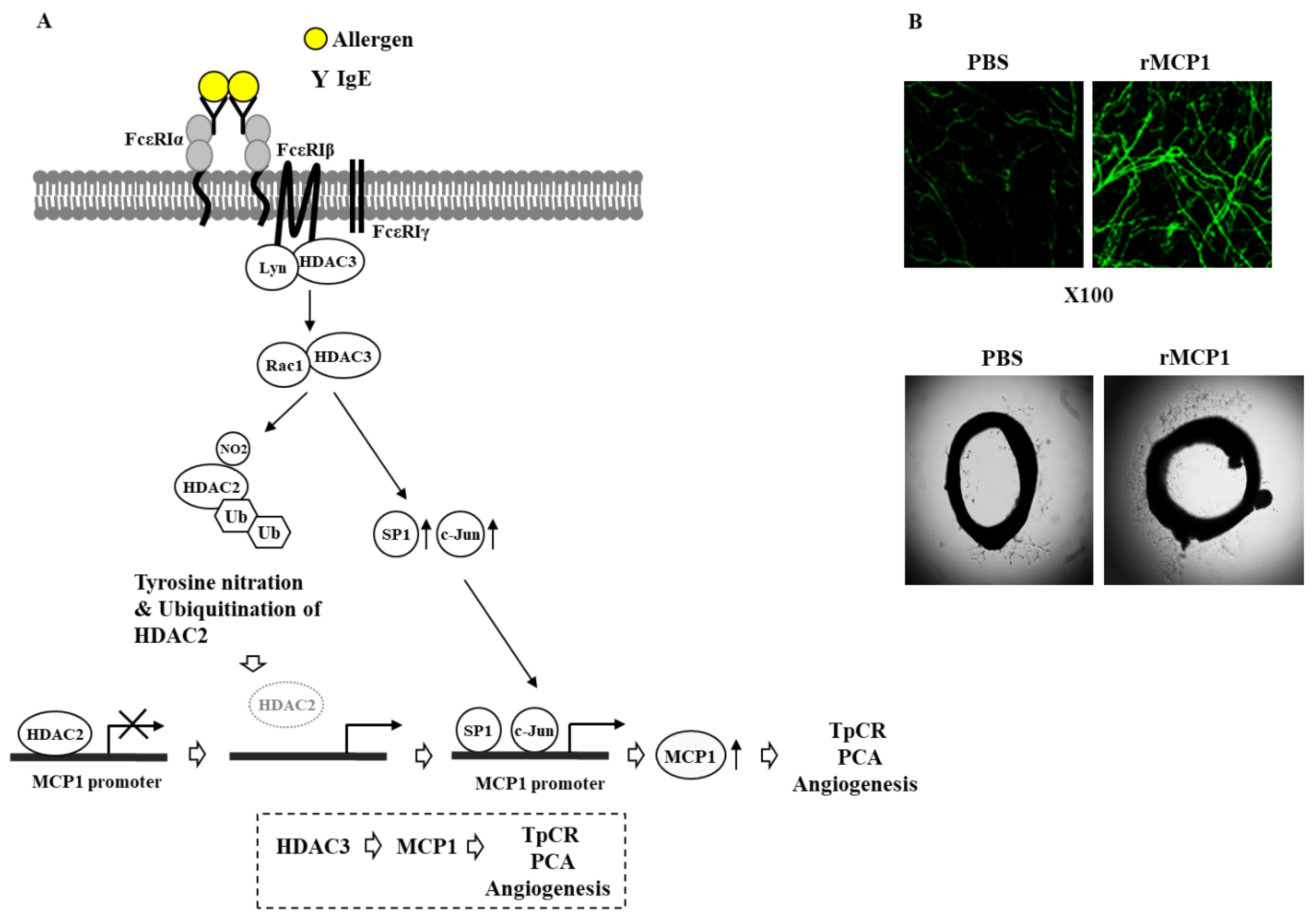
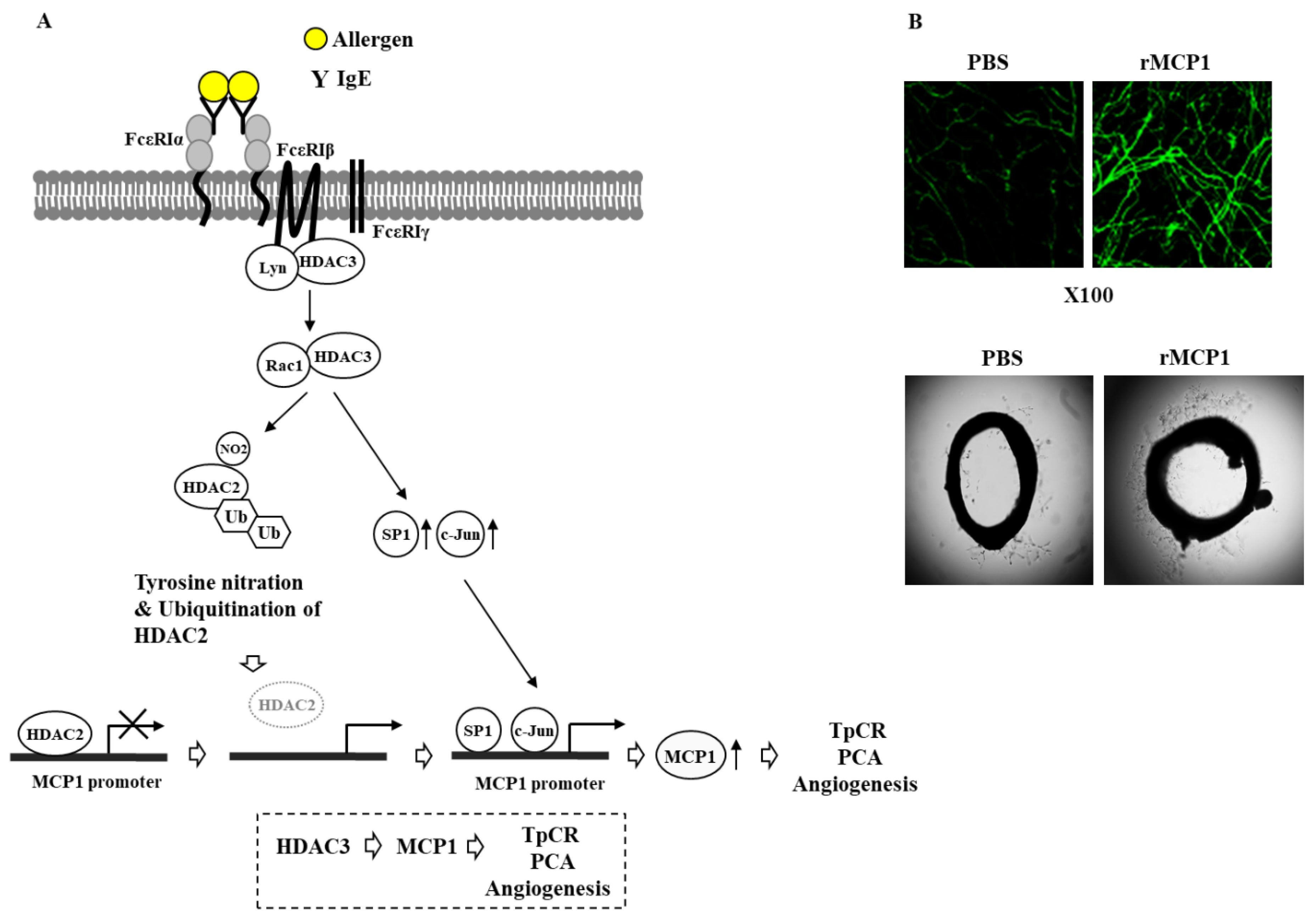
6. HDAC3 Mediates Anaphylaxis
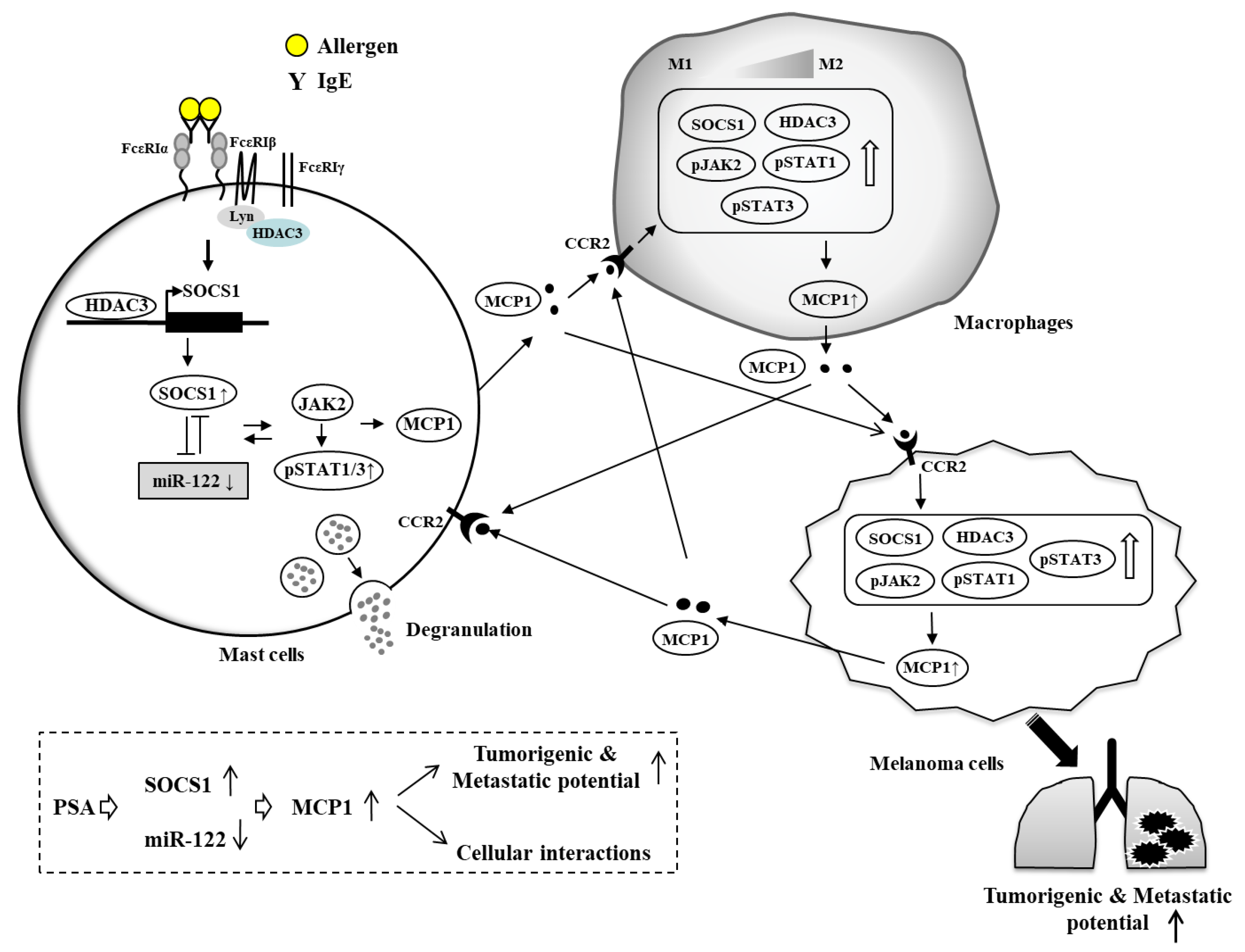
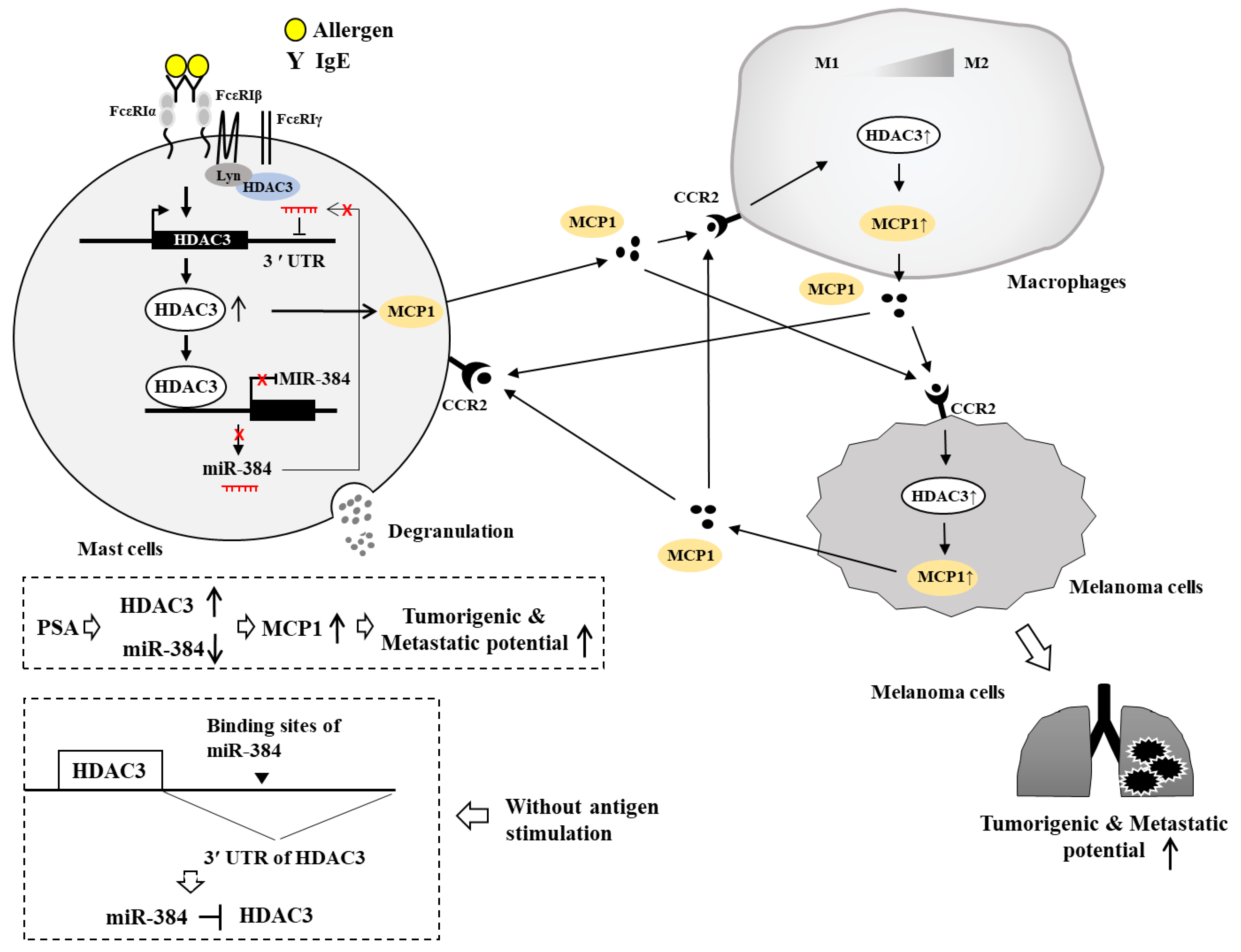
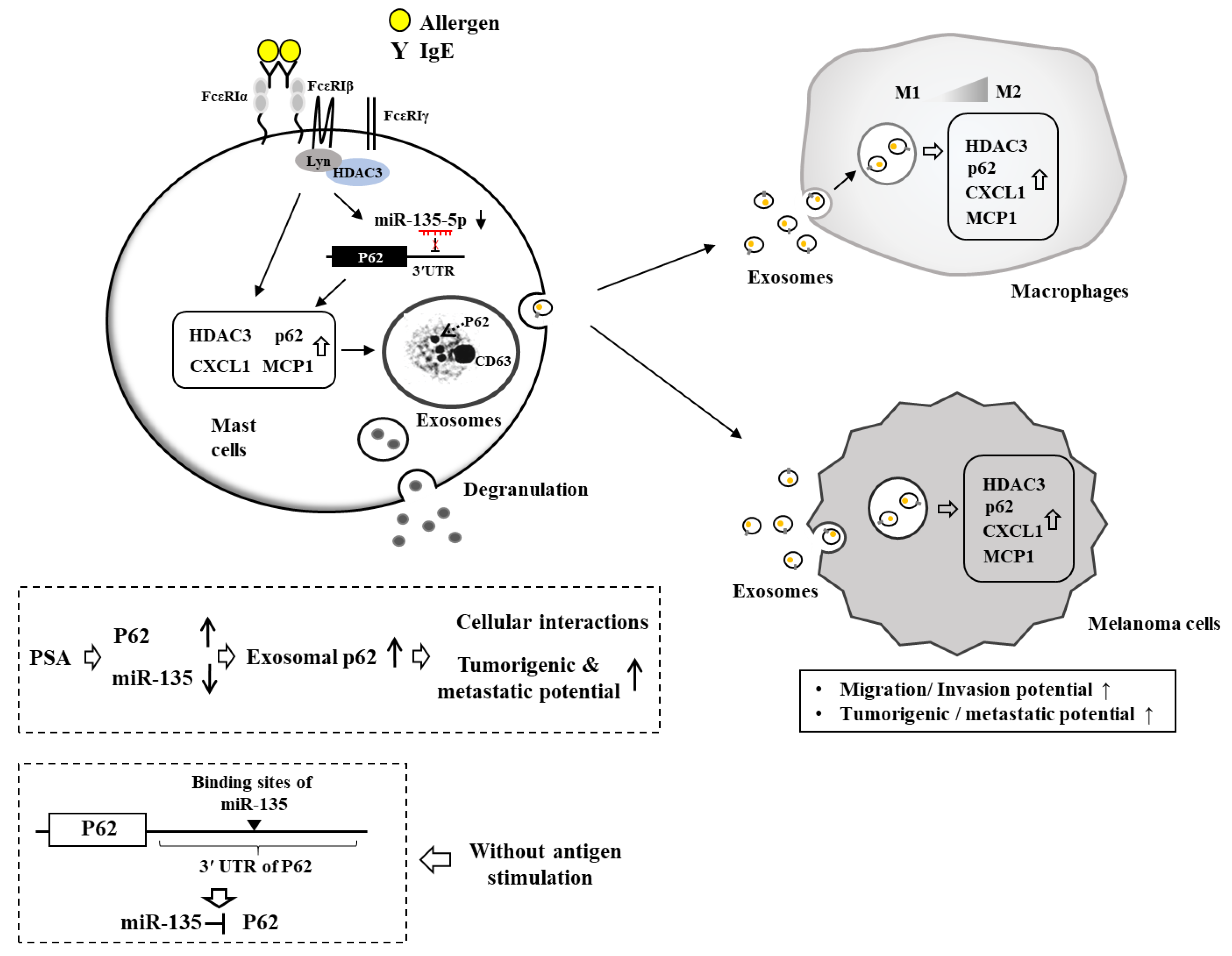
7. HDAC3, miRNAs, MCP1, and Cellular Interactions in Anaphylaxis
8. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kraft, S.; Kinet, J.P. New developments in FcepsilonRI regulation, function and inhibition. Nat. Rev. Immunol. 2007, 7, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J. Recruiting CD33 on mast cells to inhibit IgE-mediated mast cell-dependent anaphylaxis. J. Clin. Investig. 2019, 129, 955–957. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Dillahunt, S.E.; Rivera, J. Interrogation of sphingosine-1-phosphate receptor 2 function in vivo reveals a prominent role in the recovery from IgE and IgG-mediated anaphylaxis with minimal effect on its onset. Immunol. Lett. 2013, 150, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Park, Y.; Kwon, Y.; Kim, Y.; Byun, J.; Jeong, M.S.; Kim, H.U.; Jung, H.S.; Mun, J.Y.; Jeoung, D. MiR-135-5p-p62 Axis Regulates Autophagic Flux, Tumorigenic Potential, and Cellular Interactions Mediated by Extracellular Vesicles During Allergic Inflammation. Front. Immunol. 2019, 10, 738. [Google Scholar] [CrossRef] [PubMed]
- Voehringer, D. Protective and pathological roles of mast cells and basophils. Nat. Rev. Immunol. 2013, 13, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Huang, S.K. Metformin inhibits IgE- and aryl hydrocarbon receptor-mediated mast cell activation in vitro and in vivo. Eur. J. Immunol. 2018, 48, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.; Kim, M.; Kim, Y.; Kim, H.; Kim, H.; Byun, J.; Park, Y.; Lee, H.; Lee, Y.S.; Choe, J.; et al. miR-122-SOCS1-JAK2 axis regulates allergic inflammation and allergic inflammation-promoted cellular interactions. Oncotarget 2017, 8, 63155–63176. [Google Scholar] [CrossRef] [PubMed]
- Falanga, Y.T.; Chaimowitz, N.S.; Charles, N.; Finkelman, F.D.; Pullen, N.A.; Barbour, S.; Dholaria, K.; Faber, T.; Kolawole, M.; Huang, B.; et al. Lyn but not Fyn kinase controls IgG-mediated systemic anaphylaxis. J. Immunol. 2012, 188, 4360–4368. [Google Scholar] [CrossRef]
- Eom, S.; Kim, Y.; Park, D.; Lee, H.; Lee, Y.S.; Choe, J.; Kim, Y.M.; Jeoung, D. Histone deacetylase-3 mediates positive feedback relationship between anaphylaxis and tumor metastasis. J. Biol. Chem. 2014, 289, 12126–12144. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, K.; Park, D.; Lee, E.; Lee, H.; Lee, Y.S.; Choe, J.; Kim, Y.M.; Jeoung, D. DNA methyl transferase I acts as a negative regulator of allergic skin inflammation. Mol. Immunol. 2013, 53, 1–14. [Google Scholar] [CrossRef]
- Ren, Y.; Su, X.; Kong, L.; Li, M.; Zhao, X.; Yu, N.; Kang, J. Therapeutic effects of histone deacetylase inhibitors in a murine asthma model. Inflamm. Res. 2016, 65, 995–1008. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.P.; Wang, L.; Fu, J.J.; Fan, T.; Wang, Z.L.; Wang, G. Association between histone hyperacetylation status in memory T lymphocytes and allergen-induced eosinophilic airway inflammation. Respirology 2016, 21, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Grausenburger, R.; Bilic, I.; Boucheron, N.; Zupkovitz, G.; El-Housseiny, L.; Tschismarov, R.; Zhang, Y.; Rembold, M.; Gaisberger, M.; Hartl, A.; et al. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J. Immunol. 2010, 185, 3489–3497. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Xu, H.; Dai, W.; Zhu, C.; Wu, L.; Yan, S.; Ge, X.; Zhou, W.; Chen, C.; Dai, Y. The role of HDAC2 in cigarette smoke-induced airway inflammation in a murine model of asthma and the effect of intervention with roxithromycin. J. Asthma. 2018, 55, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, K.; Park, D.; Lee, E.; Lee, H.; Lee, Y.S.; Choe, J.; Jeoung, D. Histone deacetylase 3 mediates allergic skin inflammation by regulating expression of MCP1 protein. J. Biol. Chem. 2012, 287, 25844–25859. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, Y.S.; Hahn, J.H.; Choe, J.; Kwon, H.J.; Ro, J.Y.; Jeoung, D. Hyaluronic acid targets CD44 and inhibits FcepsilonRI signaling involving PKCdelta, Rac1, ROS, and MAPK to exert anti-allergic effect. Mol. Immunol. 2008, 45, 2537–2547. [Google Scholar] [CrossRef]
