New Insights into Immunotherapy Strategies for Treating Autoimmune Diabetes

 , ,
, ,
Abstract
1. Introduction
1.1. Genetics of T1D
1.2. Immunological Mechanisms Involved in T1D Pathogenesis
1.3. Animal Models of T1D
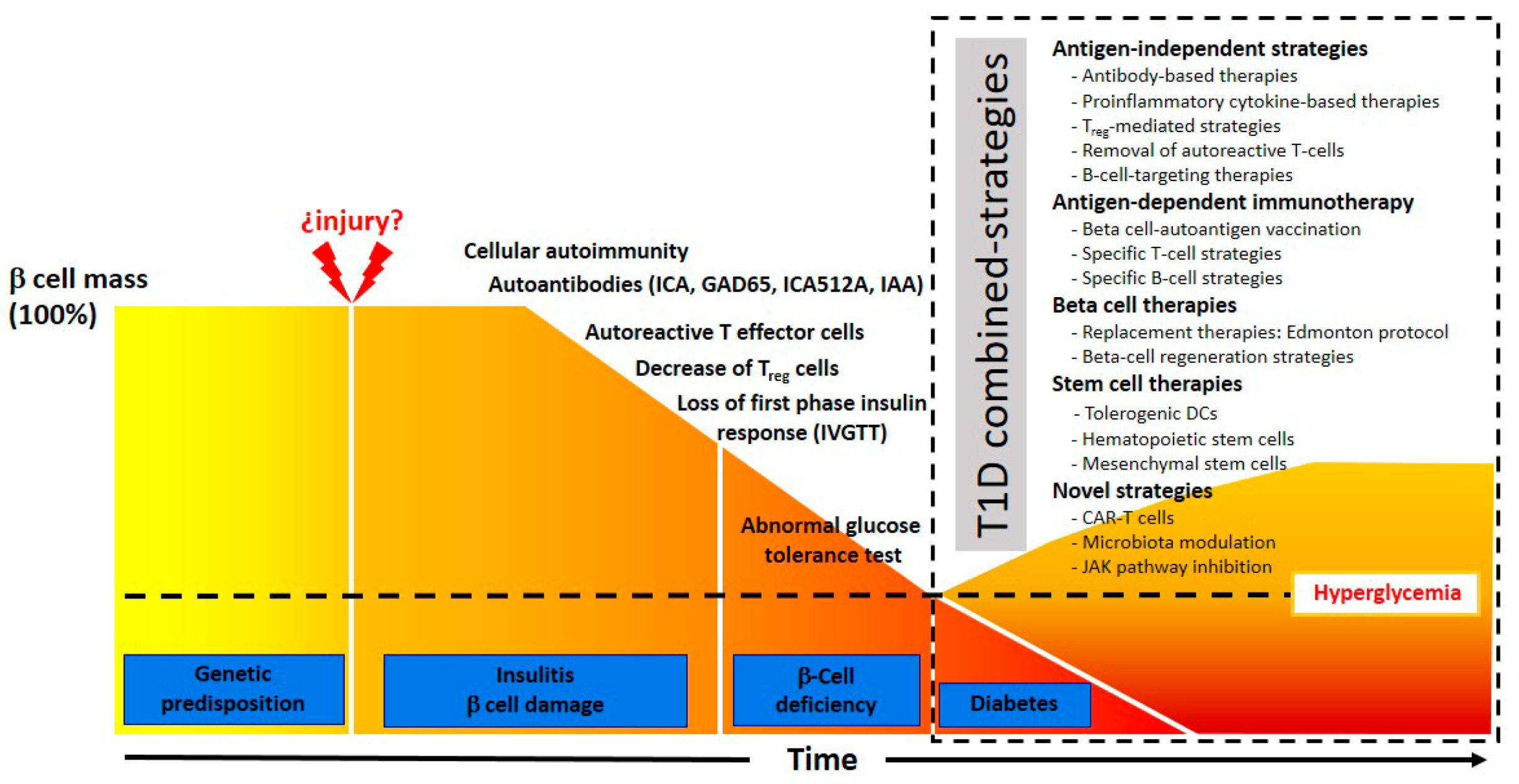
1.4. Present State of T1D Immunotherapy
2. Antigen-Independent Strategies
2.1. Antibody-Based Therapies
2.2. Proinflammatory Cytokine-Based Treatments
2.3. Treg-Mediated Strategies
2.4. Removal of Autoreactive T-cells
2.5. B-Cell-Targeting Therapies
3. Antigen-Dependent Immunotherapy
3.1. Beta Cell-Autoantigen Vaccination
3.2. Specific T-Cell Strategies
3.3. Specific B-Cell Strategies
4. Beta Cell Therapies
4.1. Replacement Therapies: Edmonton Protocol
4.2. Beta-Cell Regeneration Strategies
5. Stem Cell Therapy Strategies
5.1. Tolerogenic DCs
5.2. Hematopoietic Stem Cells (HSC)
5.3. Mesenchymal Stem Cells
6. Novel Strategies
6.1. CAR-T-Cell Therapy
6.1.1. Introduction
6.1.2. CAR-T-Cells and T1D
6.1.3. Challenges
6.1.4. Summary
6.2. Microbiota Modulation
6.2.1. Introduction
6.2.2. Microbiota and T1D
6.2.3. Summary
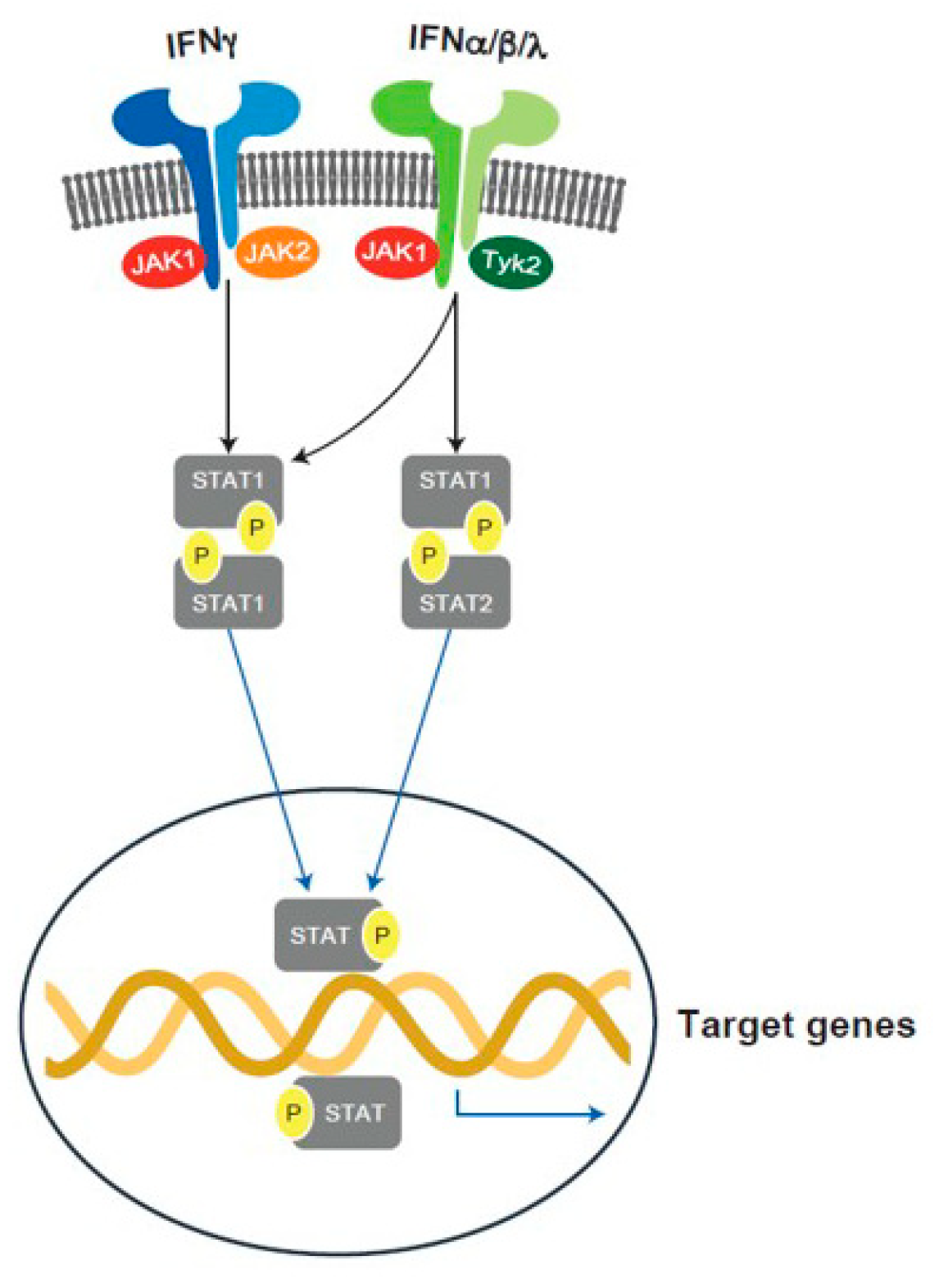
6.3. JAK Pathway Inhibition
6.3.1. Introduction
6.3.2. JAKs and T1D
6.3.3. Summary
7. Concluding Remarks and Outlook
Funding
Acknowledgments
Conflicts of Interest
References
- Patterson, C.; Guariguata, L.; Dahlquist, G.; Soltesz, G.; Ogle, G.; Silink, M. Diabetes in the young—A global view and worldwide estimates of numbers of children with type 1 diabetes. Diabetes Res. Clin. Pract. 2014, 103, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Maahs, D.M.; West, N.A.; Lawrence, J.M.; Mayer-Davis, E.J. Epidemiology of type 1 diabetes. Endocrinol. Metab. Clin. N. Am. 2010, 39, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Pociot, F.; Akolkar, B.; Concannon, P.; Erlich, H.A.; Julier, C.; Morahan, G.; Nierras, C.R.; Todd, J.A.; Rich, S.S.; Nerup, J. Genetics of type 1 diabetes: What’s next? Diabetes 2010, 59, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Nerup, J.; Platz, P.; Andersen, O.O.; Christy, M.; Lyngsoe, J.; Poulsen, J.E.; Ryder, L.P.; Nielsen, L.S.; Thomsen, M.; Svejgaard, A. HL-A antigens and diabetes mellitus. Lancet 1974, 2, 864–866. [Google Scholar] [CrossRef]
- Clark, M.; Kroger, C.J.; Tisch, R.M. Type 1 Diabetes: A Chronic Anti-Self-Inflammatory Response. Front. Immunol. 2017, 8, 1898. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.M.; Ricordi, C.; Hering, B.J.; Auchincloss, H.; Lindblad, R.; Robertson, R.P.; Secchi, A.; Brendel, M.D.; Berney, T.; Brennan, D.C.; et al. International trial of the Edmonton protocol for islet transplantation. N. Engl. J. Med. 2006, 355, 1318–1330. [Google Scholar] [CrossRef]
- Perseghin, G.; Fiorina, P.; De Cobelli, F.; Scifo, P.; Esposito, A.; Canu, T.; Danna, M.; Gremizzi, C.; Secchi, A.; Luzi, L.; et al. Cross-sectional assessment of the effect of kidney and kidney-pancreas transplantation on resting left ventricular energy metabolism in type 1 diabetic-uremic patients: A phosphorous-31 magnetic resonance spectroscopy study. J. Am. Coll. Cardiol. 2005, 46, 1085–1092. [Google Scholar] [CrossRef][Green Version]
- Simmons, K.M.; Michels, A.W. Type 1 diabetes: A predictable disease. World J. Diabetes 2015, 6, 380–390. [Google Scholar] [CrossRef]
- Regnell, S.E.; Lernmark, A. Early prediction of autoimmune (type 1) diabetes. Diabetologia 2017, 60, 1370–1381. [Google Scholar] [CrossRef]
- Di Lorenzo, T.P.; Peakman, M.; Roep, B.O. Translational mini-review series on type 1 diabetes: Systematic analysis of T cell epitopes in autoimmune diabetes. Clin. Exp. Immunol. 2007, 148, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wherrett, D.K.; Chiang, J.L.; Delamater, A.M.; DiMeglio, L.A.; Gitelman, S.E.; Gottlieb, P.A.; Herold, K.C.; Lovell, D.J.; Orchard, T.J.; Ryan, C.M.; et al. Defining pathways for development of disease-modifying therapies in children with type 1 diabetes: A consensus report. Diabetes Care 2015, 38, 1975–1985. [Google Scholar] [CrossRef] [PubMed]
- Parikka, V.; Nanto-Salonen, K.; Saarinen, M.; Simell, T.; Ilonen, J.; Hyoty, H.; Veijola, R.; Knip, M.; Simell, O. Early seroconversion and rapidly increasing autoantibody concentrations predict prepubertal manifestation of type 1 diabetes in children at genetic risk. Diabetologia 2012, 55, 1926–1936. [Google Scholar] [CrossRef]
- Krischer, J.P.; Lynch, K.F.; Schatz, D.A.; Ilonen, J.; Lernmark, A.; Hagopian, W.A.; Rewers, M.J.; She, J.X.; Simell, O.G.; Toppari, J.; et al. The 6 year incidence of diabetes-associated autoantibodies in genetically at-risk children: The TEDDY study. Diabetologia 2015, 58, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Bosi, E.; Boulware, D.C.; Becker, D.J.; Buckner, J.H.; Geyer, S.; Gottlieb, P.A.; Henderson, C.; Kinderman, A.; Sosenko, J.M.; Steck, A.K.; et al. Impact of Age and Antibody Type on Progression From Single to Multiple Autoantibodies in Type 1 Diabetes Relatives. J. Clin. Endocrinol. Metab. 2017, 102, 2881–2886. [Google Scholar] [CrossRef]
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2019. Diabetes Care 2019, 42, S13–S28. [Google Scholar] [CrossRef]
- Herold, K.C.; Gitelman, S.E.; Ehlers, M.R.; Gottlieb, P.A.; Greenbaum, C.J.; Hagopian, W.; Boyle, K.D.; Keyes-Elstein, L.; Aggarwal, S.; Phippard, D.; et al. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: Metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes 2013, 62, 3766–3774. [Google Scholar] [CrossRef]
