Tumour-Derived Extracellular Vesicles (EVs): A Dangerous “Message in A Bottle” for Bone



Abstract
1. Introduction
2. Bone and Tumours: Switching from the Virtuous to the Vicious Cycle
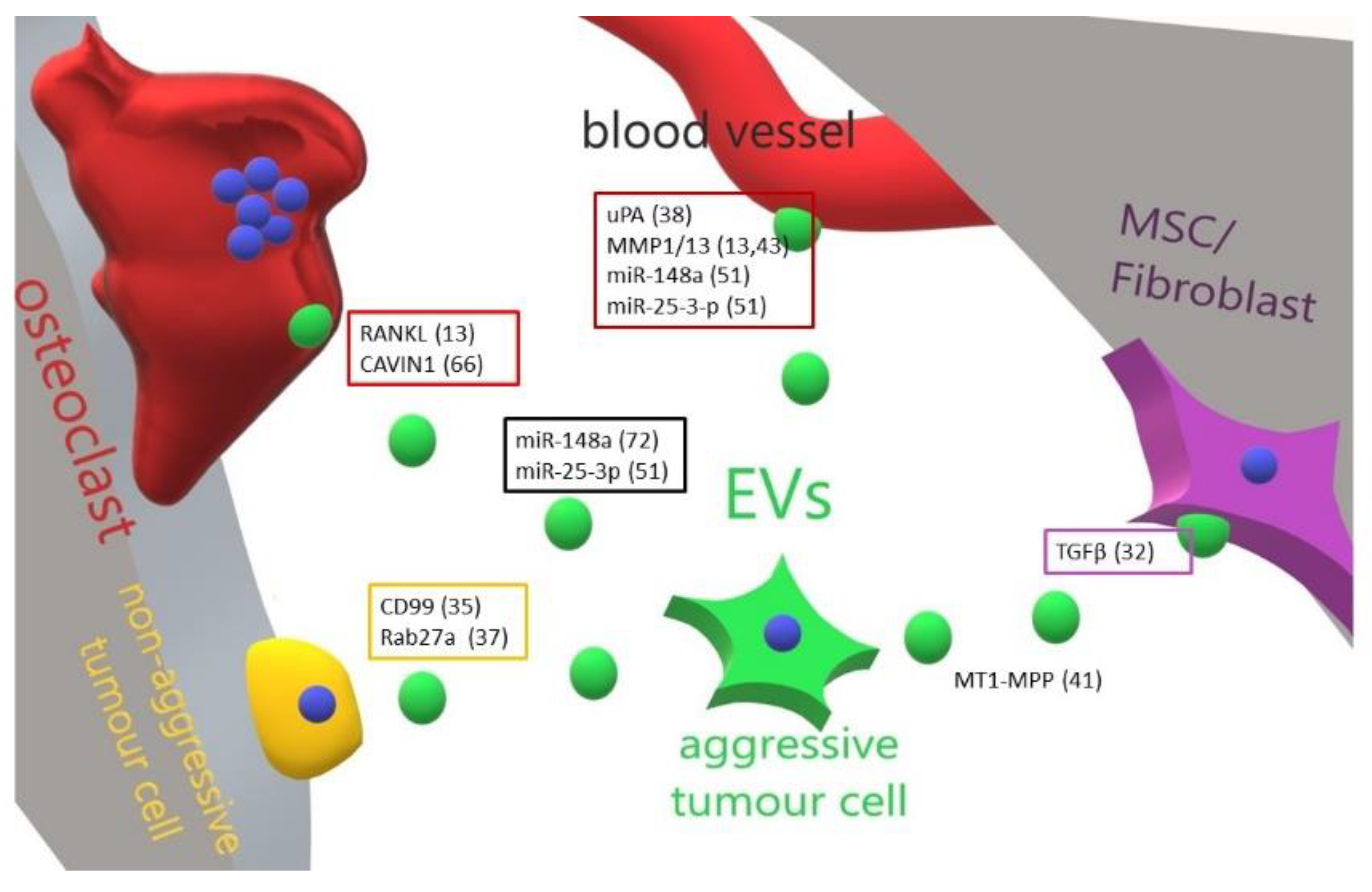
3. Role of Extracellular Vesicles in the Progression of Primary Bone Tumours
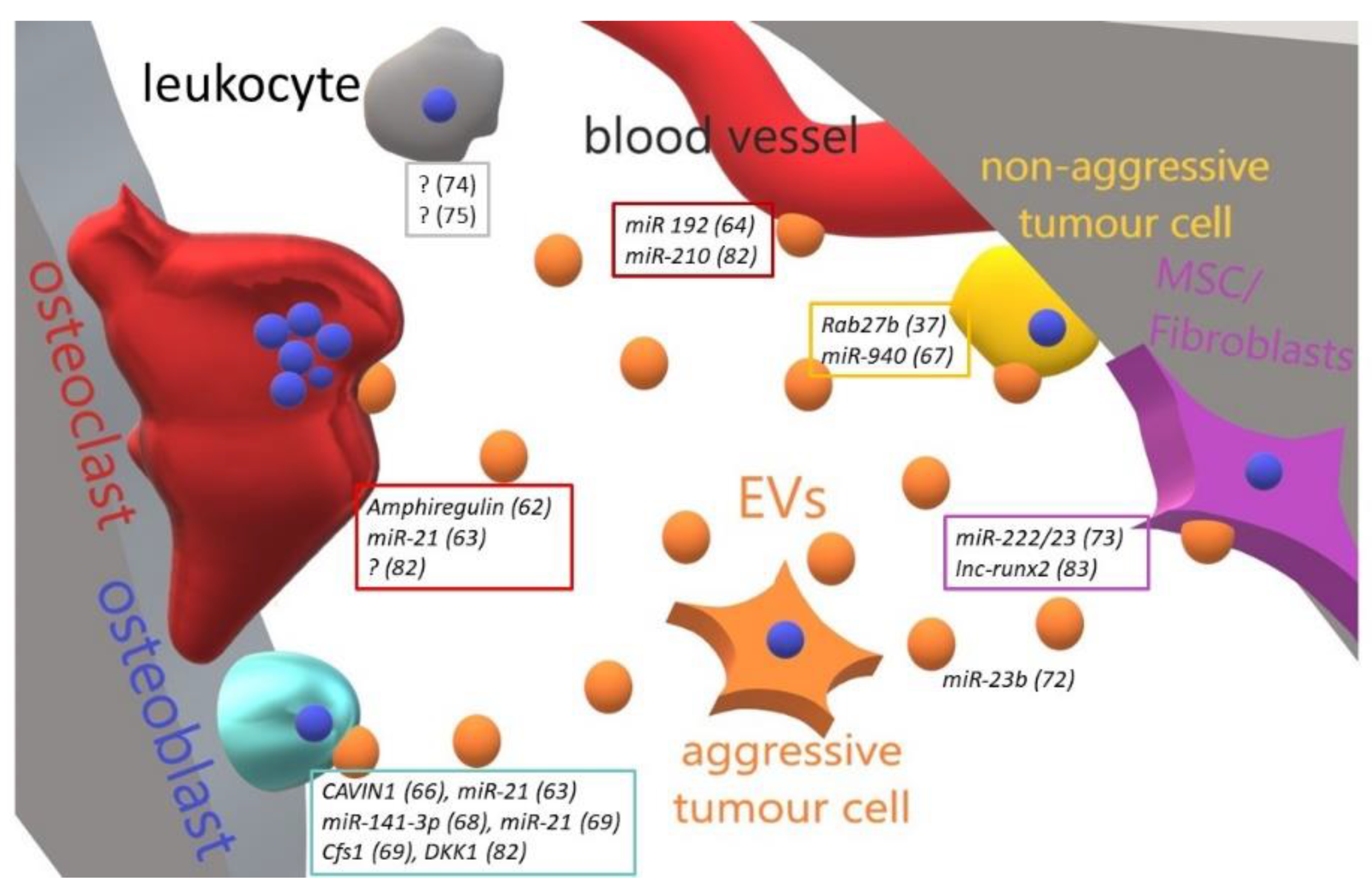
4. Bone Metastases and Extracellular Vesicles
5. Therapeutic Perspectives of Extracellular Vesicles
5.1. Targeting the Release of Cancer-Derived EVs
5.2. EVs as Biological Vehicles for Active Molecules
6. Conclusions
Funding
Conflicts of Interest
References
- Chargaff, E.; West, R. The biological significance of the thromboplastic protein of blood. J. Biol. Chem. 1946, 166, 189–197. [Google Scholar] [PubMed]
- Wolf, P. The Nature and Significance of Platelet Products in Human Plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.V.; Raposo, G.; Welsch, U.; Prazeres da Costa, O.; Thiel, U.; Lebar, M.; Maurer, M.; Bender, H.U.; von Luettichau, I.; Richter, G.H.; et al. First identification of Ewing’s sarcoma-derived extracellular vesicles and exploration of their biological and potential diagnostic implications. Biol. Cell 2013, 105, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Bobrie, A.; Théry, C. Unraveling the physiological functions of exosome secretion by tumors. Oncoimmunology 2013, 2, e22565. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, S.; EL Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Cicero, A.L.; Stahl, P.D.; Raposo, G. Extracellular vesicles shuffling intercellular messages: For good or for bad. Curr. Opin. Cell Biol. 2015, 35, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Kroemer, G.; Zitvogel, L. Extracellular vesicles: Masters of intercellular communication and potential clinical interventions. J. Clin. Investig. 2016, 126, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Cappariello, A.; Loftus, A.; Muraca, M.; Maurizi, A.; Rucci, N.; Teti, A. Osteoblast-Derived Extracellular Vesicles Are Biological Tools for the Delivery of Active Molecules to Bone. J. Bone Miner. Res. 2018, 33, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H. Vesicles associated with calcification in the matrix of epiphyseal cartilage. J. Cell 1969, 41, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Pieters, B.C.H.; Cappariello, A.; van den Bosch, M.H.J.; van Lent, P.L.E.M.; Teti, A.; van de Loo, F.A.J. Macrophage-Derived Extracellular Vesicles as Carriers of Alarmins and Their Potential Involvement in Bone Homeostasis. Front. Immunol. 2019, 10, 1901. [Google Scholar] [CrossRef] [PubMed]
- Benz, E.W., Jr.; Moses, H.L. Small, Virus-Like Particles Detected in Bovine Sera by Electron Microscopy. J. Natl. Cancer Inst. 1974, 52, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.; Quay, S.C.; Orenstein, N.S.; Dvorak, A.M.; Hahn, P.; Bitzer, A.M.; Carvalho, A.C. Tumor shedding and coagulation. Science 1981, 212, 923–924. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.; Adam, M.; Hammond, J.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Clancy, J.W.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123, 1603–1611. [Google Scholar] [CrossRef]
- Kawamoto, T.; Ohga, N.; Akiyama, K.; Hirata, N.; Kitahara, S.; Maishi, N.; Osawa, T.; Yamamoto, K.; Kondoh, M.; Shindoh, M.; et al. Tumor-derived microvesicles induce proangiogenic phenotype in endothelial cells via endocytosis. PLoS ONE 2012, 7, e34045. [Google Scholar] [CrossRef]
