Polyamine Catabolism in Acute Kidney Injury


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
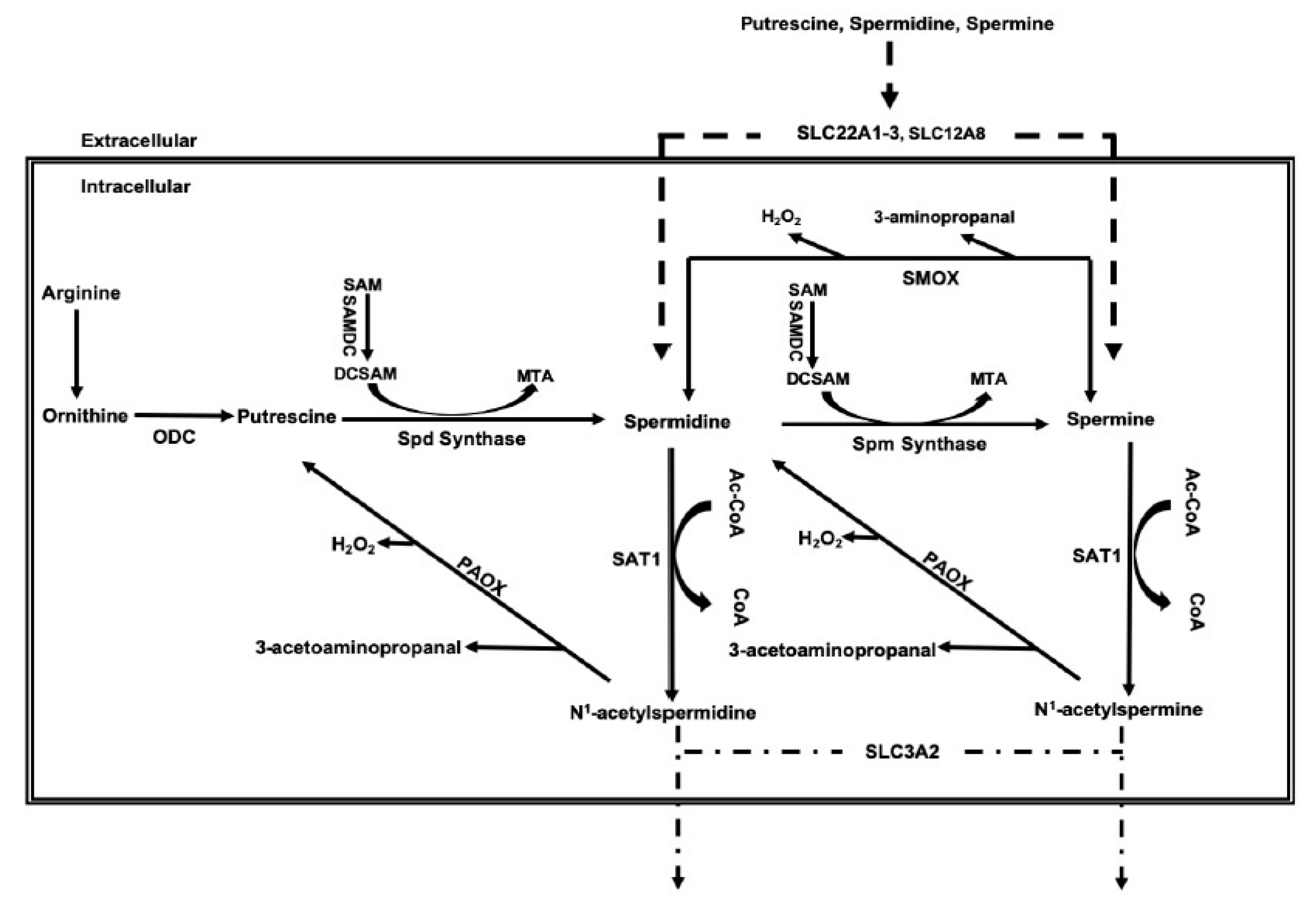
2. Regulation of Polyamines and the Polyamine Metabolic Pathway
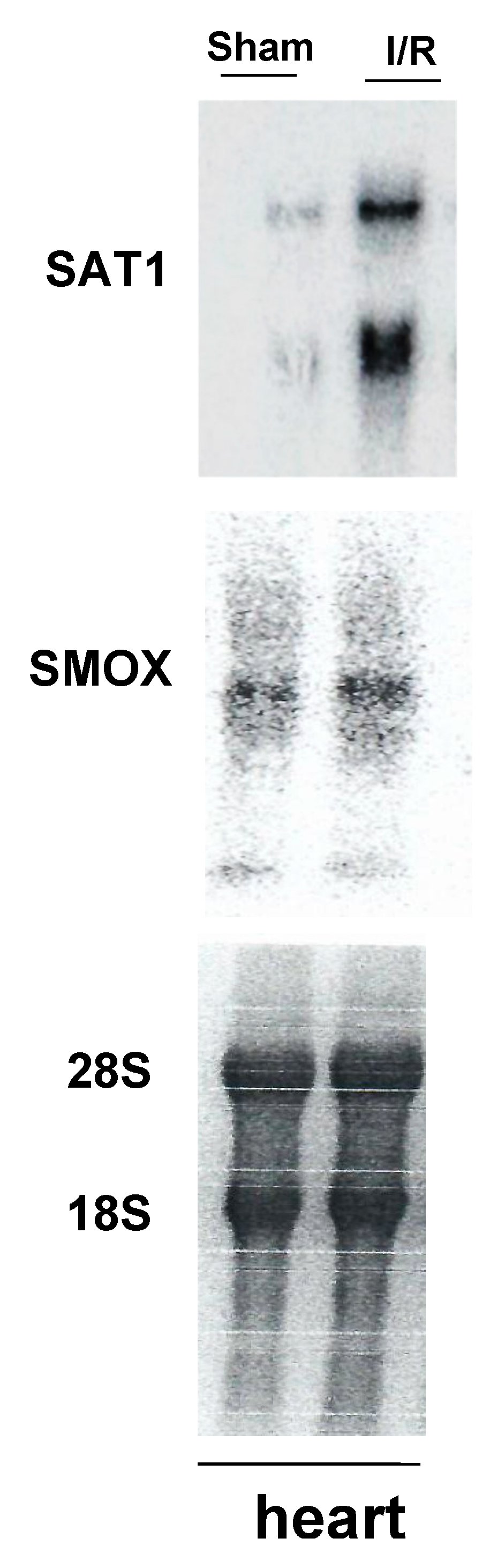
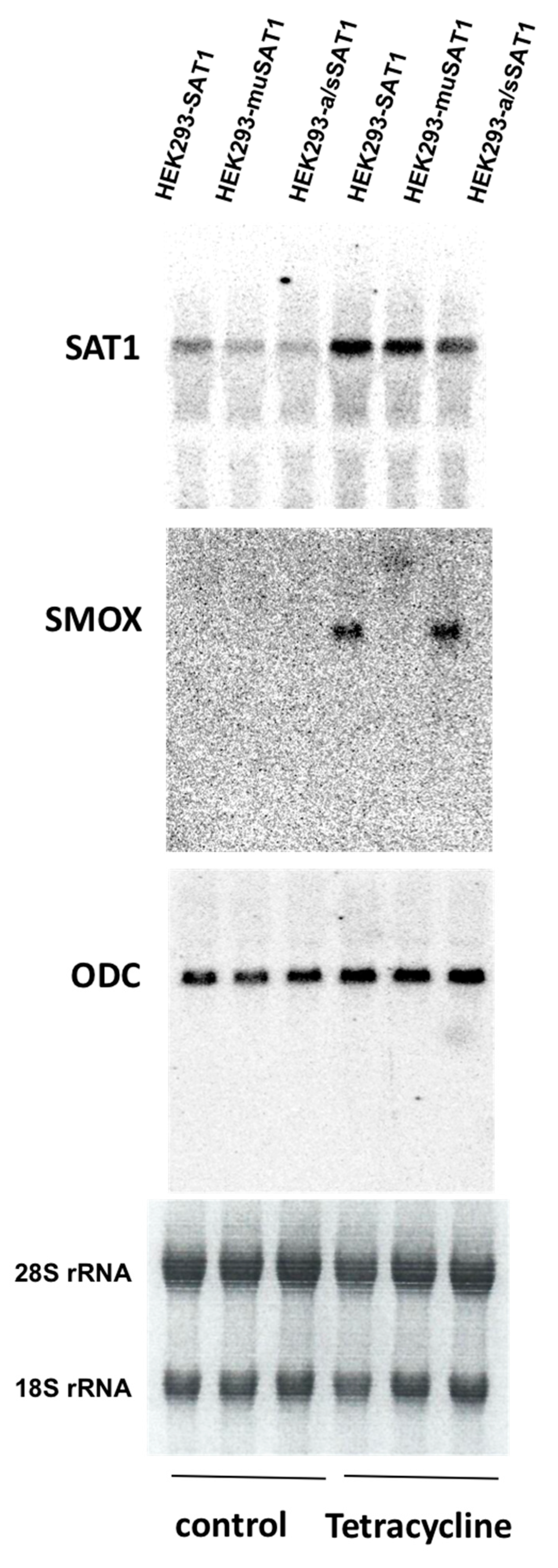
3. Expression and Activity of Polyamine Catabolic Enzymes, SAT1, and SMOX Increases in Injured Tissues
4. Expression and Activity of Polyamine Catabolic Enzymes in AKI
5. In Vivo and In Vitro Effects of Enhanced Polyamine Catabolism
6. Biochemical Basis of Renal Injury Caused by Enhanced Polyamine Catabolism
7. Mechanistic Basis of Cell Injury and Tissue Damage Caused by the Upregulation of Polyamine Catabolism
8. Conclusions
Funding
Conflicts of Interest
References
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [PubMed]
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar] [PubMed]
- Martin, R.K. Acute kidney injury: Advances in definition, pathophysiology, and diagnosis. AACN Adv. Crit. Care 2010, 21, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Zahedi, K.; Wang, Z.; Barone, S.; Prada, A.E.; Kelly, C.N.; Casero, R.A.; Yokota, N.; Porter, C.W.; Rabb, H.; Soleimani, M. Expression of SSAT, a novel biomarker of tubular cell damage, increases in kidney ischemia-reperfusion injury. Am. J. Physiol. Renal Physiol. 2003, 284, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zahedi, K.; Barone, S.; Tehrani, K.; Rabb, H.; Matlin, K.; Casero, R.A.; Soleimani, M. Overexpression of SSAT in kidney cells recapitulates various phenotypic aspects of kidney ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2004, 15, 1844–1852. [Google Scholar] [CrossRef] [PubMed]
- Barone, S.; Okaya, T.; Rudich, S.; Petrovic, S.; Tenrani, K.; Wang, Z.; Zahedi, K.; Casero, R.A.; Lentsch, A.B.; Soleimani, M. Distinct and sequential upregulation of genes regulating cell growth and cell cycle progression during hepatic ischemia-reperfusion injury. Am. J. Physiol. Cell Physiol. 2005, 289, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Zahedi, K.; Barone, S.; Destefano-Shields, C.; Brooks, M.; Murray-Stewart, T.; Dunworth, M.; Li, W.; Doherty, J.R.; Hall, M.A.; Smith, R.D.; et al. Activation of endoplasmic reticulum stress response by enhanced polyamine catabolism is important in the mediation of cisplatin-induced acute kidney injury. PLoS ONE 2017, 12, e0184570. [Google Scholar] [CrossRef]
- Zahedi, K.; Barone, S.; Kramer, D.L.; Amlal, H.; Alhonen, L.; Janne, J.; Porter, C.W.; Soleimani, M. The role of spermidine/spermine N1-acetyltransferase in endotoxin-induced acute kidney injury. Am. J. Physiol. Cell Physiol. 2010, 299, 164–174. [Google Scholar] [CrossRef]