- Rigby, M.R.; DiMeglio, L.A.; Rendell, M.S.; Felner, E.I.; Dostou, J.M.; Gitelman, S.E.; Patel, C.M.; Griffin, K.J.; Tsalikian, E.; Gottlieb, P.A.; et al. Targeting of memory T cells with alefacept in new-onset type 1 diabetes (T1DAL study): 12 Month results of a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Diabetes Endocrinol. 2013, 1, 284–294. [Google Scholar] [CrossRef]
- Mahon, J.L.; Sosenko, J.M.; Rafkin-Mervis, L.; Krause-Steinrauf, H.; Lachin, J.M.; Thompson, C.; Bingley, P.J.; Bonifacio, E.; Palmer, J.P.; Eisenbarth, G.S.; et al. The TrialNet Natural History Study of the Development of Type 1 Diabetes: Objectives, design, and initial results. Pediatr. Diabetes 2009, 10, 97–104. [Google Scholar] [CrossRef]
- Zhao, Z.; Miao, D.; Michels, A.; Steck, A.; Dong, F.; Rewers, M.; Yu, L. A multiplex assay combining insulin, GAD, IA-2 and transglutaminase autoantibodies to facilitate screening for pre-type 1 diabetes and celiac disease. J. Immunol. Methods 2016, 430, 28–32. [Google Scholar] [CrossRef]
- Slim, I.B. Cardiovascular risk in type 1 diabetes mellitus. Indian J. Endocrinol. Metab. 2013, 17, S7–S13. [Google Scholar] [CrossRef] [PubMed]
- Makino, S.; Kunimoto, K.; Muraoka, Y.; Mizushima, Y.; Katagiri, K.; Tochino, Y. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu 1980, 29, 1–13. [Google Scholar] [PubMed]
- Nakhooda, A.F.; Like, A.A.; Chappel, C.I.; Murray, F.T.; Marliss, E.B. The spontaneously diabetic Wistar rat: Metabolic and morphologic studies. Diabetes 1977, 26, 100–112. [Google Scholar] [CrossRef]
- Jackson, R.; Rassi, N.; Crump, T.; Haynes, B.; Eisenbarth, G.S. The BB diabetic rat: Profound T-cell lymphocytopenia. Diabetes 1981, 30, 887–889. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Yagi, H.; Kunimoto, K.; Kawaguchi, J.; Makino, S.; Harada, M. Transfer of autoimmune diabetes from diabetic NOD mice to NOD athymic nude mice: The roles of T cell subsets in the pathogenesis. Cell. Immunol. 1993, 148, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Matsumoto, M.; Kunimoto, K.; Kawaguchi, J.; Makino, S.; Harada, M. Analysis of the roles of CD4+ and CD8+ T cells in autoimmune diabetes of NOD mice using transfer to NOD athymic nude mice. Eur. J. Immunol. 1992, 22, 2387–2393. [Google Scholar] [CrossRef] [PubMed]
- Pontesilli, O.; Carotenuto, P.; Gazda, L.S.; Pratt, P.F.; Prowse, S.J. Circulating lymphocyte populations and autoantibodies in non-obese diabetic (NOD) mice: A longitudinal study. Clin. Exp. Immunol. 1987, 70, 84–93. [Google Scholar] [PubMed]
- Shoda, L.K.; Young, D.L.; Ramanujan, S.; Whiting, C.C.; Atkinson, M.A.; Bluestone, J.A.; Eisenbarth, G.S.; Mathis, D.; Rossini, A.A.; Campbell, S.E.; et al. A comprehensive review of interventions in the NOD mouse and implications for translation. Immunity 2005, 23, 115–126. [Google Scholar] [CrossRef]
- Pow Sang, L.; Majji, S.; Casares, S.; Brumeanu, T.D. Long-term silencing of autoimmune diabetes and improved life expectancy by a soluble pHLA-DR4 chimera in a newly-humanized NOD/DR4/B7 mouse. Hum. Vaccines Immunother. 2014, 10, 693–699. [Google Scholar] [CrossRef]
- Wen, L.; Ley, R.E.; Volchkov, P.Y.; Stranges, P.B.; Avanesyan, L.; Stonebraker, A.C.; Hu, C.; Wong, F.S.; Szot, G.L.; Bluestone, J.A.; et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008, 455, 1109–1113. [Google Scholar] [CrossRef]
- Hansen, C.H.; Krych, L.; Nielsen, D.S.; Vogensen, F.K.; Hansen, L.H.; Sorensen, S.J.; Buschard, K.; Hansen, A.K. Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia 2012, 55, 2285–2294. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Narasimhan, S.; Marchesi, J.R.; Benson, A.; Wong, F.S.; Wen, L. Long term effect of gut microbiota transfer on diabetes development. J. Autoimmun. 2014, 53, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Wesley, J.D.; Sather, B.D.; Perdue, N.R.; Ziegler, S.F.; Campbell, D.J. Cellular requirements for diabetes induction in DO11.10xRIPmOVA mice. J. Immunol. 2010, 185, 4760–4768. [Google Scholar] [CrossRef] [PubMed]
- Nagy, N.; Kaber, G.; Johnson, P.Y.; Gebe, J.A.; Preisinger, A.; Falk, B.A.; Sunkari, V.G.; Gooden, M.D.; Vernon, R.B.; Bogdani, M.; et al. Inhibition of hyaluronan synthesis restores immune tolerance during autoimmune insulitis. J. Clin. Investig. 2015, 125, 3928–3940. [Google Scholar] [CrossRef] [PubMed]
- Makhlouf, L.; Grey, S.T.; Dong, V.; Csizmadia, E.; Arvelo, M.B.; Auchincloss, H., Jr.; Ferran, C.; Sayegh, M.H. Depleting anti-CD4 monoclonal antibody cures new-onset diabetes, prevents recurrent autoimmune diabetes, and delays allograft rejection in nonobese diabetic mice. Transplantation 2004, 77, 990–997. [Google Scholar] [CrossRef]
- Cameron, M.J.; Arreaza, G.A.; Waldhauser, L.; Gauldie, J.; Delovitch, T.L. Immunotherapy of spontaneous type 1 diabetes in nonobese diabetic mice by systemic interleukin-4 treatment employing adenovirus vector-mediated gene transfer. Gene Ther. 2000, 7, 1840–1846. [Google Scholar] [CrossRef]
- Feili-Hariri, M.; Falkner, D.H.; Gambotto, A.; Papworth, G.D.; Watkins, S.C.; Robbins, P.D.; Morel, P.A. Dendritic cells transduced to express interleukin-4 prevent diabetes in nonobese diabetic mice with advanced insulitis. Hum. Gene Ther. 2003, 14, 13–23. [Google Scholar] [CrossRef]
- Clemente-Casares, X.; Tsai, S.; Huang, C.; Santamaria, P. Antigen-specific therapeutic approaches in Type 1 diabetes. Cold Spring Harb. Perspect. Med. 2012, 2, a007773. [Google Scholar] [CrossRef]
- Miller, S.D.; Turley, D.M.; Podojil, J.R. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat. Rev. Immunol. 2007, 7, 665–677. [Google Scholar] [CrossRef]
- Bone, R.N.; Evans-Molina, C. Combination Immunotherapy for Type 1 Diabetes. Curr. Diabetes Rep. 2017, 17, 50. [Google Scholar] [CrossRef]