- D’Souza-Schorey, C.; Clancy, J.W. Tumor-derived microvesicles: Shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012, 26, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Katzmann, D.J.; Babst, M.; Emr, S.D. Ubiquitin-Dependent Sorting into the Multivesicular Body Pathway Requires the Function of a Conserved Endosomal Protein Sorting Complex, ESCRT-I. Cell 2001, 106, 145–155. [Google Scholar] [CrossRef]
- Cappariello, A.; Ponzetti, M.; Rucci, N. The ‘soft’ side of the bone: Unveiling its endocrine functions. Horm. Mol. Biol. Clin. Investig. 2016, 28, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Crockett, J.C.; Rogers, M.J.; Coxon, F.P.; Hocking, L.J.; Helfrich, M.H. Bone remodelling at a glance. J. Cell Sci. 2011, 124, 991–998. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Garimella, R.; Washington, L.; Isaacson, J.; Vallejo, J.; Spence, M.; Tawfik, O.; Rowe, P.; Brotto, M.; Perez, R. Extracellular Membrane Vesicles Derived from 143B Osteosarcoma Cells Contain Pro-Osteoclastogenic Cargo: A Novel Communication Mechanism in Osteosarcoma Bone Microenvironment. Transl. Oncol. 2014, 7, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323–328. [Google Scholar] [CrossRef]
- Portal-Núñez, S.; Lozano, D.; Esbrit, P. Role of angiogenesis on bone formation. Histol. Histopathol. 2012, 27, 559–566. [Google Scholar]
- Loupakis, F.; Cremolini, C.; Masi, G.; Lonardi, S.; Zagonel, V.; Salvatore, L.; Cortesi, E.; Tomasello, G.; Ronzoni, M.; Spadi, R.; et al. Initial Therapy with FOLFOXIRI and Bevacizumab for Metastatic Colorectal Cancer. N. Engl. J. Med. 2014, 371, 1609–1618. [Google Scholar] [CrossRef]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of Bevacizumab in the Primary Treatment of Ovarian Cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef]
- Bear, H.D.; Tang, G.; Rastogi, P.; Geyer, C.E., Jr.; Robidoux, A.; Atkins, J.N.; Baez-Diaz, L.; Brufsky, A.M.; Mehta, R.S.; Fehrenbacher, L.; et al. Bevacizumab Added to Neoadjuvant Chemotherapy for Breast Cancer. N. Engl. J. Med. 2012, 366, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.K.; Greening, D.W.; Hanssen, E.G.; Zhu, H.-J.; Simpson, R.J.; Mathias, R.A. Oncogenic epithelial cell-derived exosomes containing Rac1 and PAK2 induce angiogenesis in recipient endothelial cells. Oncotarget 2016, 7, 19709–19722. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.-J.; Lin, X.-J.; Tang, X.-Y.; Zheng, T.-T.; Lin, Y.-Y.; Hua, K.-Q. Exosomal Metastasis-Associated Lung Adenocarcinoma Transcript 1 Promotes Angiogenesis and Predicts Poor Prognosis in Epithelial Ovarian Cancer. Int. J. Biol. Sci. 2018, 14, 1960. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Yerneni, S.S.; Razzo, B.M.; Whiteside, T.L. Exosomes from HNSCC Promote Angiogenesis through Reprogramming of Endothelial Cells. Mol. Cancer Res. 2018, 16, 1798–1808. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Ren, J.; Bai, Y.; Pei, X.; Han, Y. Exosomes from hypoxia-treated human adipose-derived mesenchymal stem cells enhance angiogenesis through VEGF/VEGF-R. Int. J. Biochem. Cell Biol. 2019, 109, 59–68. [Google Scholar] [CrossRef]
- Rozeman, L.B.; Cleton-Jansen, A.M.; Hogendoorn, P.C.W. Pathology of primary malignant bone and cartilage tumours. Int. Orthop. 2006, 30, 437–444. [Google Scholar] [CrossRef]
- von Eisenhart-Rothe, R.; Toepfer, A.; Salzmann, M.; Schauwecker, J.; Gollwitzer, H.; Rechl, H. Primary malignant bone tumors. Orthopade 2011, 40, 1121–1142. [Google Scholar] [CrossRef]
- Nazeri, E.; Savadkoohi, M.G.; Majidzadeh-A, K.; Esmaeili, R. Chondrosarcoma: An overview of clinical behavior, molecular mechanisms mediated drug resistance and potential therapeutic targets. Crit. Rev. Oncol. Hematol. 2018, 131, 102–109. [Google Scholar] [CrossRef]