- Park, D.; Kim, Y.; Kim, H.; Kim, K.; Lee, Y.S.; Choe, J.; Hahn, J.H.; Lee, H.; Jeon, J.; Choi, C.; et al. Hyaluronic acid promotes angiogenesis by inducing RHAMM-TGFbeta receptor interaction via CD44-PKCdelta. Mol. Cells 2012, 33, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.R.; Ando, A.; Takeishi, T.; Katona, I.M.; Drazen, J.M.; Galli, S.J. Mast cells contribute to the changes in heart rate, but not hypotension or death, associated with active anaphylaxis in mice. J. Immunol. 1993, 151, 367–376. [Google Scholar] [PubMed]
- Ji, N.; Pan, S.; Shao, C.; Chen, Y.; Zhang, Z.; Wang, R.; Qiu, Y.; Jin, M.; Kong, D. Spinacetin Suppresses the Mast Cell Activation and Passive Cutaneous Anaphylaxis in Mouse Model. Front. Pharmacol. 2018, 9, 824. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Li, X.; Deng, Y.; Timilshina, M.; Huang, B.; Kim, D.Y.; Chang, J.H.; Ichinose, H.; Baek, S.H.; Murakami, M.; et al. The orphan nuclear receptor NR4A1 promotes FcepsilonRI-stimulated mast cell activation and anaphylaxis by counteracting the inhibitory LKB1/AMPK axis. Allergy 2019, 74, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; McKell, M.; Dang, A.; Yamani, A.; Waggoner, L.; Vanoni, S.; Noah, T.; Wu, D.; Kordowski, A.; Kohl, J.; et al. Lipopolysaccharide suppresses IgE-mast cell-mediated reactions. Clin. Exp. Allergy 2017, 47, 1574–1585. [Google Scholar] [CrossRef] [PubMed]
- Abebayehu, D.; Spence, A.J.; Caslin, H.; Taruselli, M.; Haque, T.T.; Kiwanuka, K.N.; Kolawole, E.M.; Chumanevich, A.P.; Sell, S.A.; Oskeritzian, C.A.; et al. Lactic acid suppresses IgE-mediated mast cell function in vitro and in vivo. Cell Immunol. 2019, 341, 103918. [Google Scholar] [CrossRef] [PubMed]
- Broesby-Olsen, S.; Vestergaard, H.; Mortz, C.G.; Jensen, B.; Havelund, T.; Hermann, A.P.; Siebenhaar, F.; Moller, M.B.; Kristensen, T.K.; Bindslev-Jensen, C. Omalizumab prevents anaphylaxis and improves symptoms in systemic mastocytosis: Efficacy and safety observations. Allergy 2018, 73, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Constantine, G.M.; Bressler, P.B.; Petroni, D.; Metcalfe, D.D.; Carter, M.C. Twelve-year follow-up of omalizumab therapy for anaphylaxis in 2 patients with systemic mastocytosis. J. Allergy Clin. Immunol. Pract. 2019, 7, 1314–1316. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.J.; Bindslev-Jensen, C. Successful treatment with omalizumab in challenge confirmed exercise-induced anaphylaxis. J. Allergy Clin. Immunol. Pract. 2017, 5, 204–206. [Google Scholar] [CrossRef] [PubMed]
- McLeod, J.J.A.; Caslin, H.L.; Spence, A.J.; Kolawole, E.M.; Qayum, A.A.; Paranjape, A.; Taruselli, M.; Haque, T.T.; Kiwanuka, K.N.; Elford, H.L.; et al. Didox (3,4-dihydroxybenzohydroxamic acid) suppresses IgE-mediated mast cell activation through attenuation of NFkappaB and AP-1 transcription. Cell Immunol. 2017, 322, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Bahri, R.; Custovic, A.; Korosec, P.; Tsoumani, M.; Barron, M.; Wu, J.; Sayers, R.; Weimann, A.; Ruiz-Garcia, M.; Patel, N.; et al. Mast cell activation test in the diagnosis of allergic disease and anaphylaxis. J. Allergy Clin. Immunol. 2018, 142, 485–496.e416. [Google Scholar] [CrossRef]
- Yamazaki, T.; Inui, M.; Hiemori, K.; Tomono, S.; Itoh, M.; Ichimonji, I.; Nakashima, A.; Takagi, H.; Biswas, M.; Izawa, K.; et al. Receptor-destroying enzyme (RDE) from Vibrio cholerae modulates IgE activity and reduces the initiation of anaphylaxis. J. Biol. Chem. 2019, 294, 6659–6669. [Google Scholar] [CrossRef]
- Sharma, S.; Tomar, S.; Dharne, M.; Ganesan, V.; Smith, A.; Yang, Y.; Waggoner, L.; Wang, Y.H.; Hogan, S.P. Deletion of DeltadblGata motif leads to increased predisposition and severity of IgE-mediated food-induced anaphylaxis response. PLoS ONE 2019, 14, e0219375. [Google Scholar] [CrossRef]
- Qian, F.; Zhang, L.; Lu, S.; Mao, G.; Guo, F.; Liu, P.; Xu, J.; Li, Y. Scrodentoid A Inhibits Mast Cell-Mediated Allergic Response by Blocking the Lyn-FcepsilonRIbeta Interaction. Front. Immunol. 2019, 10, 1103. [Google Scholar] [CrossRef]
- Dombrowicz, D.; Flamand, V.; Brigman, K.K.; Koller, B.H.; Kinet, J.P. Abolition of anaphylaxis by targeted disruption of the high affinity immunoglobulin E receptor alpha chain gene. Cell 1993, 75, 969–976. [Google Scholar] [CrossRef]