- Orban, T.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Marks, J.B.; Monzavi, R.; et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: A randomised, double-blind, placebo-controlled trial. Lancet 2011, 378, 412–419. [Google Scholar] [CrossRef]
- Herold, K.C.; Hagopian, W.; Auger, J.A.; Poumian-Ruiz, E.; Taylor, L.; Donaldson, D.; Gitelman, S.E.; Harlan, D.M.; Xu, D.; Zivin, R.A.; et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N. Engl. J. Med. 2002, 346, 1692–1698. [Google Scholar] [CrossRef] [PubMed]
- Haller, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Michels, A.W.; Rosenthal, S.M.; Shuster, J.J.; Zou, B.; Brusko, T.M.; Hulme, M.A.; Wasserfall, C.H.; et al. Anti-thymocyte globulin/G-CSF treatment preserves beta cell function in patients with established type 1 diabetes. J. Clin. Investig. 2015, 125, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Kutlu, B.; Miani, M.; Bang-Berthelsen, C.H.; Storling, J.; Pociot, F.; Goodman, N.; Hood, L.; Welsh, N.; Bontempi, G.; et al. Temporal profiling of cytokine-induced genes in pancreatic beta-cells by meta-analysis and network inference. Genomics 2014, 103, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, S.M.; Wang, X.; Chen, Y.G.; Jia, S.; Kaldunski, M.L.; Greenbaum, C.J.; Type 1 Diabetes TrialNet Canakinumab Study Group; Mandrup-Poulsen, T.; Group, A.S.; Hessner, M.J. Interleukin-1 antagonism moderates the inflammatory state associated with Type 1 diabetes during clinical trials conducted at disease onset. Eur. J. Immunol. 2016, 46, 1030–1046. [Google Scholar] [CrossRef]
- Den Broeder, A.A.; de Jong, E.; Franssen, M.J.; Jeurissen, M.E.; Flendrie, M.; van den Hoogen, F.H. Observational study on efficacy, safety, and drug survival of anakinra in rheumatoid arthritis patients in clinical practice. Ann. Rheum. Dis. 2006, 65, 760–762. [Google Scholar] [CrossRef]
- Mandrup-Poulsen, T.; Pickersgill, L.; Donath, M.Y. Blockade of interleukin 1 in type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2010, 6, 158–166. [Google Scholar] [CrossRef]
- Mastrandrea, L.; Yu, J.; Behrens, T.; Buchlis, J.; Albini, C.; Fourtner, S.; Quattrin, T. Etanercept treatment in children with new-onset type 1 diabetes: Pilot randomized, placebo-controlled, double-blind study. Diabetes Care 2009, 32, 1244–1249. [Google Scholar] [CrossRef]
- Kuhn, C.; Weiner, H.L. Therapeutic anti-CD3 monoclonal antibodies: From bench to bedside. Immunotherapy 2016, 8, 889–906. [Google Scholar] [CrossRef]
- Pescovitz, M.D.; Greenbaum, C.J.; Krause-Steinrauf, H.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; McGee, P.F.; Moran, A.M.; et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N. Engl. J. Med. 2009, 361, 2143–2152. [Google Scholar] [CrossRef]
- Polychronakos, C.; Li, Q. Understanding type 1 diabetes through genetics: Advances and prospects. Nat. Rev. Genet. 2011, 12, 781–792. [Google Scholar] [CrossRef]
- Wherrett, D.K.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Marks, J.B.; et al. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: A randomised double-blind trial. Lancet 2011, 378, 319–327. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 2010, 327, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, A.N.; Naziruddin, B.; Lockridge, A.; Tiwari, M.; Loganathan, G.; Takita, M.; Matsumoto, S.; Papas, K.; Trieger, M.; Rainis, H.; et al. Islet product characteristics and factors related to successful human islet transplantation from the Collaborative Islet Transplant Registry (CITR) 1999–2010. Am. J. Transplant. 2014, 14, 2595–2606. [Google Scholar] [CrossRef]
- Faradji, R.N.; Tharavanij, T.; Messinger, S.; Froud, T.; Pileggi, A.; Monroy, K.; Mineo, D.; Baidal, D.A.; Cure, P.; Ponte, G.; et al. Long-term insulin independence and improvement in insulin secretion after supplemental islet infusion under exenatide and etanercept. Transplantation 2008, 86, 1658–1665. [Google Scholar] [CrossRef]
- Suarez-Pinzon, W.L.; Cembrowski, G.S.; Rabinovitch, A. Combination therapy with a dipeptidyl peptidase-4 inhibitor and a proton pump inhibitor restores normoglycaemia in non-obese diabetic mice. Diabetologia 2009, 52, 1680–1682. [Google Scholar] [CrossRef]
- Suarez-Pinzon, W.L.; Rabinovitch, A. Combination therapy with a dipeptidyl peptidase-4 inhibitor and a proton pump inhibitor induces beta-cell neogenesis from adult human pancreatic duct cells implanted in immunodeficient mice. Cell Transplant. 2011, 20, 1343–1349. [Google Scholar] [CrossRef]
- Suarez-Pinzon, W.L.; Power, R.F.; Yan, Y.; Wasserfall, C.; Atkinson, M.; Rabinovitch, A. Combination therapy with glucagon-like peptide-1 and gastrin restores normoglycemia in diabetic NOD mice. Diabetes 2008, 57, 3281–3288. [Google Scholar] [CrossRef]
- Vigouroux, S.; Yvon, E.; Biagi, E.; Brenner, M.K. Antigen-induced regulatory T cells. Blood 2004, 104, 26–33. [Google Scholar] [CrossRef][Green Version]
- Griffin, K.J.; Thompson, P.A.; Gottschalk, M.; Kyllo, J.H.; Rabinovitch, A. Combination therapy with sitagliptin and lansoprazole in patients with recent-onset type 1 diabetes (REPAIR-T1D): 12-Month results of a multicentre, randomised, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol. 2014, 2, 710–718. [Google Scholar] [CrossRef]
- Giannoukakis, N.; Phillips, B.; Finegold, D.; Harnaha, J.; Trucco, M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 2011, 34, 2026–2032. [Google Scholar] [CrossRef]