- Zahedi, K.; Barone, S.L.; Xu, J.; Steinbergs, N.; Schuster, R.; Lentsch, A.B.; Amlal, H.; Wang, J.; Casero, R.A., Jr.; Soleimani, M. Hepatocyte-specific ablation of spermine/spermidine-N1-acetyltransferase gene reduces the severity of CCl4-induced acute liver injury. Am. J. Physiol. Gastrointest Liver Physiol. 2012, 303, 546–560. [Google Scholar] [CrossRef] [PubMed]
- Zahedi, K.; Huttinger, F.; Morrison, R.; Murray-Stewart, T.; Casero, R.A.; Strauss, K.I. Polyamine catabolism is enhanced after traumatic brain injury. J. Neurotrauma 2010, 27, 515–525. [Google Scholar] [CrossRef]
- Igarashi, K.; Kashiwagi, K. Modulation of protein synthesis by polyamines. IUBMB Life 2015, 67, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Mandal, A.; Johansson, H.E.; Orjalo, A.V.; Park, M.H. Depletion of cellular polyamines, spermidine and spermine, causes a total arrest in translation and growth in mammalian cells. Proc. Natl Acad. Sci. USA 2013, 110, 2169–2174. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.; Alam, M.K.; Ali, R. Polyamine induced Z-conformation of native calf thymus DNA. FEBS Lett. 1995, 368, 27–30. [Google Scholar] [CrossRef]
- Sun, H.; Xiang, J.; Liu, Y.; Li, L.; Li, Q.; Xu, G.; Tang, Y. A stabilizing and denaturing dual-effect for natural polyamines interacting with G-quadruplexes depending on concentration. Biochimie 2011, 93, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Brooks, W.H. Increased polyamines alter chromatin and stabilize autoantigens in autoimmune diseases. Front. Immunol. 2013, 4, 91. [Google Scholar] [CrossRef] [PubMed]
- Perez-Leal, O.; Barrero, C.A.; Clarkson, A.B.; Casero, R.A., Jr.; Merali, S. Polyamine-regulated translation of spermidine/spermine-N1-acetyltransferase. Mol. Cell. Biol. 2012, 32, 1453–1467. [Google Scholar] [CrossRef] [PubMed]
- Antony, T.; Hoyer, W.; Cherny, D.; Heim, G.; Jovin, T.M.; Subramaniam, V. Cellular polyamines promote the aggregation of alpha-synuclein. J. Biol. Chem. 2003, 278, 3235–3240. [Google Scholar] [CrossRef] [PubMed]
- Cordella-Miele, E.; Miele, L.; Beninati, S.; Mukherjee, A.B. Transglutaminase-catalyzed incorporation of polyamines into phospholipase A2. J. Biochem. 1993, 113, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Krasnoslobodtsev, A.V.; Peng, J.; Asiago, J.M.; Hindupur, J.; Rochet, J.C.; Lyubchenko, Y.L. Effect of spermidine on misfolding and interactions of alpha-synuclein. PLoS ONE 2012, 7, e38099. [Google Scholar] [CrossRef] [PubMed]
- Abdulhussein, A.A.; Wallace, H.M. Polyamines and membrane transporters. Amino Acids 2014, 46, 655–660. [Google Scholar] [CrossRef]