- Feyerabend, T.B.; Weiser, A.; Tietz, A.; Stassen, M.; Harris, N.; Kopf, M.; Radermacher, P.; Moller, P.; Benoist, C.; Mathis, D.; et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity 2011, 35, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Wershil, B.K.; Wang, Z.S.; Gordon, J.R.; Galli, S.J. Recruitment of neutrophils during IgE-dependent cutaneous late phase reactions in the mouse is mast cell-dependent. Partial inhibition of the reaction with antiserum against tumor necrosis factor-alpha. J. Clin. Invest. 1991, 87, 446–453. [Google Scholar] [CrossRef]
- Kim, D.E.; Min, K.J.; Kim, M.J.; Kim, S.H.; Kwon, T.K. Hispidulin Inhibits Mast Cell-Mediated Allergic Inflammation through Down-Regulation of Histamine Release and Inflammatory Cytokines. Molecules 2019, 24, 2131. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, A.; Fernandez-Hernando, C.; Cirino, G.; Sessa, W.C. Akt1 is critical for acute inflammation and histamine-mediated vascular leakage. Proc. Natl. Acad. Sci. USA 2009, 106, 14552–14557. [Google Scholar] [CrossRef] [Green Version]
- Oschatz, C.; Maas, C.; Lecher, B.; Jansen, T.; Bjorkqvist, J.; Tradler, T.; Sedlmeier, R.; Burfeind, P.; Cichon, S.; Hammerschmidt, S.; et al. Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity 2011, 34, 258–268. [Google Scholar] [CrossRef]
- Bender, L.; Weidmann, H.; Rose-John, S.; Renne, T.; Long, A.T. Factor XII-Driven Inflammatory Reactions with Implications for Anaphylaxis. Front. Immunol. 2017, 8, 1115. [Google Scholar] [CrossRef]
- Silwal, P.; Shin, K.; Choi, S.; Kang, S.W.; Park, J.B.; Lee, H.J.; Koo, S.J.; Chung, K.H.; Namgung, U.; Lim, K.; et al. Adenine suppresses IgE-mediated mast cell activation. Mol. Immunol. 2015, 65, 242–249. [Google Scholar] [CrossRef]
- Jonsson, F.; de Chaisemartin, L.; Granger, V.; Gouel-Cheron, A.; Gillis, C.M.; Zhu, Q.; Dib, F.; Nicaise-Roland, P.; Ganneau, C.; Hurtado-Nedelec, M.; et al. An IgG-induced neutrophil activation pathway contributes to human drug-induced anaphylaxis. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Kow, A.S.F.; Chik, A.; Soo, K.M.; Khoo, L.W.; Abas, F.; Tham, C.L. Identification of Soluble Mediators in IgG-Mediated Anaphylaxis via Fcgamma Receptor: A Meta-Analysis. Front. Immunol. 2019, 10, 190. [Google Scholar] [CrossRef]
- Kajiwara, N.; Sasaki, T.; Bradding, P.; Cruse, G.; Sagara, H.; Ohmori, K.; Saito, H.; Ra, C.; Okayama, Y. Activation of human mast cells through the platelet-activating factor receptor. J. Allergy Clin. Immunol. 2010, 125, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Schafer, B.; Piliponsky, A.M.; Oka, T.; Song, C.H.; Gerard, N.P.; Gerard, C.; Tsai, M.; Kalesnikoff, J.; Galli, S.J. Mast cell anaphylatoxin receptor expression can enhance IgE-dependent skin inflammation in mice. J. Allergy Clin. Immunol. 2013, 131, 541–548.e541–549. [Google Scholar] [CrossRef] [PubMed]
- Kordowski, A.; Reinicke, A.T.; Wu, D.; Orinska, Z.; Hagemann, P.; Huber-Lang, M.; Lee, J.B.; Wang, Y.H.; Hogan, S.P.; Kohl, J. C5a receptor 1(-/-) mice are protected from the development of IgE-mediated experimental food allergy. Allergy 2019, 74, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Arias, K.; Chu, D.K.; Flader, K.; Botelho, F.; Walker, T.; Arias, N.; Humbles, A.A.; Coyle, A.J.; Oettgen, H.C.; Chang, H.D.; et al. Distinct immune effector pathways contribute to the full expression of peanut-induced anaphylactic reactions in mice. J. Allergy Clin. Immunol. 2011, 127, 1552–1561.e1551. [Google Scholar] [CrossRef]
- Smit, J.J.; Willemsen, K.; Hassing, I.; Fiechter, D.; Storm, G.; van Bloois, L.; Leusen, J.H.; Pennings, M.; Zaiss, D.; Pieters, R.H. Contribution of classic and alternative effector pathways in peanut-induced anaphylactic responses. PLoS ONE 2011, 6, e28917. [Google Scholar] [CrossRef]
- Beutier, H.; Gillis, C.M.; Iannascoli, B.; Godon, O.; England, P.; Sibilano, R.; Reber, L.L.; Galli, S.J.; Cragg, M.S.; Van Rooijen, N.; et al. IgG subclasses determine pathways of anaphylaxis in mice. J. Allergy Clin. Immunol. 2017, 139, 269–280.e267. [Google Scholar] [CrossRef]
- Strait, R.T.; Morris, S.C.; Yang, M.; Qu, X.W.; Finkelman, F.D. Pathways of anaphylaxis in the mouse. J. Allergy Clin. Immunol. 2002, 109, 658–668. [Google Scholar] [CrossRef]
- Guilliams, M.; Bruhns, P.; Saeys, Y.; Hammad, H.; Lambrecht, B.N. The function of Fcgamma receptors in dendritic cells and macrophages. Nat. Rev. Immunol. 2014, 14, 94–108. [Google Scholar] [CrossRef]
- Bohlson, S.S.; O’Conner, S.D.; Hulsebus, H.J.; Ho, M.M.; Fraser, D.A. Complement, c1q, and c1q-related molecules regulate macrophage polarization. Front. Immunol. 2014, 5, 402. [Google Scholar] [CrossRef]