- D’Addio, F.; Valderrama Vasquez, A.; Ben Nasr, M.; Franek, E.; Zhu, D.; Li, L.; Ning, G.; Snarski, E.; Fiorina, P. Autologous nonmyeloablative hematopoietic stem cell transplantation in new-onset type 1 diabetes: A multicenter analysis. Diabetes 2014, 63, 3041–3046. [Google Scholar] [CrossRef]
- Vital, E.M.; Emery, P. Abatacept in the treatment of rheumatoid arthritis. Ther. Clin. Risk Manag. 2006, 2, 365–375. [Google Scholar] [CrossRef][Green Version]
- Haller, M.J.; Schatz, D.A.; Skyler, J.S.; Krischer, J.P.; Bundy, B.N.; Miller, J.L.; Atkinson, M.A.; Becker, D.J.; Baidal, D.; DiMeglio, L.A.; et al. Low-Dose Anti-Thymocyte Globulin (ATG) Preserves beta-Cell Function and Improves HbA1c in New-Onset Type 1 Diabetes. Diabetes Care 2018, 41, 1917–1925. [Google Scholar] [CrossRef]
- Nepom, G.T.; Ehlers, M.; Mandrup-Poulsen, T. Anti-cytokine therapies in T1D: Concepts and strategies. Clin. Immunol. 2013, 149, 279–285. [Google Scholar] [CrossRef]
- Sumpter, K.M.; Adhikari, S.; Grishman, E.K.; White, P.C. Preliminary studies related to anti-interleukin-1beta therapy in children with newly diagnosed type 1 diabetes. Pediatr. Diabetes 2011, 12, 656–667. [Google Scholar] [CrossRef]
- Crino, A.; Schiaffini, R.; Manfrini, S.; Mesturino, C.; Visalli, N.; Beretta Anguissola, G.; Suraci, C.; Pitocco, D.; Spera, S.; Corbi, S.; et al. A randomized trial of nicotinamide and vitamin E in children with recent onset type 1 diabetes (IMDIAB IX). Eur. J. Endocrinol. 2004, 150, 719–724. [Google Scholar] [CrossRef]
- Hundhausen, C.; Roth, A.; Whalen, E.; Chen, J.; Schneider, A.; Long, S.A.; Wei, S.; Rawlings, R.; Kinsman, M.; Evanko, S.P.; et al. Enhanced T cell responses to IL-6 in type 1 diabetes are associated with early clinical disease and increased IL-6 receptor expression. Sci. Transl. Med. 2016, 8, 356ra119. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Interleukin (IL-6) Immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef]
- Hartemann, A.; Bensimon, G.; Payan, C.A.; Jacqueminet, S.; Bourron, O.; Nicolas, N.; Fonfrede, M.; Rosenzwajg, M.; Bernard, C.; Klatzmann, D. Low-dose interleukin 2 in patients with type 1 diabetes: A phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2013, 1, 295–305. [Google Scholar] [CrossRef]
- Bogdani, M.; Johnson, P.Y.; Potter-Perigo, S.; Nagy, N.; Day, A.J.; Bollyky, P.L.; Wight, T.N. Hyaluronan and hyaluronan-binding proteins accumulate in both human type 1 diabetic islets and lymphoid tissues and associate with inflammatory cells in insulitis. Diabetes 2014, 63, 2727–2743. [Google Scholar] [CrossRef]
- Aronson, R.; Gottlieb, P.A.; Christiansen, J.S.; Donner, T.W.; Bosi, E.; Bode, B.W.; Pozzilli, P.; Group, D.I. Low-dose otelixizumab anti-CD3 monoclonal antibody DEFEND-1 study: Results of the randomized phase III study in recent-onset human type 1 diabetes. Diabetes Care 2014, 37, 2746–2754. [Google Scholar] [CrossRef]
- Marino, E.; Silveira, P.A.; Stolp, J.; Grey, S.T. B cell-directed therapies in type 1 diabetes. Trends Immunol. 2011, 32, 287–294. [Google Scholar] [CrossRef]
- Townsend, M.J.; Monroe, J.G.; Chan, A.C. B-cell targeted therapies in human autoimmune diseases: An updated perspective. Immunol. Rev. 2010, 237, 264–283. [Google Scholar] [CrossRef]
- Coppieters, K.; von Herrath, M. Antigen-Specific Peptide Immunotherapy for Type 1 Diabetes: Proof of Safety, Hope for Efficacy. Cell Metab. 2017, 26, 595–597. [Google Scholar] [CrossRef][Green Version]
- Thrower, S.L.; James, L.; Hall, W.; Green, K.M.; Arif, S.; Allen, J.S.; Van-Krinks, C.; Lozanoska-Ochser, B.; Marquesini, L.; Brown, S.; et al. Proinsulin peptide immunotherapy in type 1 diabetes: Report of a first-in-man Phase I safety study. Clin. Exp. Immunol. 2009, 155, 156–165. [Google Scholar] [CrossRef]
- Alhadj Ali, M.; Liu, Y.F.; Arif, S.; Tatovic, D.; Shariff, H.; Gibson, V.B.; Yusuf, N.; Baptista, R.; Eichmann, M.; Petrov, N.; et al. Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Tian, J.; Clare-Salzler, M.; Herschenfeld, A.; Middleton, B.; Newman, D.; Mueller, R.; Arita, S.; Evans, C.; Atkinson, M.A.; Mullen, Y.; et al. Modulating autoimmune responses to GAD inhibits disease progression and prolongs islet graft survival in diabetes-prone mice. Nat. Med. 1996, 2, 1348–1353. [Google Scholar] [CrossRef]
- Yu, H.; Paiva, R.; Flavell, R.A. Harnessing the power of regulatory T-cells to control autoimmune diabetes: Overview and perspective. Immunology 2018, 153, 161–170. [Google Scholar] [CrossRef]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef]
- Henry, R.A.; Kendall, P.L.; Thomas, J.W. Autoantigen-specific B-cell depletion overcomes failed immune tolerance in type 1 diabetes. Diabetes 2012, 61, 2037–2044. [Google Scholar] [CrossRef]
- Gangemi, A.; Salehi, P.; Hatipoglu, B.; Martellotto, J.; Barbaro, B.; Kuechle, J.B.; Qi, M.; Wang, Y.; Pallan, P.; Owens, C.; et al. Islet transplantation for brittle type 1 diabetes: The UIC protocol. Am. J. Transplant. 2008, 8, 1250–1261. [Google Scholar] [CrossRef]
- Bell, G.M.; Anderson, A.E.; Diboll, J.; Reece, R.; Eltherington, O.; Harry, R.A.; Fouweather, T.; MacDonald, C.; Chadwick, T.; McColl, E.; et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann. Rheum. Dis. 2017, 76, 227–234. [Google Scholar] [CrossRef]
- Van de Pavert, S.A.; Mebius, R.E. New insights into the development of lymphoid tissues. Nat. Rev. Immunol. 2010, 10, 664–674. [Google Scholar] [CrossRef]
- Di Caro, V.; Phillips, B.; Engman, C.; Harnaha, J.; Trucco, M.; Giannoukakis, N. Retinoic acid-producing, ex-vivo-generated human tolerogenic dendritic cells induce the proliferation of immunosuppressive B lymphocytes. Clin. Exp. Immunol. 2013, 174, 302–317. [Google Scholar] [CrossRef]