- Damron, T.A.; Ward, W.G.; Stewart, A. Osteosarcoma, Chondrosarcoma, and Ewing’s Sarcoma. Clin. Orthop. Relat. Res. 2007, 459, 40–47. [Google Scholar] [CrossRef]
- Anderson, H.C.; Mulhall, D.; Garimella, R. Role of extracellular membrane vesicles in the pathogenesis of various diseases, including cancer, renal diseases, atherosclerosis and arthritis. Lab. Investig. 2010, 90, 1549–1557. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Cortini, M.; Avnet, S.; Baldini, N. Mesenchymal stroma: Role in osteosarcoma progression. Cancer Lett. 2017, 405, 90–99. [Google Scholar] [CrossRef]
- Vallabhaneni, K.C.; Hassler, M.Y.; Abraham, A.; Whitt., J.; Mo, Y.Y.; Atfi, A.; Pochampally, R. Mesenchymal Stem/Stromal Cells under Stress Increase Osteosarcoma Migration and Apoptosis Resistance via Extracellular Vesicle Mediated Communication. PLoS ONE 2016, 11, e0166027. [Google Scholar] [CrossRef]
- Cruz, F.; Matushansky, I. MicroRNAs in chromosomal translocation-associated solid tumors: Learning from sarcomas. Discov. Med. 2011, 12, 307–317. [Google Scholar]
- Lulla, R.R.; Costa, F.F.; Bischof, J.M.; Chou, P.M.; de F Bonaldo, M.; Vanin, E.F.; Soares, M.B. Identification of Differentially Expressed MicroRNAs in Osteosarcoma. Sarcoma 2011, 2011, 732690. [Google Scholar] [CrossRef]
- Baglio, S.R.; Lagerweij, T.; Pérez-Lanzón, M.; Ho, X.D.; Léveillé, N.; Melo, S.A.; Cleton-Jansen, A.M.; Jordanova, E.S.; Roncuzzi, L.; Greco, M.; et al. Blocking tumor-educated MSC paracrine activity halts osteosarcoma progression. Clin. Cancer Res. 2017, 23, 3721–3733. [Google Scholar] [CrossRef]
- Bernstein, M.; Kovar, H.; Paulussen, M.; Randall, R.L.; Schuck, A.; Teot, L.A.; Juergens, H. Ewing’s Sarcoma Family of Tumors: Current Management. Oncologist 2006, 11, 503–519. [Google Scholar] [CrossRef]
- Lizard-Nacol, S.; Volk, C.; Lizard, G.; Turc-Carel, C. Abnormal expression of neurofilament proteins in Ewing’s sarcoma cell cultures. Tumour Biol. 1992, 13, 36–43. [Google Scholar] [CrossRef]
- Rocchi, A.; Manara, M.C.; Sciandra, M.; Zambelli, D.; Nardi, F.; Nicoletti, G.; Garofalo, C.; Meschini, S.; Astolfi, A.; Colombo, M.P.; et al. CD99 inhibits neural differentiation of human Ewing sarcoma cells and thereby contributes to oncogenesis. J. Clin. Investig. 2010, 120, 668–680. [Google Scholar] [CrossRef]
- Roussos, E.T.; Condeelis, J.S.; Patsialou, A. Chemotaxis in cancer. Nat. Rev. Cancer 2011, 11, 573–587. [Google Scholar] [CrossRef]
- Sung, B.H.; Weaver, A.M. Exosome secretion promotes chemotaxis of cancer cells. Cell Adhes. Migr. 2017, 11, 187–195. [Google Scholar] [CrossRef]
- Endo-Munoz, L.; Cai, N.; Cumming, A.; Macklin, R.; Merida de Long, L.; Topkas, E.; Mukhopadhyay, P.; Hill, M.; Saunders, N.A. Progression of Osteosarcoma from a Non-Metastatic to a Metastatic Phenotype Is Causally Associated with Activation of an Autocrine and Paracrine uPA Axis. PLoS ONE 2015, 10, e0133592. [Google Scholar] [CrossRef]
- Bjørnland, K.; Flatmark, K.; Pettersen, S.; Aaasen, A.O.; Fodstad, Ø.; Mælandsmo, G.M. Matrix Metalloproteinases Participate in Osteosarcoma Invasion. J. Surg. Res. 2005, 127, 151–156. [Google Scholar] [CrossRef]
- Uchibori, N.; Nishida, M.; Nagasaka, Y.; Yamada, T.; Nakanishi, Y.; Ishiguro, K. Increased expression of membrane-type matrix metalloproteinase-1 is correlated with poor prognosis in patients with osteosarcoma. Int. J. Oncol. 2006, 28, 33–42. [Google Scholar] [CrossRef][Green Version]
- Hakulinen, J.; Sankkila, L.; Sugiyama, N.; Lehti, K.; Keski-Oja, J. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J. Cell. Biochem. 2008, 105, 1211–1218. [Google Scholar] [CrossRef]