- Balbino, B.; Sibilano, R.; Starkl, P.; Marichal, T.; Gaudenzio, N.; Karasuyama, H.; Bruhns, P.; Tsai, M.; Reber, L.L.; Galli, S.J. Pathways of immediate hypothermia and leukocyte infiltration in an adjuvant-free mouse model of anaphylaxis. J. Allergy Clin. Immunol. 2017, 139, 584–596.e510. [Google Scholar] [CrossRef]
- Jiao, D.; Liu, Y.; Lu, X.; Liu, B.; Pan, Q.; Liu, Y.; Liu, Y.; Zhu, P.; Fu, N. Macrophages are the dominant effector cells responsible for IgG-mediated passive systemic anaphylaxis challenged by natural protein antigen in BALB/c and C57BL/6 mice. Cell Immunol. 2014, 289, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Kandhare, A.D.; Liu, Z.; Mukherjee, A.A.; Bodhankar, S.L. Therapeutic Potential of Morin in Ovalbumin-induced Allergic Asthma Via Modulation of SUMF2/IL-13 and BLT2/NF-κB Signaling Pathway. Curr. Mol. Pharmacol. 2019, 12, 122–138. [Google Scholar] [CrossRef] [PubMed]
- Kinney, S.R.; Carlson, L.; Ser-Dolansky, J.; Thompson, C.; Shah, S.; Gambrah, A.; Xing, W.; Schneider, S.S.; Mathias, C.B. Curcumin Ingestion Inhibits Mastocytosis and Suppresses Intestinal Anaphylaxis in a Murine Model of Food Allergy. PLoS ONE 2015, 10, e0132467. [Google Scholar] [CrossRef] [PubMed]
- Uto, T.; Takagi, H.; Fukaya, T.; Nasu, J.; Fukui, T.; Miyanaga, N.; Arimura, K.; Nakamura, T.; Choijookhuu, N.; Hishikawa, Y.; et al. Critical role of plasmacytoid dendritic cells in induction of oral tolerance. J. Allergy Clin. Immunol. 2018, 141, 2156–2167.e2159. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, A.; Kawamura, T.; Kanbe, K.; Kanamaru, Y.; Ogawa, H.; Okumura, K.; Nakao, A. Suppression of serum IgE response and systemic anaphylaxis in a food allergy model by orally administered high-dose TGF-beta. Int. Immunol. 2005, 17, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Tordesillas, L.; Mondoulet, L.; Blazquez, A.B.; Benhamou, P.H.; Sampson, H.A.; Berin, M.C. Epicutaneous immunotherapy induces gastrointestinal LAP(+) regulatory T cells and prevents food-induced anaphylaxis. J. Allergy Clin. Immunol. 2017, 139, 189–201.e184. [Google Scholar] [CrossRef] [PubMed]
- Schiavi, E.; Barletta, B.; Butteroni, C.; Corinti, S.; Boirivant, M.; Di Felice, G. Oral therapeutic administration of a probiotic mixture suppresses established Th2 responses and systemic anaphylaxis in a murine model of food allergy. Allergy 2011, 66, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Ganeshan, K.; Neilsen, C.V.; Hadsaitong, A.; Schleimer, R.P.; Luo, X.; Bryce, P.J. Impairing oral tolerance promotes allergy and anaphylaxis: a new murine food allergy model. J. Allergy Clin. Immunol. 2009, 123, 231–238.e234. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, M.; Cervantes-Garcia, D.; Munoz, Y.H.; Garcia, A.; Haro, L.M., Jr.; Salinas, E. Novel Mechanisms Underlying the Therapeutic Effect of Glycomacropeptide on Allergy: Change in Gut Microbiota, Upregulation of TGF-beta, and Inhibition of Mast Cells. Int. Arch. Allergy Immunol. 2016, 171, 217–226. [Google Scholar] [CrossRef]
- Kim, M.; Lee, S.H.; Kim, Y.; Kwon, Y.; Park, Y.; Lee, H.K.; Jung, H.S.; Jeoung, D. Human Adipose Tissue-Derived Mesenchymal Stem Cells Attenuate Atopic Dermatitis by Regulating the Expression of MIP-2, miR-122a-SOCS1 Axis, and Th1/Th2 Responses. Front. Pharmacol 2018, 9, 1175. [Google Scholar] [CrossRef] [Green Version]
- Nico, B.; Mangieri, D.; Crivellato, E.; Vacca, A.; Ribatti, D. Mast cells contribute to vasculogenic mimicry in multiple myeloma. Stem Cells Dev. 2008, 17, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Eissmann, M.F.; Dijkstra, C.; Jarnicki, A.; Phesse, T.; Brunnberg, J.; Poh, A.R.; Etemadi, N.; Tsantikos, E.; Thiem, S.; Huntington, N.D.; et al. IL-33-mediated mast cell activation promotes gastric cancer through macrophage mobilization. Nat. Commun. 2019, 10, 2735. [Google Scholar] [CrossRef] [PubMed]
- Gatla, H.R.; Muniraj, N.; Thevkar, P.; Yavvari, S.; Sukhavasi, S.; Makena, M.R. Regulation of Chemokines and Cytokines by Histone Deacetylases and an Update on Histone Decetylase Inhibitors in Human Diseases. Int. J. Mol. Sci. 2019, 20, 1110. [Google Scholar] [CrossRef] [PubMed]