- Daigle, N.D.; Carpentier, G.A.; Frenette-Cotton, R.; Simard, M.G.; Lefoll, M.H.; Noel, M.; Caron, L.; Noel, J.; Isenring, P. Molecular characterization of a human cation-Cl- cotransporter (SLC12A8A, CCC9A) that promotes polyamine and amino acid transport. J. Cell. Physiol. 2009, 220, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Roy, U.K.; Rial, N.S.; Kachel, K.L.; Gerner, E.W. Activated K-RAS increases polyamine uptake in human colon cancer cells through modulation of caveolar endocytosis. Mol. Carcinog. 2008, 47, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Stringer, D.E.; Blohm-Mangone, K.A.; Gerner, E.W. Polyamine transport is mediated by both endocytic and solute carrier transport mechanisms in the gastrointestinal tract. Am. J. Physiol. Gastrointest Liver Physiol. 2010, 299, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Yerushalmi, H.F.; Tsaprailis, G.; Stringer, D.E.; Pastorian, K.E.; Hawel, L., 3rd; Byus, C.V.; Gerner, E.W. Identification and characterization of a diamine exporter in colon epithelial cells. J. Biol. Chem. 2008, 283, 26428–26435. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A.; Pegg, A.E. Polyamine catabolism and disease. Biochem. J. 2009, 421, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Bupp, C.P.; Schultz, C.R.; Uhl, K.L.; Rajasekaran, S.; Bachmann, A.S. Novel de novo pathogenic variant in the ODC1 gene in a girl with developmental delay, alopecia, and dysmorphic features. Am. J. Med. Genet. A 2018, 176, 2548–2553. [Google Scholar] [CrossRef]
- Li, C.; Brazill, J.M.; Liu, S.; Bello, C.; Zhu, Y.; Morimoto, M.; Cascio, L.; Pauly, R.; Diaz-Perez, Z.; Malicdan, M.C.V.; et al. Spermine synthase deficiency causes lysosomal dysfunction and oxidative stress in models of Snyder-Robinson syndrome. Nat. Commun. 2017, 8, 1257. [Google Scholar] [CrossRef]
- Murray-Stewart, T.; Dunworth, M.; Foley, J.R.; Schwartz, C.E.; Casero, R.A., Jr. Polyamine Homeostasis in Snyder-Robinson Syndrome. Med. Sci. 2018, 6, 112. [Google Scholar] [CrossRef]
- Chaturvedi, R.; de Sablet, T.; Asim, M.; Piazuelo, M.B.; Barry, D.P.; Verriere, T.G.; Sierra, J.C.; Hardbower, D.M.; Delgado, A.G.; Schneider, B.G.; et al. Increased Helicobacter pylori-associated gastric cancer risk in the Andean region of Colombia is mediated by spermine oxidase. Oncogene 2015, 34, 3429–3440. [Google Scholar] [CrossRef]
- Goodwin, A.C.; Destefano Shields, C.E.; Wu, S.; Huso, D.L.; Wu, X.; Murray-Stewart, T.R.; Hacker-Prietz, A.; Rabizadeh, S.; Woster, P.M.; Sears, C.L.; et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15354–15359. [Google Scholar] [CrossRef]