- Cappariello, A.; Paone, R.; Maurizi, A.; Capulli, M.; Rucci, N.; Muraca, M.; Teti, A. Biotechnological approach for systemic delivery of membrane Receptor Activator of NF-κB Ligand (RANKL) active domain into the circulation. Biomaterials 2015, 46, 58–69. [Google Scholar] [CrossRef]
- Husmann, K.; Arlt, M.J.; Muff, R.; Langsam, B.; Bertz, J.; Born, W.; Fuchs, B. Matrix Metalloproteinase 1 promotes tumor formation and lung metastasis in an intratibial injection osteosarcoma mouse model. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 347–354. [Google Scholar] [CrossRef]
- Ferrari, C.; Benassi, S.; Ponticelli, F.; Gamberi, G.; Ragazzini, P.; Pazzaglia, L.; Balladelli, A.; Bertoni, F.; Picci, P. Role of MMP-9 and its tissue inhibitor TIMP-1 in human osteosarcoma. Findings in 42 patients followed for 1–16 years. Acta Orthop. Scand. 2004, 75, 487–491. [Google Scholar] [CrossRef]
- Osaki, M.; Takeshita, F.; Sugimoto, Y.; Kosaka, N.; Yamamoto, Y.; Yoshioka, Y.; Kobayashi, E.; Yamada, T.; Kawai, A.; Inoue, T.; et al. MicroRNA-143 regulates human osteosarcoma metastasis by regulating matrix metalloprotease-13 expression. Mol. Ther. 2011, 19, 1123–1130. [Google Scholar] [CrossRef]
- Troyer, R.M.R.; Ruby, C.E.; Goodall, C.P.; Yang, L.; Maier, C.S.; Albarqi, H.A.; Brady, J.V.; Bathke, K.; Taratula, O.; Mourich, D.; et al. Exosomes from Osteosarcoma and normal osteoblast differ in proteomic cargo and immunomodulatory effects on T cells. Exp. Cell Res. 2017, 358, 369–376. [Google Scholar] [CrossRef]
- Jerez, S.; Araya, H.; Thaler, R.; Charlesworth, M.C.; López-Solís, R.; Kalergis, A.M.; Céspedes, P.F.; Dudakovic, A.; Stein, G.S.; van Wijnen, A.J.; et al. Proteomic Analysis of Exosomes and Exosome-Free Conditioned Media From Human Osteosarcoma Cell Lines Reveals Secretion of Proteins Related to Tumor Progression. J. Cell. Biochem. 2017, 118, 351–360. [Google Scholar] [CrossRef]
- Savitskaya, Y.A.; Rico-Martínez, G.; Linares-González, L.M.; Delgado-Cedillo, E.A.; Téllez-Gastelum, R.; Alfaro-Rodríguez, A.B.; Redón-Tavera, A.; Ibarra-Ponce de León, J.C. Serum tumor markers in pediatric osteosarcoma: A summary review. Clin. Sarcoma Res. 2012, 2, 9. [Google Scholar] [CrossRef]
- Cai, H.; Zhao, H.; Tang, J.; Wu, H. Serum miR-195 is a diagnostic and prognostic marker for osteosarcoma. J. Surg. Res. 2015, 194, 505–510. [Google Scholar] [CrossRef]
- Ma, W.; Zhang, X.; Chai, J.; Chen, P.; Ren, P.; Gong, M. Circulating miR-148a is a significant diagnostic and prognostic biomarker for patients with osteosarcoma. Tumor Biol. 2014, 35, 12467–12472. [Google Scholar] [CrossRef]
- Fujiwara, T.; Uotani, K.; Yoshida, A.; Morita, T.; Nezu, Y.; Kobayashi, E.; Yoshida, A.; Uehara, T.; Omori, T.; Sugiu, K.; et al. Clinical significance of circulating miR-25-3p as a novel diagnostic and prognostic biomarker in osteosarcoma. Oncotarget 2017, 8, 33375–33392. [Google Scholar] [CrossRef]
- Coleman, R.E. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin. Cancer Res. 2006, 12, 6243s–6249s. [Google Scholar] [CrossRef]
- Capulli, M.; Angelucci, A.; Driouch, K.; Garcia, T.; Clement-Lacroix, P.; Martella, F.; Ventura, L.; Bologna, M.; Flamini, S.; Moreschini, O.; et al. Increased expression of a set of genes enriched in oxygen binding function discloses a predisposition of breast cancer bone metastases to generate metastasis spread in multiple organs. J. Bone Miner. Res. 2012, 27, 2387–2398. [Google Scholar] [CrossRef]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Goncalves, F. Bone metastases: An overview. Oncol. Rev. 2017, 11, 321. [Google Scholar] [CrossRef]