- Chun, P. Therapeutic effects of histone deacetylase inhibitors on kidney disease. Arch. Pharm. Res. 2018, 41, 162–183. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Mahlknecht, U.; Emiliani, S.; Najfeld, V.; Young, S.; Verdin, E. Genomic organization and chromosomal localization of the human histone deacetylase 3 gene. Genomics 1999, 56, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Longworth, M.S.; Laimins, L.A. Histone deacetylase 3 localizes to the plasma membrane and is a substrate of Src. Oncogene 2006, 25, 4495–4500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yuan, Z.; Zhang, Y.; Yong, S.; Salas-Burgos, A.; Koomen, J.; Olashaw, N.; Parsons, J.T.; Yang, X.J.; Dent, S.R.; et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell 2007, 27, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Soriano, F.X.; Chawla, S.; Skehel, P.; Hardingham, G.E. SMRT-mediated co-shuttling enables export of class IIa HDACs independent of their CaM kinase phosphorylation sites. J. Neurochem. 2013, 124, 26–35. [Google Scholar] [CrossRef]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Ocker, M. Deacetylase inhibitors - focus on non-histone targets and effects. World J. Biol. Chem. 2010, 1, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Ververis, K.; Hiong, A.; Karagiannis, T.C.; Licciardi, P.V. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biologics 2013, 7, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Yang, C.; Mine, Y. Multiple T cell epitope peptides suppress allergic responses in an egg allergy mouse model by the elicitation of forkhead box transcription factor 3- and transforming growth factor-beta-associated mechanisms. Clin. Exp. Allergy 2010, 40, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, S.; Xiao, L.; Nie, J.; Zhang, X.; Xu, M.; Li, J.; Wong, J.; Seto, E.; Yang, X.J. Histone deacetylase 3 interacts with and deacetylates myocyte enhancer factor 2. Mol. Cell Biol. 2007, 27, 1280–1295. [Google Scholar] [CrossRef] [PubMed]
- Steelant, B.; Wawrzyniak, P.; Martens, K.; Jonckheere, A.C.; Pugin, B.; Schrijvers, R.; Bullens, D.M.; Vanoirbeek, J.A.; Krawczyk, K.; Dreher, A.; et al. Blocking histone deacetylase activity as a novel target for epithelial barrier defects in patients with allergic rhinitis. J. Allergy Clin. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, P.; Ahmad, T.; Adcock, I.M. The role of histone deacetylases in asthma and allergic diseases. J. Allergy Clin. Immunol. 2008, 121, 580–584. [Google Scholar] [CrossRef]
- Choi, J.H.; Oh, S.W.; Kang, M.S.; Kwon, H.J.; Oh, G.T.; Kim, D.Y. Trichostatin A attenuates airway inflammation in mouse asthma model. Clin. Exp. Allergy 2005, 35, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Stefanowicz, D.; Lee, J.Y.; Lee, K.; Shaheen, F.; Koo, H.K.; Booth, S.; Knight, D.A.; Hackett, T.L. Elevated H3K18 acetylation in airway epithelial cells of asthmatic subjects. Respir Res. 2015, 16, 95. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, F.; Li, X.; Wang, L.W.; Chu, X.G.; Zhang, H.; Gong, Z.J. Trichostatin A Protects Against Experimental Acute-on-Chronic Liver Failure in Rats Through Regulating the Acetylation of Nuclear Factor-kappaB. Inflammation 2015, 38, 1364–1373. [Google Scholar] [CrossRef]
- Imre, G.; Gekeler, V.; Leja, A.; Beckers, T.; Boehm, M. Histone deacetylase inhibitors suppress the inducibility of nuclear factor-kappaB by tumor necrosis factor-alpha receptor-1 down-regulation. Cancer Res. 2006, 66, 5409–5418. [Google Scholar] [CrossRef]
- Magalhaes, G.S.; Barroso, L.C.; Reis, A.C.; Rodrigues-Machado, M.G.; Gregorio, J.F.; Motta-Santos, D.; Oliveira, A.C.; Perez, D.A.; Barcelos, L.S.; Teixeira, M.M.; et al. Angiotensin-(1-7) Promotes Resolution of Eosinophilic Inflammation in an Experimental Model of Asthma. Front. Immunol. 2018, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Meng, J.X.; Liu, Z.; Liu, X.W.; Huang, Y.G.; Zhao, J. Propofol Attenuates Airway Inflammation in a Mast Cell-Dependent Mouse Model of Allergic Asthma by Inhibiting the Toll-like Receptor 4/Reactive Oxygen Species/Nuclear Factor kappaB Signaling Pathway. Inflammation 2018, 41, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Piao, H.; Jiang, J.; Jin, G.; Zheng, M.; Yang, J.; Jin, X.; Sun, T.; Choi, Y.H.; Li, L.; et al. Polydatin inhibits mast cell-mediated allergic inflammation by targeting PI3K/Akt, MAPK, NF-kappaB and Nrf2/HO-1 pathways. Sci. Rep. 2017, 7, 11895. [Google Scholar] [CrossRef] [PubMed]