- Murray-Stewart, T.; Sierra, J.C.; Piazuelo, M.B.; Mera, R.M.; Chaturvedi, R.; Bravo, L.E.; Correa, P.; Schneider, B.G.; Wilson, K.T.; Casero, R.A. Epigenetic silencing of miR-124 prevents spermine oxidase regulation: Implications for Helicobacter pylori-induced gastric cancer. Oncogene 2016, 35, 5480–5488. [Google Scholar] [CrossRef]
- Zahedi, K.; Lentsch, A.B.; Okaya, T.; Barone, S.; Sakai, N.; Witte, D.P.; Arend, L.J.; Alhonen, L.; Jell, J.; Janne, J.; et al. Spermidine/spermine-N1-acetyltransferase ablation protects against liver and kidney ischemia-reperfusion injury in mice. Am. J. Physiol. Gastrointest Liver Physiol. 2009, 296, G899–G909. [Google Scholar] [CrossRef]
- Golab, F.; Kadkhodaee, M.; Zahmatkesh, M.; Hedayati, M.; Arab, H.; Schuster, R.; Zahedi, K.; Lentsch, A.B.; Soleimani, M. Ischemic and non-ischemic acute kidney injury cause hepatic damage. Kidney Int. 2009, 75, 783–792. [Google Scholar] [CrossRef]
- Cervelli, M.; Bellavia, G.; D’Amelio, M.; Cavallucci, V.; Moreno, S.; Berger, J.; Nardacci, R.; Marcoli, M.; Maura, G.; Piacentini, M.; et al. A New Transgenic Mouse Model for Studying the Neurotoxicity of Spermine Oxidase Dosage in the Response to Excitotoxic Injury. PLoS ONE 2013, 8, e64810. [Google Scholar] [CrossRef]
- Dogan, A.; Rao, A.M.; Baskaya, M.K.; Hatcher, J.; Temiz, C.; Rao, V.L.; Dempsey, R.J. Contribution of polyamine oxidase to brain injury after trauma. J. Neurosurg. 1999, 90, 1078–1082. [Google Scholar] [CrossRef]
- Ivanova, S.; Batliwalla, F.; Mocco, J.; Kiss, S.; Huang, J.; Mack, W.; Coon, A.; Eaton, J.W.; Al-Abed, Y.; Gregersen, P.K.; et al. Neuroprotection in cerebral ischemia by neutralization of 3-aminopropanal. Proc. Natl. Acad. Sci. USA 2002, 99, 5579–5584. [Google Scholar] [CrossRef]
- Zoli, M.; Pedrazzi, P.; Zini, I.; Agnati, L.F. Spermidine/spermine N1-acetyltransferase mRNA levels show marked and region-specific changes in the early phase after transient forebrain ischemia. Brain Res. Mol. Brain Res. 1996, 38, 122–134. [Google Scholar] [CrossRef]
- Tomitori, H.; Usui, T.; Saeki, N.; Ueda, S.; Kase, H.; Nishimura, K.; Kashiwagi, K.; Igarashi, K. Polyamine oxidase and acrolein as novel biochemical markers for diagnosis of cerebral stroke. Stroke 2005, 36, 2609–2613. [Google Scholar] [CrossRef]
- Patel, C.; Xu, Z.; Shosha, E.; Xing, J.; Lucas, R.; Caldwell, R.W.; Caldwell, R.B.; Narayanan, S.P. Treatment with polyamine oxidase inhibitor reduces microglial activation and limits vascular injury in ischemic retinopathy. Biochim. Biophys. Acta 2016, 1862, 1628–1639. [Google Scholar] [CrossRef]
- Pichavaram, P.; Palani, C.D.; Patel, C.; Xu, Z.; Shosha, E.; Fouda, A.Y.; Caldwell, R.B.; Narayanan, S.P. Targeting Polyamine Oxidase to Prevent Excitotoxicity-Induced Retinal Neurodegeneration. Front. Neurosci. 2018, 12, 956. [Google Scholar] [CrossRef]