- Morelli, A.E.; Thomson, A.W. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat. Rev. Immunol. 2007, 7, 610–621. [Google Scholar] [CrossRef]
- Voltarelli, J.C.; Couri, C.E.; Stracieri, A.B.; Oliveira, M.C.; Moraes, D.A.; Pieroni, F.; Coutinho, M.; Malmegrim, K.C.; Foss-Freitas, M.C.; Simoes, B.P.; et al. Autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA 2007, 297, 1568–1576. [Google Scholar] [CrossRef]
- Snarski, E.; Milczarczyk, A.; Halaburda, K.; Torosian, T.; Paluszewska, M.; Urbanowska, E.; Krol, M.; Boguradzki, P.; Jedynasty, K.; Franek, E.; et al. Immunoablation and autologous hematopoietic stem cell transplantation in the treatment of new-onset type 1 diabetes mellitus: Long-term observations. Bone Marrow Transplant. 2016, 51, 398–402. [Google Scholar] [CrossRef]
- Malmegrim, K.C.; de Azevedo, J.T.; Arruda, L.C.; Abreu, J.R.; Couri, C.E.; de Oliveira, G.L.; Palma, P.V.; Scortegagna, G.T.; Stracieri, A.B.; Moraes, D.A.; et al. Immunological Balance Is Associated with Clinical Outcome after Autologous Hematopoietic Stem Cell Transplantation in Type 1 Diabetes. Front. Immunol. 2017, 8, 167. [Google Scholar] [CrossRef]
- Murphy, M.B.; Moncivais, K.; Caplan, A.I. Mesenchymal stem cells: Environmentally responsive therapeutics for regenerative medicine. Exp. Mol. Med. 2013, 45, e54. [Google Scholar] [CrossRef]
- Bianco, P.; Cao, X.; Frenette, P.S.; Mao, J.J.; Robey, P.G.; Simmons, P.J.; Wang, C.Y. The meaning, the sense and the significance: Translating the science of mesenchymal stem cells into medicine. Nat. Med. 2013, 19, 35–42. [Google Scholar] [CrossRef]
- Eckert, M.A.; Vu, Q.; Xie, K.; Yu, J.; Liao, W.; Cramer, S.C.; Zhao, W. Evidence for high translational potential of mesenchymal stromal cell therapy to improve recovery from ischemic stroke. J. Cereb. Blood Flow Metab. 2013, 33, 1322–1334. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X.; Cao, W.; Shi, Y. Plasticity of mesenchymal stem cells in immunomodulation: Pathological and therapeutic implications. Nat. Immunol. 2014, 15, 1009–1016. [Google Scholar] [CrossRef]
- Moreira, A.; Kahlenberg, S.; Hornsby, P. Therapeutic potential of mesenchymal stem cells for diabetes. J. Mol. Endocrinol. 2017, 59, R109–R120. [Google Scholar] [CrossRef]
- Carlsson, P.O.; Schwarcz, E.; Korsgren, O.; Le Blanc, K. Preserved beta-cell function in type 1 diabetes by mesenchymal stromal cells. Diabetes 2015, 64, 587–592. [Google Scholar] [CrossRef]
- Jurewicz, M.; Yang, S.; Augello, A.; Godwin, J.G.; Moore, R.F.; Azzi, J.; Fiorina, P.; Atkinson, M.; Sayegh, M.H.; Abdi, R. Congenic mesenchymal stem cell therapy reverses hyperglycemia in experimental type 1 diabetes. Diabetes 2010, 59, 3139–3147. [Google Scholar] [CrossRef]
- Cai, J.; Wu, Z.; Xu, X.; Liao, L.; Chen, J.; Huang, L.; Wu, W.; Luo, F.; Wu, C.; Pugliese, A.; et al. Umbilical Cord Mesenchymal Stromal Cell With Autologous Bone Marrow Cell Transplantation in Established Type 1 3Diabetes: A Pilot Randomized Controlled Open-Label Clinical Study to Assess Safety and Impact on Insulin Secretion. Diabetes Care 2016, 39, 149–157. [Google Scholar] [CrossRef]
- Maude, S.; Barrett, D.M. Current status of chimeric antigen receptor therapy for haematological malignancies. Br. J. Haematol. 2016, 172, 11–22. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Yoon, J.; Schmidt, A.; Zhang, A.H.; Konigs, C.; Kim, Y.C.; Scott, D.W. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood 2017, 129, 238–245. [Google Scholar] [CrossRef]
- MacDonald, K.G.; Hoeppli, R.E.; Huang, Q.; Gillies, J.; Luciani, D.S.; Orban, P.C.; Broady, R.; Levings, M.K. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J. Clin. Investig. 2016, 126, 1413–1424. [Google Scholar] [CrossRef]
- Pierini, A.; Iliopoulou, B.P.; Peiris, H.; Perez-Cruz, M.; Baker, J.; Hsu, K.; Gu, X.; Zheng, P.P.; Erkers, T.; Tang, S.W.; et al. T cells expressing chimeric antigen receptor promote immune tolerance. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Noyan, F.; Zimmermann, K.; Hardtke-Wolenski, M.; Knoefel, A.; Schulde, E.; Geffers, R.; Hust, M.; Huehn, J.; Galla, M.; Morgan, M.; et al. Prevention of Allograft Rejection by Use of Regulatory T Cells with an MHC-Specific Chimeric Antigen Receptor. Am. J. Transplant. 2017, 17, 917–930. [Google Scholar] [CrossRef]
- Fransson, M.; Piras, E.; Burman, J.; Nilsson, B.; Essand, M.; Lu, B.; Harris, R.A.; Magnusson, P.U.; Brittebo, E.; Loskog, A.S. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J. Neuroinflamm. 2012, 9, 112. [Google Scholar] [CrossRef]
- Skuljec, J.; Chmielewski, M.; Happle, C.; Habener, A.; Busse, M.; Abken, H.; Hansen, G. Chimeric Antigen Receptor-Redirected Regulatory T Cells Suppress Experimental Allergic Airway Inflammation, a Model of Asthma. Front. Immunol. 2017, 8, 1125. [Google Scholar] [CrossRef]
- Kim, Y.C.; Zhang, A.H.; Su, Y.; Rieder, S.A.; Rossi, R.J.; Ettinger, R.A.; Pratt, K.P.; Shevach, E.M.; Scott, D.W. Engineered antigen-specific human regulatory T cells: Immunosuppression of FVIII-specific T- and B-cell responses. Blood 2015, 125, 1107–1115. [Google Scholar] [CrossRef]
- Hull, C.M.; Nickolay, L.E.; Estorninho, M.; Richardson, M.W.; Riley, J.L.; Peakman, M.; Maher, J.; Tree, T.I. Generation of human islet-specific regulatory T cells by TCR gene transfer. J. Autoimmun. 2017, 79, 63–73. [Google Scholar] [CrossRef]
- Tenspolde, M.; Zimmermann, K.; Weber, L.C.; Hapke, M.; Lieber, M.; Dywicki, J.; Frenzel, A.; Hust, M.; Galla, M.; Buitrago-Molina, L.E.; et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J. Autoimmun. 2019. [Google Scholar] [CrossRef]