- Grabiec, A.M.; Krausz, S.; de Jager, W.; Burakowski, T.; Groot, D.; Sanders, M.E.; Prakken, B.J.; Maslinski, W.; Eldering, E.; Tak, P.P.; et al. Histone deacetylase inhibitors suppress inflammatory activation of rheumatoid arthritis patient synovial macrophages and tissue. J. Immunol. 2010, 184, 2718–2728. [Google Scholar] [CrossRef] [PubMed]
- Adcock, I.M.; Ford, P.; Ito, K.; Barnes, P.J. Epigenetics and airways disease. Respir Res. 2006, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wen, L.; Wang, Y.; Chen, F. Therapeutic Effect of Histone Deacetylase Inhibitor, Sodium Butyrate, on Allergic Rhinitis In Vivo. DNA Cell Biol. 2016, 35, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Xian, Z.; Jin, G.; Li, H.; Jiang, J.; Wang, C.; Zhu, L.; Jin, Z.; Li, L.; Piao, H.; Zheng, M.; et al. Imperatorin Suppresses Anaphylactic Reaction and IgE-Mediated Allergic Responses by Inhibiting Multiple Steps of FceRI Signaling in Mast Cells: IMP Alleviates Allergic Responses in PCA. Biomed. Res. Int. 2019, 2019, 7823761. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lee, Y.J.; Jin, F.; Park, Y.N.; Deng, Y.; Kang, Y.; Yang, J.H.; Chang, J.H.; Kim, D.Y.; Kim, J.A.; et al. Sirt1 negatively regulates FcepsilonRI-mediated mast cell activation through AMPK- and PTP1B-dependent processes. Sci. Rep. 2017, 7, 6444. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Park, S.J.; Jin, F.; Deng, Y.; Yang, J.H.; Chang, J.H.; Kim, D.Y.; Kim, J.A.; Lee, Y.J.; Murakami, M.; et al. Tanshinone IIA suppresses FcepsilonRI-mediated mast cell signaling and anaphylaxis by activation of the Sirt1/LKB1/AMPK pathway. Biochem. Pharmacol. 2018, 152, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Li, L.B.; Leung, D.Y.; Martin, R.J.; Goleva, E. Inhibition of histone deacetylase 2 expression by elevated glucocorticoid receptor beta in steroid-resistant asthma. Am. J. Respir. Crit. Care Med. 2010, 182, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.Y.; Horvat, J.C.; Pinkerton, J.W.; Starkey, M.R.; Essilfie, A.T.; Mayall, J.R.; Nair, P.M.; Hansbro, N.G.; Jones, B.; Haw, T.J.; et al. MicroRNA-21 drives severe, steroid-insensitive experimental asthma by amplifying phosphoinositide 3-kinase-mediated suppression of histone deacetylase 2. J. Allergy Clin. Immunol. 2017, 139, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Caramori, G.; Lim, S.; Oates, T.; Chung, K.F.; Barnes, P.J.; Adcock, I.M. Expression and activity of histone deacetylases in human asthmatic airways. Am. J. Respir Crit. Care Med. 2002, 166, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.; Wu, M.; Zhang, C.; Che, L.; Xu, F.; Wang, Y.; Wu, Y.; Xuan, N.; Cao, C.; Du, X.; et al. HDAC2 attenuates airway inflammation by suppressing IL-17A production in HDM-challenged mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L269–L279. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Kang, J.H.; Shin, J.M.; Lee, H.M. Trichostatin A Inhibits Epithelial Mesenchymal Transition Induced by TGF-beta1 in Airway Epithelium. PLoS ONE 2016, 11, e0162058. [Google Scholar] [CrossRef]
- Wen, Y.D.; Perissi, V.; Staszewski, L.M.; Yang, W.M.; Krones, A.; Glass, C.K.; Rosenfeld, M.G.; Seto, E. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc. Natl. Acad. Sci. USA 2000, 97, 7202–7207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, S.; Hu, Q.; Zhang, H.; Yang, F.; Peng, C.; Huang, C. HDAC3 Inhibition Upregulates PD-L1 Expression in B-Cell Lymphomas and Augments the Efficacy of Anti-PD-L1 Therapy. Mol. Cancer Ther. 2019, 18, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Angiolilli, C.; Kabala, P.A.; Grabiec, A.M.; Van Baarsen, I.M.; Ferguson, B.S.; Garcia, S.; Malvar Fernandez, B.; McKinsey, T.A.; Tak, P.P.; Fossati, G.; et al. Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast-like synoviocytes. Ann. Rheum Dis. 2017, 76, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.; Du, R.; Zhu, X.; Yin, S.; Wang, J.; Cui, H.; Cao, W.; Lowenstein, C.J. Histone deacetylase isoforms regulate innate immune responses by deacetylating mitogen-activated protein kinase phosphatase-1. J. Leukoc Biol. 2014, 95, 651–659. [Google Scholar] [CrossRef]
- Chen, X.; Barozzi, I.; Termanini, A.; Prosperini, E.; Recchiuti, A.; Dalli, J.; Mietton, F.; Matteoli, G.; Hiebert, S.; Natoli, G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E2865–E2874. [Google Scholar] [CrossRef] [Green Version]