- Hyvonen, M.T.; Herzig, K.H.; Sinervirta, R.; Albrecht, E.; Nordback, I.; Sand, J.; Keinanen, T.A.; Vepsalainen, J.; Grigorenko, N.; Khomutov, A.R.; et al. Activated polyamine catabolism in acute pancreatitis: Alpha-methylated polyamine analogues prevent trypsinogen activation and pancreatitis-associated mortality. Am. J. Pathol. 2006, 168, 115–122. [Google Scholar] [CrossRef]
- Hyvonen, M.T.; Sinervirta, R.; Grigorenko, N.; Khomutov, A.R.; Vepsalainen, J.; Keinanen, T.A.; Alhonen, L. alpha-Methylspermidine protects against carbon tetrachloride-induced hepatic and pancreatic damage. Amino Acids 2010, 38, 575–581. [Google Scholar] [CrossRef]
- Duan, Q.; Yang, W.; Jiang, D.; Tao, K.; Dong, A.; Cheng, H. Spermine ameliorates ischemia/reperfusion injury in cardiomyocytes via regulation of autophagy. Am. J. Transl. Res. 2016, 8, 3976–3985. [Google Scholar]
- Jell, J.; Merali, S.; Hensen, M.L.; Mazurchuk, R.; Spernyak, J.A.; Diegelman, P.; Kisiel, N.D.; Barrero, C.; Deeb, K.K.; Alhonen, L.; et al. Genetically altered expression of spermidine/spermine N1-acetyltransferase affects fat metabolism in mice via acetyl-CoA. J. Biol. Chem. 2007, 282, 8404–8413. [Google Scholar] [CrossRef]
- Yuan, F.; Zhang, L.; Cao, Y.; Gao, W.; Zhao, C.; Fang, Y.; Zahedi, K.; Soleimani, M.; Lu, X.; Fang, Z.; et al. Spermidine/spermine N1-acetyltransferase-mediated polyamine catabolism regulates beige adipocyte biogenesis. Metabolism 2018, 85, 298–304. [Google Scholar] [CrossRef]
- Jain, V.; Raina, S.; Gheware, A.P.; Singh, R.; Rehman, R.; Negi, V.; Murray Stewart, T.; Mabalirajan, U.; Mishra, A.K.; Casero, R.A., Jr.; et al. Reduction in polyamine catabolism leads to spermine-mediated airway epithelial injury and induces asthma features. Allergy 2018, 73, 2033–2045. [Google Scholar] [CrossRef]
- Zahedi, K.; Barone, S.; Wang, Y.; Murray-Stewart, T.; Roy-Chaudhury, P.; Smith, R.D.; Casero, R.A., Jr.; Soleimani, M. Proximal tubule epithelial cell specific ablation of the spermidine/spermine N1-acetyltransferase gene reduces the severity of renal ischemia/reperfusion injury. PLoS ONE 2014, 9, e110161. [Google Scholar] [CrossRef]
- Pirnes-Karhu, S.; Sironen, R.; Alhonen, L.; Uimari, A. Lipopolysaccharide-induced anti-inflammatory acute phase response is enhanced in spermidine/spermine N1-acetyltransferase (SSAT) overexpressing mice. Amino Acids 2012, 42, 473–484. [Google Scholar] [CrossRef]