- Balhuizen, A.; Massa, S.; Mathijs, I.; Turatsinze, J.V.; De Vos, J.; Demine, S.; Xavier, C.; Villate, O.; Millard, I.; Egrise, D.; et al. A nanobody-based tracer targeting DPP6 for non-invasive imaging of human pancreatic endocrine cells. Sci. Rep. 2017, 7, 15130. [Google Scholar] [CrossRef]
- Burtea, C.; Laurent, S.; Crombez, D.; Delcambre, S.; Sermeus, C.; Millard, I.; Rorive, S.; Flamez, D.; Beckers, M.C.; Salmon, I.; et al. Development of a peptide-functionalized imaging nanoprobe for the targeting of (FXYD2)gammaa as a highly specific biomarker of pancreatic beta cells. Contrast Media Mol. Imaging 2015, 10, 398–412. [Google Scholar] [CrossRef]
- Saunders, D.C.; Brissova, M.; Phillips, N.; Shrestha, S.; Walker, J.T.; Aramandla, R.; Poffenberger, G.; Flaherty, D.K.; Weller, K.P.; Pelletier, J.; et al. Ectonucleoside Triphosphate Diphosphohydrolase-3 Antibody Targets Adult Human Pancreatic beta Cells for In Vitro and In Vivo Analysis. Cell Metab. 2019, 29, 745–754. [Google Scholar] [CrossRef]
- Brown, K.; DeCoffe, D.; Molcan, E.; Gibson, D.L. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients 2012, 4, 1095–1119. [Google Scholar] [CrossRef]
- Macdonald, T.T.; Monteleone, G. Immunity, inflammation, and allergy in the gut. Science 2005, 307, 1920–1925. [Google Scholar] [CrossRef]
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef]
- Quigley, E.M. Gut bacteria in health and disease. Gastroenterol. Hepatol. 2013, 9, 560–569. [Google Scholar]
- Wichmann, A.; Allahyar, A.; Greiner, T.U.; Plovier, H.; Lunden, G.O.; Larsson, T.; Drucker, D.J.; Delzenne, N.M.; Cani, P.D.; Backhed, F. Microbial modulation of energy availability in the colon regulates intestinal transit. Cell Host Microbe 2013, 14, 582–590. [Google Scholar] [CrossRef]
- Sanz, Y.; Olivares, M.; Moya-Perez, A.; Agostoni, C. Understanding the role of gut microbiome in metabolic disease risk. Pediatr. Res. 2015, 77, 236–244. [Google Scholar] [CrossRef]
- Aljutaily, T.; Consuegra-Fernandez, M.; Aranda, F.; Lozano, F.; Huarte, E. Gut microbiota metabolites for sweetening type I diabetes. Cell. Mol. Immunol. 2018, 15, 92–95. [Google Scholar] [CrossRef]
- Vaishnava, S.; Behrendt, C.L.; Ismail, A.S.; Eckmann, L.; Hooper, L.V. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. USA 2008, 105, 20858–20863. [Google Scholar] [CrossRef]
- Barnes, M.J.; Powrie, F. Regulatory T cells reinforce intestinal homeostasis. Immunity 2009, 31, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed]
- Paun, A.; Yau, C.; Danska, J.S. The Influence of the Microbiome on Type 1 Diabetes. J. Immunol. 2017, 198, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Vaarala, O.; Atkinson, M.A.; Neu, J. The “perfect storm” for type 1 diabetes: The complex interplay between intestinal microbiota, gut permeability, and mucosal immunity. Diabetes 2008, 57, 2555–2562. [Google Scholar] [CrossRef]
- Pellegrini, S.; Sordi, V.; Bolla, A.M.; Saita, D.; Ferrarese, R.; Canducci, F.; Clementi, M.; Invernizzi, F.; Mariani, A.; Bonfanti, R.; et al. Duodenal Mucosa of Patients With Type 1 Diabetes Shows Distinctive Inflammatory Profile and Microbiota. J. Clin. Endocrinol. Metab. 2017, 102, 1468–1477. [Google Scholar] [CrossRef]
- Allin, K.H.; Tremaroli, V.; Caesar, R.; Jensen, B.A.H.; Damgaard, M.T.F.; Bahl, M.I.; Licht, T.R.; Hansen, T.H.; Nielsen, T.; Dantoft, T.M.; et al. Aberrant intestinal microbiota in individuals with prediabetes. Diabetologia 2018, 61, 810–820. [Google Scholar] [CrossRef]
- Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Novelo, L.L.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; Hyoty, H.; et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011, 5, 82–91. [Google Scholar] [CrossRef]
- Huang, Y.; Li, S.C.; Hu, J.; Ruan, H.B.; Guo, H.M.; Zhang, H.H.; Wang, X.; Pei, Y.F.; Pan, Y.; Fang, C. Gut microbiota profiling in Han Chinese with type 1 diabetes. Diabetes Res. Clin. Pract. 2018, 141, 256–263. [Google Scholar] [CrossRef]
- Brown, C.T.; Davis-Richardson, A.G.; Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE 2011, 6, e25792. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef]
- Derrien, M.; Belzer, C.; de Vos, W.M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef]
- Tanca, A.; Palomba, A.; Fraumene, C.; Manghina, V.; Silverman, M.; Uzzau, S. Clostridial Butyrate Biosynthesis Enzymes Are Significantly Depleted in the Gut Microbiota of Nonobese Diabetic Mice. mSphere 2018, 3. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyotylainen, T.; Hamalainen, A.M.; Peet, A.; Tillmann, V.; Poho, P.; Mattila, I.; et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015, 17, 260–273. [Google Scholar] [CrossRef]
- Kriegel, M.A.; Sefik, E.; Hill, J.A.; Wu, H.J.; Benoist, C.; Mathis, D. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc. Natl. Acad. Sci. USA 2011, 108, 11548–11553. [Google Scholar] [CrossRef]
- Marco, M.L.; Heeney, D.; Binda, S.; Cifelli, C.J.; Cotter, P.D.; Foligne, B.; Ganzle, M.; Kort, R.; Pasin, G.; Pihlanto, A.; et al. Health benefits of fermented foods: Microbiota and beyond. Curr. Opin. Biotechnol. 2017, 44, 94–102. [Google Scholar] [CrossRef]
- Macia, L.; Thorburn, A.N.; Binge, L.C.; Marino, E.; Rogers, K.E.; Maslowski, K.M.; Vieira, A.T.; Kranich, J.; Mackay, C.R. Microbial influences on epithelial integrity and immune function as a basis for inflammatory diseases. Immunol. Rev. 2012, 245, 164–176. [Google Scholar] [CrossRef]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916. [Google Scholar] [CrossRef]