- Leus, N.G.; van der Wouden, P.E.; van den Bosch, T.; Hooghiemstra, W.T.R.; Ourailidou, M.E.; Kistemaker, L.E.; Bischoff, R.; Gosens, R.; Haisma, H.J.; Dekker, F.J. HDAC 3-selective inhibitor RGFP966 demonstrates anti-inflammatory properties in RAW 264.7 macrophages and mouse precision-cut lung slices by attenuating NF-kappaB p65 transcriptional activity. Biochem. Pharmacol. 2016, 108, 58–74. [Google Scholar] [CrossRef]
- Macglashan, D., Jr.; Moore, G.; Muchhal, U. Regulation of IgE-mediated signalling in human basophils by CD32b and its role in Syk down-regulation: basic mechanisms in allergic disease. Clin. Exp. Allergy 2014, 44, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Eom, S.; Kim, K.; Lee, Y.S.; Choe, J.; Hahn, J.H.; Lee, H.; Kim, Y.M.; Ha, K.S.; Ro, J.Y.; et al. Transglutaminase II interacts with rac1, regulates production of reactive oxygen species, expression of snail, secretion of Th2 cytokines and mediates in vitro and in vivo allergic inflammation. Mol. Immunol. 2010, 47, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Thullberg, M.; Zhang, H.; Onischenko, A.; Stromblad, S. Cell attachment to the extracellular matrix induces proteasomal degradation of p21(CIP1) via Cdc42/Rac1 signaling. Mol. Cell Biol. 2002, 22, 4587–4597. [Google Scholar] [CrossRef] [PubMed]
- Lores, P.; Visvikis, O.; Luna, R.; Lemichez, E.; Gacon, G. The SWI/SNF protein BAF60b is ubiquitinated through a signalling process involving Rac GTPase and the RING finger protein Unkempt. FEBS J. 2010, 277, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Adenuga, D.; Yao, H.; March, T.H.; Seagrave, J.; Rahman, I. Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am. J. Respir Cell Mol. Biol. 2009, 40, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Tu, H.Q.; Chang, Y.; Tan, B.; Wang, G.; Zhou, J.; Wang, L.; Mu, R.; Zhang, W.N. USP19 deubiquitinates HDAC1/2 to regulate DNA damage repair and control chromosomal stability. Oncotarget 2017, 8, 2197–2208. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Ebaugh, S.; Martens, A.; Gao, H.; Olson, E.; Ng, P.K.W.; Gangur, V. A Mouse Model of Anaphylaxis and Atopic Dermatitis to Salt-Soluble Wheat Protein Extract. Int. Arch. Allergy Immunol. 2017, 174, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, Z.; Xie, B.; Song, Y.; Ye, X.; Liu, P. hsa-miR-20a-5p attenuates allergic inflammation in HMC-1 cells by targeting HDAC4. Mol. Immunol. 2019, 107, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Bartel, S.; Carraro, G.; Alessandrini, F.; Krauss-Etschmann, S.; Ricciardolo, F.L.M.; Bellusci, S. miR-142-3p is associated with aberrant WNT signaling during airway remodeling in asthma. Am. J. Physiol Lung Cell Mol. Physiol 2018, 315, L328–L333. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Kim, Y.; Eom, S.; Kim, M.; Park, D.; Kim, H.; Noh, K.; Lee, H.; Lee, Y.S.; Choe, J.; et al. MicroRNA-26a/-26b-COX-2-MIP-2 Loop Regulates Allergic Inflammation and Allergic Inflammation-promoted Enhanced Tumorigenic and Metastatic Potential of Cancer Cells. J. Biol. Chem. 2015, 290, 14245–14266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qayum, A.A.; Paranjape, A.; Abebayehu, D.; Kolawole, E.M.; Haque, T.T.; McLeod, J.J.; Spence, A.J.; Caslin, H.L.; Taruselli, M.T.; Chumanevich, A.P.; et al. IL-10-Induced miR-155 Targets SOCS1 To Enhance IgE-Mediated Mast Cell Function. J. Immunol. 2016, 196, 4457–4467. [Google Scholar] [CrossRef]
- Li, Z.; Liang, Y.; Tang, H.; Luo, B.; Chen, Z.; Wu, J.; Yang, Q.; Ma, Z. Effect of anaphylactic shock on suppressors of cytokine signaling. Immunol. Invest. 2010, 39, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, K.; Park, D.; Eom, S.; Park, H.; Lee, H.; Lee, Y.S.; Choe, J.; Hahn, J.H.; Kim, Y.M.; et al. Integrin alpha(5) interacts with EGFR, is necessary for FcvarepsilonRI signaling and is necessary for allergic inflammation in relation with angiogenesis. Mol. Immunol. 2011, 48, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Pushparaj, P.N.; Tay, H.K.; H’Ng S, C.; Pitman, N.; Xu, D.; McKenzie, A.; Liew, F.Y.; Melendez, A.J. The cytokine interleukin-33 mediates anaphylactic shock. Proc. Natl. Acad. Sci. USA 2009, 106, 9773–9778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodoun, M.V.; Tomar, S.; Tocker, J.E.; Wang, Y.H.; Finkelman, F.D. Prevention of food allergy development and suppression of established food allergy by neutralization of thymic stromal lymphopoietin, IL-25, and IL-33. J. Allergy Clin. Immunol. 2018, 141, 171–179.e171. [Google Scholar] [CrossRef] [PubMed]