- Cerrada-Gimenez, M.; Pietila, M.; Loimas, S.; Pirinen, E.; Hyvonen, M.T.; Keinanen, T.A.; Janne, J.; Alhonen, L. Continuous oxidative stress due to activation of polyamine catabolism accelerates aging and protects against hepatotoxic insults. Transgenic Res. 2011, 20, 387–396. [Google Scholar] [CrossRef]
- Ha, H.C.; Woster, P.M.; Yager, J.D.; Casero, R.A., Jr. The role of polyamine catabolism in polyamine analogue-induced programmed cell death. Proc. Natl. Acad. Sci. USA 1997, 94, 11557–11562. [Google Scholar] [CrossRef]
- Hussain, T.; Tan, B.; Ren, W.; Rahu, N.; Dad, R.; Kalhoro, D.H.; Yin, Y. Polyamines: Therapeutic perspectives in oxidative stress and inflammatory diseases. Amino Acids 2017, 49, 1457–1468. [Google Scholar] [CrossRef]
- Murray Stewart, T.; Dunston, T.T.; Woster, P.M.; Casero, R.A., Jr. Polyamine catabolism and oxidative damage. J. Biol. Chem. 2018, 293, 18736–18745. [Google Scholar] [CrossRef]
- Pegg, A.E. Spermidine/spermine-N(1)-acetyltransferase: A key metabolic regulator. Am. J. Physiol. Endocrinol. Metab. 2008, 294, 995–1010. [Google Scholar] [CrossRef]
- Johnson, A.C.M.; Zager, R.A. Mechanisms and consequences of oxidant-induced renal preconditioning: An Nrf2-dependent, P21-independent, anti-senescence pathway. Nephrol. Dial. Transplant. 2018, 33, 1927–1941. [Google Scholar] [CrossRef]
- Pan, T.; Jia, P.; Chen, N.; Fang, Y.; Liang, Y.; Guo, M.; Ding, X. Delayed Remote Ischemic Preconditioning ConfersRenoprotection against Septic Acute Kidney Injury via Exosomal miR-21. Theranostics 2019, 9, 405–423. [Google Scholar] [CrossRef]
- Babbar, N.; Casero, R.A., Jr. Tumor necrosis factor-alpha increases reactive oxygen species by inducing spermine oxidase in human lung epithelial cells: A potential mechanism for inflammation-induced carcinogenesis. Cancer Res. 2006, 66, 11125–11130. [Google Scholar] [CrossRef]
- Babbar, N.; Hacker, A.; Huang, Y.; Casero, R.A., Jr. Tumor necrosis factor alpha induces spermidine/spermine N1-acetyltransferase through nuclear factor kappaB in non-small cell lung cancer cells. J. Biol. Chem. 2006, 281, 24182–24192. [Google Scholar] [CrossRef]
- Chopra, S.; Wallace, H.M. Induction of spermidine/spermine N1-acetyltransferase in human cancer cells in response to increased production of reactive oxygen species. Biochem. Pharmacol. 1998, 55, 1119–1123. [Google Scholar] [CrossRef]
- Smirnova, O.A.; Isaguliants, M.G.; Hyvonen, M.T.; Keinanen, T.A.; Tunitskaya, V.L.; Vepsalainen, J.; Alhonen, L.; Kochetkov, S.N.; Ivanov, A.V. Chemically induced oxidative stress increases polyamine levels by activating the transcription of ornithine decarboxylase and spermidine/spermine-N1-acetyltransferase in human hepatoma HUH7 cells. Biochimie 2012, 94, 1876–1883. [Google Scholar] [CrossRef]