- Smits, L.P.; Bouter, K.E.; de Vos, W.M.; Borody, T.J.; Nieuwdorp, M. Therapeutic potential of fecal microbiota transplantation. Gastroenterology 2013, 145, 946–953. [Google Scholar] [CrossRef]
- He, C.; Shan, Y.; Song, W. Targeting gut microbiota as a possible therapy for diabetes. Nutr. Res. 2015, 35, 361–367. [Google Scholar] [CrossRef]
- Gomes, A.C.; Bueno, A.A.; de Souza, R.G.; Mota, J.F. Gut microbiota, probiotics and diabetes. Nutr. J. 2014, 13, 60. [Google Scholar] [CrossRef]
- Mishra, S.P.; Wang, S.; Nagpal, R.; Miller, B.; Singh, R.; Taraphder, S.; Yadav, H. Probiotics and Prebiotics for the Amelioration of Type 1 Diabetes: Present and Future Perspectives. Microorganisms 2019, 7, 67. [Google Scholar] [CrossRef]
- Zhao, S.; Liu, W.; Wang, J.; Shi, J.; Sun, Y.; Wang, W.; Ning, G.; Liu, R.; Hong, J. Akkermansia muciniphila improves metabolic profiles by reducing inflammation in chow diet-fed mice. J. Mol. Endocrinol. 2017, 58, 1–14. [Google Scholar] [CrossRef]
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; Van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Delzenne, N.M.; et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: A proof-of-concept exploratory study. Nat. Med. 2019, 25, 1096–1103. [Google Scholar] [CrossRef]
- Wang, L.; Wu, Y.; Zhuang, L.; Chen, X.; Min, H.; Song, S.; Liang, Q.; Li, A.D.; Gao, Q. Puerarin prevents high-fat diet-induced obesity by enriching Akkermansia muciniphila in the gut microbiota of mice. PLoS ONE 2019, 14, e0218490. [Google Scholar] [CrossRef]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef]
- Howell, M.D.; Fitzsimons, C.; Smith, P.A. JAK/STAT inhibitors and other small molecule cytokine antagonists for the treatment of allergic disease. Ann. Allergy Asthma Immunol. 2018, 120, 367–375. [Google Scholar] [CrossRef]
- Ferreira, R.C.; Guo, H.; Coulson, R.M.; Smyth, D.J.; Pekalski, M.L.; Burren, O.S.; Cutler, A.J.; Doecke, J.D.; Flint, S.; McKinney, E.F.; et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes 2014, 63, 2538–2550. [Google Scholar] [CrossRef]
- Huang, X.; Yuang, J.; Goddard, A.; Foulis, A.; James, R.F.; Lernmark, A.; Pujol-Borrell, R.; Rabinovitch, A.; Somoza, N.; Stewart, T.A. Interferon expression in the pancreases of patients with type I diabetes. Diabetes 1995, 44, 658–664. [Google Scholar] [CrossRef]
- Kallionpaa, H.; Elo, L.L.; Laajala, E.; Mykkanen, J.; Ricano-Ponce, I.; Vaarma, M.; Laajala, T.D.; Hyoty, H.; Ilonen, J.; Veijola, R.; et al. Innate immune activity is detected prior to seroconversion in children with HLA-conferred type 1 diabetes susceptibility. Diabetes 2014, 63, 2402–2414. [Google Scholar] [CrossRef] [PubMed]
- Foulis, A.K.; Farquharson, M.A.; Meager, A. Immunoreactive alpha-interferon in insulin-secreting beta cells in type 1 diabetes mellitus. Lancet 1987, 2, 1423–1427. [Google Scholar] [CrossRef]
- Richardson, S.J.; Rodriguez-Calvo, T.; Gerling, I.C.; Mathews, C.E.; Kaddis, J.S.; Russell, M.A.; Zeissler, M.; Leete, P.; Krogvold, L.; Dahl-Jorgensen, K.; et al. Islet cell hyperexpression of HLA class I antigens: A defining feature in type 1 diabetes. Diabetologia 2016, 59, 2448–2458. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; Dos Santos, R.S.; Op de Beeck, A.; Coomans de Brachene, A.; Marselli, L.; Marchetti, P.; Eizirik, D.L. Interferon-alpha mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia 2017, 60, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.H.; Zou, Y.F.; Feng, X.L.; Li, J.; Wang, F.; Pan, F.M.; Ye, D.Q. Meta-analysis of TYK2 gene polymorphisms association with susceptibility to autoimmune and inflammatory diseases. Mol. Biol. Rep. 2011, 38, 4663–4672. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.; Smyth, D.J.; Maisuria-Armer, M.; Walker, N.M.; Todd, J.A.; Clayton, D.G. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat. Genet. 2010, 42, 68–71. [Google Scholar] [CrossRef]
- Roep, B.O.; Kleijwegt, F.S.; van Halteren, A.G.; Bonato, V.; Boggi, U.; Vendrame, F.; Marchetti, P.; Dotta, F. Islet inflammation and CXCL10 in recent-onset type 1 diabetes. Clin. Exp. Immunol. 2010, 159, 338–343. [Google Scholar] [CrossRef]
- Sarkar, S.A.; Lee, C.E.; Victorino, F.; Nguyen, T.T.; Walters, J.A.; Burrack, A.; Eberlein, J.; Hildemann, S.K.; Homann, D. Expression and regulation of chemokines in murine and human type 1 diabetes. Diabetes 2012, 61, 436–446. [Google Scholar] [CrossRef]
- Morimoto, J.; Yoneyama, H.; Shimada, A.; Shigihara, T.; Yamada, S.; Oikawa, Y.; Matsushima, K.; Saruta, T.; Narumi, S. CXC chemokine ligand 10 neutralization suppresses the occurrence of diabetes in nonobese diabetic mice through enhanced beta cell proliferation without affecting insulitis. J. Immunol. 2004, 173, 7017–7024. [Google Scholar] [CrossRef]
- Savinov, A.Y.; Wong, F.S.; Chervonsky, A.V. IFN-gamma affects homing of diabetogenic T cells. J. Immunol. 2001, 167, 6637–6643. [Google Scholar] [CrossRef]
- Barroso-Sousa, R.; Ott, P.A.; Hodi, F.S.; Kaiser, U.B.; Tolaney, S.M.; Min, L. Endocrine dysfunction induced by immune checkpoint inhibitors: Practical recommendations for diagnosis and clinical management. Cancer 2018, 124, 1111–1121. [Google Scholar] [CrossRef]
- Cukier, P.; Santini, F.C.; Scaranti, M.; Hoff, A.O. Endocrine side effects of cancer immunotherapy. Endocr. Relat. Cancer 2017, 24, T331–T347. [Google Scholar] [CrossRef]
- Stamatouli, A.M.; Quandt, Z.; Perdigoto, A.L.; Clark, P.L.; Kluger, H.; Weiss, S.A.; Gettinger, S.; Sznol, M.; Young, A.; Rushakoff, R.; et al. Collateral Damage: Insulin-Dependent Diabetes Induced With Checkpoint Inhibitors. Diabetes 2018, 67, 1471–1480. [Google Scholar] [CrossRef]