- Luksch, R.; Castellani, M.R.; Collini, P.; De Bernardi, B.; Conte, M.; Gambini, C.; Gandola, L.; Garaventa, A.; Biasoni, D.; Podda, M.; et al. Neuroblastoma (Peripheral neuroblastic tumours). Crit. Rev. Oncol. Hematol. 2016, 107, 163–181. [Google Scholar] [CrossRef]
- D’Oronzo, S.; Coleman, R.; Brown, J.; Silvestris, F. Metastatic bone disease: Pathogenesis and therapeutic options. J. Bone Oncol. 2019, 15, 100205. [Google Scholar] [CrossRef]
- Galson, D.L.; Silbermann, R.; Roodman, G.D. Mechanisms of multiple myeloma bone disease. Bonekey Rep. 2012, 1, 135. [Google Scholar] [CrossRef]
- Taube, T.; Beneton, M.N.; McCloskey, E.V.; Rogers, S.; Greaves, M.; Kanis, J.A. Abnormal bone remodelling in patients with myelomatosis and normal biochemical indices of bone resorption. Eur. J. Haematol. 1992, 49, 192–198. [Google Scholar] [CrossRef]
- Saad, F.; Lipton, A.; Cook, R.; Chen, Y.-M.; Smith, M.; Coleman, R. Pathologic fractures correlate with reduced survival in patients with malignant bone disease. Cancer 2007, 110, 1860–1867. [Google Scholar] [CrossRef]
- Kuwahara, K.; Kudo, K.; Yashima-Abo, A.; Katayama, K.; Kojima, K.; Tone, K.; Ito, E.; Nakazawa, A.; Iwafuchi, H.; Kurose, A. Classic Hodgkin lymphoma with osseous involvement mimicking Langerhans cell histiocytosis in a child. Hum. Pathol. 2018, 77, 147–151. [Google Scholar] [CrossRef]
- Seymour, J.F.; Gagel, R.F.; Hagemeister, F.B.; Dimopoulos, M.A.; Cabanillas, F. Calcitriol Production in Hypercalcemic and Normocalcemic Patients with Non-Hodgkin Lymphoma. Ann. Intern. Med. 1994, 121, 633. [Google Scholar] [CrossRef]
- Taverna, S.; Pucci, M.; Giallombardo, M.; Di Bella, M.A.; Santarpia, M.; Reclusa, P.; Gil-Bazo, I.; Rolfo, C.; Alessandro, R. Amphiregulin contained in NSCLC-exosomes induces osteoclast differentiation through the activation of EGFR pathway. Sci. Rep. 2017, 7, 3170. [Google Scholar] [CrossRef]
- Xu, Z.; Liu, X.; Wang, H.; Li, J.; Dai, L.; Li, J.; Dong, C. Lung adenocarcinoma cell-derived exosomal miR-21 facilitates osteoclastogenesis. Gene 2018, 666, 116–122. [Google Scholar] [CrossRef]
- Valencia, K.; Luis-Ravelo, D.; Bovy, N.; Antón, I.; Martínez-Canarias, S.; Zandueta, C.; Ormazábal, C.; Struman, I.; Tabruyn, S.; Rebmann, V.; et al. miRNA cargo within exosome-like vesicle transfer influences metastatic bone colonization. Mol. Oncol. 2014, 8, 689–703. [Google Scholar] [CrossRef]
- Karlsson, T.; Lundholm, M.; Widmark, A.; Persson, E. Tumor Cell-Derived Exosomes from the Prostate Cancer Cell Line TRAMP-C1 Impair Osteoclast Formation and Differentiation. PLoS ONE 2016, 11, e0166284. [Google Scholar] [CrossRef]
- Inder, K.L.; Ruelcke, J.E.; Petelin, L.; Moon, H.; Choi, E.; Rae, J.; Blumenthal, A.; Hutmacher, D.; Saunders, N.A.; Stow, J.L. Cavin-1/PTRF alters prostate cancer cell-derived extracellular vesicle content and internalization to attenuate extracellular vesicle-mediated osteoclastogenesis and osteoblast proliferation. J. Extracell. Vesicles 2014, 3, 23784. [Google Scholar] [CrossRef]
- Hashimoto, K.; Ochi, H.; Sunamura, S.; Kosaka, N.; Mabuchi, Y.; Fukuda, T.; Yao, K.; Kanda, H.; Ae, K.; Okawa, A.; et al. Cancer-secreted hsa-miR-940 induces an osteoblastic phenotype in the bone metastatic microenvironment via targeting ARHGAP1 and FAM134A. Proc. Natl. Acad. Sci. USA 2018, 115, 2204–2209. [Google Scholar] [CrossRef]
- Ye, Y.; Li, S.L.; Ma, Y.Y.; Diao, Y.J.; Yang, L.; Su, M.Q.; Li, Z.; Ji, Y.; Wang, J.; Lei, L.; et al. Exosomal miR-141-3p regulates osteoblast activity to promote the osteoblastic metastasis of prostate cancer. Oncotarget 2017, 8, 94834–94849. [Google Scholar] [CrossRef]