- Donnahoo, K.K.; Meng, X.; Ayala, A.; Cain, M.P.; Harken, A.H.; Meldrum, D.R. Early kidney TNF-alpha expression mediates neutrophil infiltration and injury after renal ischemia-reperfusion. Am. J. Physiol. 1999, 277, 922–929. [Google Scholar]
- Kielar, M.L.; John, R.; Bennett, M.; Richardson, J.A.; Shelton, J.M.; Chen, L.; Jeyarajah, D.R.; Zhou, X.J.; Zhou, H.; Chiquett, B.; et al. Maladaptive role of IL-6 in ischemic acute renal failure. J. Am. Soc. Nephrol. 2005, 16, 3315–3325. [Google Scholar] [CrossRef]
- Nechemia-Arbely, Y.; Barkan, D.; Pizov, G.; Shriki, A.; Rose-John, S.; Galun, E.; Axelrod, J.H. IL-6/IL-6R axis plays a critical role in acute kidney injury. J. Am. Soc. Nephrol. 2008, 19, 1106–1115. [Google Scholar] [CrossRef]
- Zhang, B.; Ramesh, G.; Norbury, C.C.; Reeves, W.B. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-alpha produced by renal parenchymal cells. Kidney Int. 2007, 72, 37–44. [Google Scholar] [CrossRef]
- Marko, L.; Vigolo, E.; Hinze, C.; Park, J.K.; Roel, G.; Balogh, A.; Choi, M.; Wubken, A.; Cording, J.; Blasig, I.E.; et al. Tubular Epithelial NF-kappaB Activity Regulates Ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 2658–2669. [Google Scholar] [CrossRef]
- Song, N.; Thaiss, F.; Guo, L. NFkappaB and Kidney Injury. Front. Immunol. 2019, 10, 815. [Google Scholar] [CrossRef]
- McNicholas, B.A.; Griffin, M.D. Double-edged sword: A p53 regulator mediates both harmful and beneficial effects in experimental acute kidney injury. Kidney Int. 2012, 81, 1161–1164. [Google Scholar] [CrossRef]
- Molitoris, B.A.; Dagher, P.C.; Sandoval, R.M.; Campos, S.B.; Ashush, H.; Fridman, E.; Brafman, A.; Faerman, A.; Atkinson, S.J.; Thompson, J.D.; et al. siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J. Am. Soc. Nephrol. 2009, 20, 1754–1764. [Google Scholar] [CrossRef]
- Tang, C.; Ma, Z.; Zhu, J.; Liu, Z.; Liu, Y.; Liu, Y.; Cai, J.; Dong, Z. P53 in kidney injury and repair: Mechanism and therapeutic potentials. Pharmacol. Ther. 2019, 195, 5–12. [Google Scholar] [CrossRef]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef]
- Pietila, M.; Alhonen, L.; Halmekyto, M.; Kanter, P.; Janne, J.; Porter, C.W. Activation of polyamine catabolism profoundly alters tissue polyamine pools and affects hair growth and female fertility in transgenic mice overexpressing spermidine/spermine N1-acetyltransferase. J. Biol. Chem. 1997, 272, 18746–18751. [Google Scholar] [CrossRef]
- Zahedi, K.; Bissler, J.J.; Wang, Z.; Josyula, A.; Lu, L.; Diegelman, P.; Kisiel, N.; Porter, C.W.; Soleimani, M. Spermidine/spermine N1-acetyltransferase overexpression in kidney epithelial cells disrupts polyamine homeostasis, leads to DNA damage, and causes G2 arrest. Am. J. Physiol. Cell Physiol. 2007, 292, 1204–1215. [Google Scholar] [CrossRef]