- Melgarejo, E.; Medina, M.A.; Sanchez-Jimenez, F.; Botana, L.M.; Dominguez, M.; Escribano, L.; Orfao, A.; Urdiales, J.L. (-)-Epigallocatechin-3-gallate interferes with mast cell adhesiveness, migration and its potential to recruit monocytes. Cell Mol. Life Sci. 2007, 64, 2690–2701. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Okamoto, N.; Kondo, M.; Arkwright, P.D.; Karasawa, K.; Ishizaka, S.; Yokota, S.; Matsuda, A.; Jung, K.; Oida, K.; et al. Mast cell hyperactivity underpins the development of oxygen-induced retinopathy. J. Clin. Invest. 2017, 127, 3987–4000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussens, L.M.; Raymond, W.W.; Bergers, G.; Laig-Webster, M.; Behrendtsen, O.; Werb, Z.; Caughey, G.H.; Hanahan, D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999, 13, 1382–1397. [Google Scholar] [CrossRef] [PubMed]
- Rabenhorst, A.; Schlaak, M.; Heukamp, L.C.; Forster, A.; Theurich, S.; von Bergwelt-Baildon, M.; Buttner, R.; Kurschat, P.; Mauch, C.; Roers, A.; et al. Mast cells play a protumorigenic role in primary cutaneous lymphoma. Blood 2012, 120, 2042–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.J.; Oh, H.A.; Nam, S.Y.; Han, N.R.; Kim, Y.S.; Kim, J.H.; Lee, S.J.; Kim, M.H.; Moon, P.D.; Kim, H.M.; et al. The critical role of mast cell-derived hypoxia-inducible factor-1alpha in human and mice melanoma growth. Int. J. Cancer 2013, 132, 2492–2501. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Yao, L.; Tosato, G. Mast cell-derived angiopoietin-1 plays a critical role in the growth of plasma cell tumors. J. Clin. Invest. 2004, 114, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Kobayashi, M.; Yano, K.; Miura, M.; Izumi, A.; Mataki, C.; Doi, T.; Hamakubo, T.; Reid, P.C.; Hume, D.A.; et al. Histone deacetylase inhibitor reduces monocyte adhesion to endothelium through the suppression of vascular cell adhesion molecule-1 expression. Arterioscler Thromb Vasc. Biol. 2006, 26, 2652–2659. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Wang, L.X.; Wang, M.; Cheng, C.; Li, S.; Shen, Q.; Fang, L.; Liu, R. MTOR-Mediated Autophagy Is Involved in the Protective Effect of Ketamine on Allergic Airway Inflammation. J. Immunol. Res. 2019, 2019, 5879714. [Google Scholar] [CrossRef] [PubMed]
- Ushio, H.; Ueno, T.; Kojima, Y.; Komatsu, M.; Tanaka, S.; Yamamoto, A.; Ichimura, Y.; Ezaki, J.; Nishida, K.; Komazawa-Sakon, S.; et al. Crucial role for autophagy in degranulation of mast cells. J. Allergy Clin. Immunol. 2011, 127, 1267–1276.e1266. [Google Scholar] [CrossRef] [PubMed]
- Xia, F.; Deng, C.; Jiang, Y.; Qu, Y.; Deng, J.; Cai, Z.; Ding, Y.; Guo, Z.; Wang, J. IL4 (interleukin 4) induces autophagy in B cells leading to exacerbated asthma. Autophagy 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Nakano, H.; Ushio, H. An unexpected role for autophagy in degranulation of mast cells. Autophagy 2011, 7, 657–659. [Google Scholar] [CrossRef] [Green Version]
- McAlinden, K.D.; Deshpande, D.A.; Ghavami, S.; Xenaki, D.; Sohal, S.S.; Oliver, B.G.; Haghi, M.; Sharma, P. Autophagy Activation in Asthma Airways Remodeling. Am. J. Respir Cell Mol. Biol. 2019, 60, 541–553. [Google Scholar] [CrossRef]
- Thapa, P.; Romero Arocha, S.; Chung, J.Y.; Sant’Angelo, D.B.; Shapiro, V.S. Histone deacetylase 3 is required for iNKT cell development. Sci. Rep. 2017, 7, 5784. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.; Kwon, Y.; Jung, H.S.; Kim, Y.; Jeoung, D. FcεRI-HDAC3-MCP1 Signaling Axis Promotes Passive Anaphylaxis Mediated by Cellular Interactions. Int. J. Mol. Sci. 2019, 20, 4964. https://doi.org/10.3390/ijms20194964
Kim M, Kwon Y, Jung HS, Kim Y, Jeoung D. FcεRI-HDAC3-MCP1 Signaling Axis Promotes Passive Anaphylaxis Mediated by Cellular Interactions. International Journal of Molecular Sciences. 2019; 20(19):4964. https://doi.org/10.3390/ijms20194964
Chicago/Turabian StyleKim, Misun, Yoojung Kwon, Hyun Suk Jung, Youngmi Kim, and Dooil Jeoung. 2019. "FcεRI-HDAC3-MCP1 Signaling Axis Promotes Passive Anaphylaxis Mediated by Cellular Interactions" International Journal of Molecular Sciences 20, no. 19: 4964. https://doi.org/10.3390/ijms20194964
APA StyleKim, M., Kwon, Y., Jung, H. S., Kim, Y., & Jeoung, D. (2019). FcεRI-HDAC3-MCP1 Signaling Axis Promotes Passive Anaphylaxis Mediated by Cellular Interactions. International Journal of Molecular Sciences, 20(19), 4964. https://doi.org/10.3390/ijms20194964