- Ansari, M.J.; Salama, A.D.; Chitnis, T.; Smith, R.N.; Yagita, H.; Akiba, H.; Yamazaki, T.; Azuma, M.; Iwai, H.; Khoury, S.J.; et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J. Exp. Med. 2003, 198, 63–69. [Google Scholar] [CrossRef]
- Coomans de Brachene, A.; Dos Santos, R.S.; Marroqui, L.; Colli, M.L.; Marselli, L.; Mirmira, R.G.; Marchetti, P.; Eizirik, D.L. IFN-alpha induces a preferential long-lasting expression of MHC class I in human pancreatic beta cells. Diabetologia 2018, 61, 636–640. [Google Scholar] [CrossRef]
- Trivedi, P.M.; Graham, K.L.; Scott, N.A.; Jenkins, M.R.; Majaw, S.; Sutherland, R.M.; Fynch, S.; Lew, A.M.; Burns, C.J.; Krishnamurthy, B.; et al. Repurposed JAK1/JAK2 Inhibitor Reverses Established Autoimmune Insulitis in NOD Mice. Diabetes 2017, 66, 1650–1660. [Google Scholar] [CrossRef]
- Kostromina, E.; Gustavsson, N.; Wang, X.; Lim, C.Y.; Radda, G.K.; Li, C.; Han, W. Glucose intolerance and impaired insulin secretion in pancreas-specific signal transducer and activator of transcription-3 knockout mice are associated with microvascular alterations in the pancreas. Endocrinology 2010, 151, 2050–2059. [Google Scholar] [CrossRef][Green Version]
- Kostromina, E.; Wang, X.; Han, W. Altered islet morphology but normal islet secretory function in vitro in a mouse model with microvascular alterations in the pancreas. PLoS ONE 2013, 8, e71277. [Google Scholar] [CrossRef]
- Lee, J.Y.; Gavrilova, O.; Davani, B.; Na, R.; Robinson, G.W.; Hennighausen, L. The transcription factors Stat5a/b are not required for islet development but modulate pancreatic beta-cell physiology upon aging. Biochim. Biophys. Acta 2007, 1773, 1455–1461. [Google Scholar] [CrossRef][Green Version]
- Bach, J.F. Insulin-dependent diabetes mellitus as an autoimmune disease. Endocr. Rev. 1994, 15, 516–542. [Google Scholar] [CrossRef]
- Eisenbarth, G.S. Type 1 diabetes: Molecular, cellular and clinical immunology. Adv. Exp. Med. Biol. 2004, 552, 306–310. [Google Scholar]
- Gomez-Tourino, I.; Arif, S.; Eichmann, M.; Peakman, M. T cells in type 1 diabetes: Instructors, regulators and effectors: A comprehensive review. J. Autoimmun. 2016, 66, 7–16. [Google Scholar] [CrossRef]
- Smith, E.L.; Peakman, M. Peptide Immunotherapy for Type 1 Diabetes-Clinical Advances. Front. Immunol. 2018, 9, 392. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| NOD | Human | |
|---|---|---|
| Age at onset | > 10 weeks | >6 months–late adolescence |
| Genetic susceptibility | MHC most important | HLA most important |
| Autoantigens | Insulin, GAD, IA-2, IA-2b, ZnT8, IGRP, Crhomogranin A | Insulin, GAD, IA-2, IA-2b, ZnT8, IGRP, IAPP, HSP60, Carboxypetidase H |
| Insulitis | DCs, Macrophages, B cells, NK cells, CD4 & CD8 T cells | DCs, Macrophages, B cells, NK cells, CD4 & CD8 T cells |
| Ketoacidosis | Mild | Severe |
| Gender effect | Females predominantly affected | Males and females almost equally affected |
| Antigen-Independent Strategies | References | |
|---|---|---|
| Antibody-based therapies | ||
| Anti-CTLA-4 | Clinical trial NCT01773707 | |
| Anti-CD3 | Clinical trial NCT01030861 | |
| Anti-CD2 | [18] | |
| Anti-thymocyte globulin (ATG) | [35] | |
| Proinflammatory citokine-based therapies | ||
| IL-1a/IL-1b | [41] | |
| TNF | [42] | |
| Nicotinamide | [43] | |
| IL-12/23 | Clinical trial NCT02117765 | |
| IL-6 | Clinical trial NCT02293837 | |
| Treg-mediated strategies | ||
| Treg suppression | [44,45] | |
| Removal of autoreactive T-cells | ||
| Anti-CD3 | [46,47] | |
| B-cell-targeting therapies | ||
| Anti-CD2 | [48] | |
| Antigen-dependent immunotherapy | ||
| Beta cell-autoantigen vaccination | ||
| GAD | [49] | |
| Specific T-cell strategies | ||
| Tolerized T effector cells | [50] | |
| Specific B-cell strategies | ||
| Depleting insulin-reactive B cells | [51] | |
| Beta cell therapies | ||
| Replacement therapies | ||
| Edmonton protocol | [7] | |
| Beta-cell regeneration strategies | ||
| Gastrin + GLP-1 | [52,53] | |
| Stem cell therapy strategies | ||
| Tolerogenic DCs (tDCs) | ||
| Autologous tDCs | [54,55] | |
| Combination tDC + Tregs | [56,57] | |
| Hematopoietic stem cells (HSC) | ||
| Autologous myeloablative HSC transplantation | [58] | |
| Autologous non-myeloablative HSC transplantation | [59] | |
| Mesenchymal stem cells (MSC) | ||
| Autologous MSCs | [60,61] | |
| Allogeneic adipose-derived MSCs | Clinical trial NCT02940418 | |
| Umbilical cord blood MSCs (UC-MSCs) | [62] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabello-Olmo, M.; Araña, M.; Radichev, I.; Smith, P.; Huarte, E.; Barajas, M. New Insights into Immunotherapy Strategies for Treating Autoimmune Diabetes. Int. J. Mol. Sci. 2019, 20, 4789. https://doi.org/10.3390/ijms20194789
Cabello-Olmo M, Araña M, Radichev I, Smith P, Huarte E, Barajas M. New Insights into Immunotherapy Strategies for Treating Autoimmune Diabetes. International Journal of Molecular Sciences. 2019; 20(19):4789. https://doi.org/10.3390/ijms20194789
Chicago/Turabian StyleCabello-Olmo, Miriam, Miriam Araña, Ilian Radichev, Paul Smith, Eduardo Huarte, and Miguel Barajas. 2019. "New Insights into Immunotherapy Strategies for Treating Autoimmune Diabetes" International Journal of Molecular Sciences 20, no. 19: 4789. https://doi.org/10.3390/ijms20194789
APA StyleCabello-Olmo, M., Araña, M., Radichev, I., Smith, P., Huarte, E., & Barajas, M. (2019). New Insights into Immunotherapy Strategies for Treating Autoimmune Diabetes. International Journal of Molecular Sciences, 20(19), 4789. https://doi.org/10.3390/ijms20194789