- Probert, C.; Dottorini, T.; Speakman, A.; Hunt, S.; Nafee, T.; Fazeli, A.; Wood, S.; Brown, J.E.; James, V. Communication of prostate cancer cells with bone cells via extracellular vesicle RNA; a potential mechanism of metastasis. Oncogene 2019, 38, 1751–1763. [Google Scholar] [CrossRef]
- Paez, D.; Labonte, M.J.; Bohanes, P.; Zhang, W.; Benhanim, L.; Ning, Y.; Wakatsuki, T.; Loupakis, F.; Lenz, H.J. Cancer Dormancy: A Model of Early Dissemination and Late Cancer Recurrence. Clin. Cancer Res. 2012, 18, 645–653. [Google Scholar] [CrossRef]
- Capulli, M.; Hristova, D.; Valbret, Z.; Carys, K.; Arjan, R.; Maurizi, A.; Masedu, F.; Cappariello, A.; Rucci, N.; Teti, A. Notch2 pathway mediates breast cancer cellular dormancy and mobilisation in bone and contributes to haematopoietic stem cell mimicry. Br. J. Cancer 2019, 121, 157–171. [Google Scholar] [CrossRef]
- Ono, M.; Kosaka, N.; Tominaga, N.; Yoshioka, Y.; Takeshita, F.; Takahashi, R.U.; Yoshida, M.; Tsuda, H.; Tamura, K.; Ochiya, T. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. 2014, 7, ra63. [Google Scholar] [CrossRef]
- Bliss, S.A.; Sinha, G.; Sandiford, O.A.; Williams, L.M.; Engelberth, D.J.; Guiro, K.; Isenalumhe, L.L.; Greco, S.J.; Ayer, S.; Bryan, M.; et al. Mesenchymal Stem Cell-Derived Exosomes Stimulate Cycling Quiescence and Early Breast Cancer Dormancy in Bone Marrow. Cancer Res. 2016, 76, 5832–5844. [Google Scholar] [CrossRef]
- Wen, S.W.; Sceneay, J.; Lima, L.G.; Wong, C.S.; Becker, M.; Krumeich, S.; Lobb, R.J.; Castillo, V.; Wong, K.N.; Ellis, S.; et al. The Biodistribution and Immune Suppressive Effects of Breast Cancer-Derived Exosomes. Cancer Res. 2016, 76, 6816–6827. [Google Scholar] [CrossRef]
- Plebanek, M.P.; Angeloni, N.L.; Vinokour, E.; Li, J.; Henkin, A.; Martinez-Marin, D.; Filleur, S.; Bhowmick, R.; Henkin, J.; Miller, S.D.; et al. Pre-metastatic cancer exosomes induce immune surveillance by patrolling monocytes at the metastatic niche. Nat. Commun. 2017, 8, 1319. [Google Scholar] [CrossRef]
- McLellan, A.D. Exosome release by primary B cells. Crit. Rev. Immunol. 2009, 29, 203–217. [Google Scholar] [CrossRef]
- Lu, J.; Wu, J.; Tian, J.; Wang, S. Role of T cell-derived exosomes in immunoregulation. Immunol. Res. 2018, 66, 313–322. [Google Scholar] [CrossRef]
- Raimondi, L.; De Luca, A.; Amodio, N.; Manno, M.; Raccosta, S.; Taverna, S.; Bellavia, D.; Naselli, F.; Fontana, S.; Schillaci, O.; et al. Involvement of multiple myeloma cell-derived exosomes in osteoclast differentiation. Oncotarget 2015, 6, 13772–13789. [Google Scholar] [CrossRef]
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; De Bruyne, E.; Van Valckenborgh, E.; Lahoutte, T.; De Wever, O.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 124, 555–566. [Google Scholar] [CrossRef]
- Kumar, B.; Garcia, M.; Weng, L.; Jung, X.; Murakami, J.L.; Hu, X.; McDonald, T.; Lin, A.; Kumar, A.R.; DiGiusto, D.L.; et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 2018, 32, 575–587. [Google Scholar] [CrossRef]
- Faict, S.; Muller, J.; De Veirman, K.; De Bruyne, E.; Maes, K.; Vrancken, L.; Heusschen, R.; De Raeve, H.; Schots, R.; Vanderkerken, K.; et al. Exosomes play a role in multiple myeloma bone disease and tumor development by targeting osteoclasts and osteoblasts. Blood Cancer J. 2018, 8, 105. [Google Scholar] [CrossRef]
- Li, B.; Xu, H.; Han, H.; Song, S.; Zhang, X.; Ouyang, L.; Qian, C.; Hong, Y.; Qiu, Y.; Zhou, W.; et al. Exosome-mediated transfer of lncRUNX2-AS1 from multiple myeloma cells to MSCs contributes to osteogenesis. Oncogene 2018, 37, 5508–5519. [Google Scholar] [CrossRef]
- Raimondo, S.; Saieva, L.; Vicario, E.; Pucci, M.; Toscani, D.; Manno, M.; Raccosta, S.; Giuliani, N.; Alessandro, R. Multiple myeloma-derived exosomes are enriched of amphiregulin (AREG) and activate the epidermal growth factor pathway in the bone microenvironment leading to osteoclastogenesis. J. Hematol. Oncol. 2019, 12, 2. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Essandoh, K.; Yang, L.; Wang, X.; Huang, W.; Qin, D.; Hao, J.; Wang, Y.; Zingarelli, B.; Peng, T.; Fan, G.C. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2362–2371. [Google Scholar] [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef]
- Singh, R.; Pochampally, R.; Watabe, K.; Lu, Z.; Mo, Y.-Y. Exosome-mediated transfer of miR-10b promotes cell invasion in breast cancer. Mol. Cancer 2014, 13, 256. [Google Scholar] [CrossRef]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef]
- Blanc, L.; Vidal, M. New insights into the function of Rab GTPases in the context of exosomal secretion. Small GTPases 2018, 9, 95–106. [Google Scholar] [CrossRef]
- Savina, A.; Fader, C.M.; Damiani, M.T.; Colombo, M.I. Rab11 Promotes Docking and Fusion of Multivesicular Bodies in a Calcium-Dependent Manner. Traffic 2005, 6, 131–143. [Google Scholar] [CrossRef]
- Bobrie, A.; Krumeich, S.; Reyal, F.; Recchi, C.; Moita, L.F.; Seabra, M.C.; Ostrowski, M.; Théry, C. Rab27a Supports Exosome-Dependent and -Independent Mechanisms That Modify the Tumor Microenvironment and Can Promote Tumor Progression. Cancer Res. 2012, 72, 4920–4930. [Google Scholar] [CrossRef]
- Macia, E.; Ehrlich, M.; Massol, R.; Boucrot, E.; Brunner, C.; Kirchhausen, T. Dynasore, a Cell-Permeable Inhibitor of Dynamin. Dev. Cell 2006, 10, 839–850. [Google Scholar] [CrossRef]
- Christianson, H.C.; Svensson, K.J.; van Kuppevelt, T.H.; Li, J.-P.; Belting, M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc. Natl. Acad. Sci. USA 2013, 110, 17380–17385. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef]
- Hazan-Halevy, I.; Rosenblum, D.; Weinstein, S.; Bairey, O.; Raanani, P.; Peer, D. Cell-specific uptake of mantle cell lymphoma-derived exosomes by malignant and non-malignant B-lymphocytes. Cancer Lett. 2015, 364, 59–69. [Google Scholar] [CrossRef]
- Zhu, X.; Badawi, M.; Pomeroy, S.; Sutaria, D.S.; Xie, Z.; Baek, A.; Jiang, J.; Elgamal, O.A.; Mo, X.; Perle, K. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J. Extracell. Vesicles 2017, 6, 1324730. [Google Scholar] [CrossRef]
- Pitt, J.M.; André, F.; Amigorena, S.; Soria, J.C.; Eggermont, A.; Kroemer, G.; Zitvogel, L. Dendritic cell–derived exosomes for cancer therapy. J. Clin. Investig. 2016, 126, 1224–1232. [Google Scholar] [CrossRef]
- Quah, B.J.C.; O’Neill, H.C. The immunogenicity of dendritic cell-derived exosomes. Blood Cells, Mol. Dis. 2005, 35, 94–110. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Mizrak, A.; Bolukbasi, M.F.; Ozdener, G.B.; Brenner, G.J.; Madlener, S.; Erkan, E.P.; Ströbel, T.; Breakefield, X.O.; Saydam, O. Genetically Engineered Microvesicles Carrying Suicide mRNA/Protein Inhibit Schwannoma Tumor Growth. Mol. Ther. 2013, 21, 101–108. [Google Scholar] [CrossRef]
- van den Boorn, J.G.; Schlee, M.; Coch, C.; Hartmann, G. SiRNA delivery with exosome nanoparticles. Nat. Biotechnol. 2011, 29, 325–326. [Google Scholar] [CrossRef]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of Brain Inflammatory Diseases by Delivering Exosome Encapsulated Anti-inflammatory Drugs From the Nasal Region to the Brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef]
- Andre, F.; Schartz, N.E.; Movassagh, M.; Flament, C.; Pautier, P.; Morice, P.; Pomel, C.; Lhomme, C.; Escudier, B.; Le Chevalier, T.; et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet 2002, 360, 295–305. [Google Scholar] [CrossRef]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumors using a novel cell-free vaccine: Dendritic cell derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Tumour | Educator | Target Cell | Effect | Ref. |
|---|---|---|---|---|
| Osteosarcoma | TGF beta | MSCs | Increase of IL-6 production | [47] |
| Ewing Sarcoma | CD99 | Autologous | Increase of miR-34a Inhibition of NFκB | [50] |
| Fibrosarcoma | Rab27a | Autologous | Induction of chemotaxis | [52] |
| Osteosarcoma | uPA | Lung | Pro-metastatic | [53] |
| Fibrosarcoma | MT1-MMP | Extracellular matrix | Activation of MMP2 Degradation of type 1 collagen and gelatine | [56] |
| Osteosarcoma | MMP1/MMP13 | Lung | Increase of aggressiveness | [27] [58] |
| Osteosarcoma | RANKL | Osteoclasts | Increase of osteoclastogenesis | [27] |
| Osteosarcoma | miR-148a | Unspecified | Increase of aggressiveness | [66] |
| Osteosarcoma | miR-25-3-p | Unspecified | Increase of aggressiveness | [66] |
| Tumour | Educator | Target Cell | Effect | Ref. |
|---|---|---|---|---|
| Lung cancer | Amphiregulin | Osteoclasts | Increase of osteoclastogenesis | [77] |
| Lung cancer | miR-21 | Osteoclasts | Increase of osteoclastogenesis | [78] |
| Lung cancer | miR-192 | Endothelial cells | Increase of osteoclastogenesis | [79] |
| Prostate cancer | Unspecified | Osteoclasts | Inhibition of osteoclast formation, activity and survival | [80] |
| Prostate cancer | Cavin1 | Osteoblasts | Inhibition of osteoblast proliferation | [81] |
| Prostate cancer | Cavin1 | Osteoclasts | Inhibition of osteoclast differentiation | [81] |
| Prostate/Breast cancer | hsa-miR-940 | Autologous | Inhibition of metastasis | [82] |
| Prostate cancer | miR-141-3p | Osteoblasts | Increase of osteoblast activity, decrease of OPG expression | [83] |
| Prostate cancer | miRNA (i.e., miR-21) and mRNA (i.e., CSF-1) pool | Osteoblasts | Increase of osteoblast viability | [84] |
| Breast cancer | miR-23b | HSC | Dormancy induction | [87] |
| Breast cancer | miR-222/23 | MSC | Dormancy induction | [88] |
| Breast cancer | Unspecified | Myeloid, T- and NK cells | Inhibition of immune response | [89] |
| Multiple Myeloma | Dkk1 Unspecified(?) | Osteoblasts, Osteoclasts | Decrease of osteoblast differentiation, increase of osteoclast activity | [96] |
| Multiple Myeloma | lncRNA vs RUNX2 | MCS | Silencing of RUNX2 mRNA and decrease of osteogenesis | [97] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappariello, A.; Rucci, N. Tumour-Derived Extracellular Vesicles (EVs): A Dangerous “Message in A Bottle” for Bone. Int. J. Mol. Sci. 2019, 20, 4805. https://doi.org/10.3390/ijms20194805
Cappariello A, Rucci N. Tumour-Derived Extracellular Vesicles (EVs): A Dangerous “Message in A Bottle” for Bone. International Journal of Molecular Sciences. 2019; 20(19):4805. https://doi.org/10.3390/ijms20194805
Chicago/Turabian StyleCappariello, Alfredo, and Nadia Rucci. 2019. "Tumour-Derived Extracellular Vesicles (EVs): A Dangerous “Message in A Bottle” for Bone" International Journal of Molecular Sciences 20, no. 19: 4805. https://doi.org/10.3390/ijms20194805
APA StyleCappariello, A., & Rucci, N. (2019). Tumour-Derived Extracellular Vesicles (EVs): A Dangerous “Message in A Bottle” for Bone. International Journal of Molecular Sciences, 20(19), 4805. https://doi.org/10.3390/ijms20194805