- Alhonen, L.; Pietila, M.; Halmekyto, M.; Kramer, D.L.; Janne, J.; Porter, C.W. Transgenic mice with activated polyamine catabolism due to overexpression of spermidine/spermine N1-acetyltransferase show enhanced sensitivity to the polyamine analog, N1, N11-diethylnorspermine. Mol. Pharmacol. 1999, 55, 693–698. [Google Scholar]
- Ceci, R.; Duranti, G.; Leonetti, A.; Pietropaoli, S.; Spinozzi, F.; Marcocci, L.; Amendola, R.; Cecconi, F.; Sabatini, S.; Mariottini, P.; et al. Adaptive responses of heart and skeletal muscle to spermine oxidase overexpression: Evaluation of a new transgenic mouse model. Free Radic. Biol. Med. 2017, 103, 216–225. [Google Scholar] [CrossRef]
- Mandal, S.; Mandal, A.; Park, M.H. Depletion of the polyamines spermidine and spermine by overexpression of spermidine/spermine N(1)-acetyltransferase 1 (SAT1) leads to mitochondria-mediated apoptosis in mammalian cells. Biochem. J. 2015, 468, 435–447. [Google Scholar] [CrossRef]
- Landau, G.; Bercovich, Z.; Park, M.H.; Kahana, C. The role of polyamines in supporting growth of mammalian cells is mediated through their requirement for translation initiation and elongation. J. Biol. Chem. 2010, 285, 12474–12481. [Google Scholar] [CrossRef]
- Adibhatla, R.M.; Hatcher, J.F.; Sailor, K.; Dempsey, R.J. Polyamines and central nervous system injury: Spermine and spermidine decrease following transient focal cerebral ischemia in spontaneously hypertensive rats. Brain Res. 2002, 938, 81–86. [Google Scholar] [CrossRef]
- Das, K.C.; Misra, H.P. Hydroxyl radical scavenging and singlet oxygen quenching properties of polyamines. Mol. Cell. Biochem. 2004, 262, 127–133. [Google Scholar] [CrossRef]
- Ha, H.C.; Sirisoma, N.S.; Kuppusamy, P.; Zweier, J.L.; Woster, P.M.; Casero, R.A., Jr. The natural polyamine spermine functions directly as a free radical scavenger. Proc. Natl. Acad. Sci. USA 1998, 95, 11140–11145. [Google Scholar] [CrossRef]
- Marzabadi, M.R.; Llvaas, E. Spermine prevent iron accumulation and depress lipofuscin accumulation in cultured myocardial cells. Free Radic. Biol. Med. 1996, 21, 375–381. [Google Scholar] [CrossRef]
- Kim, J. Spermidine is protective against kidney ischemia and reperfusion injury through inhibiting DNA nitration and PARP1 activation. Anat. Cell Biol. 2017, 50, 200–206. [Google Scholar] [CrossRef]
- Cohen, S.S. A Guide to the Polyamines; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Barbouti, A.; Doulias, P.T.; Zhu, B.Z.; Frei, B.; Galaris, D. Intracellular iron, but not copper, plays a critical role in hydrogen peroxide-induced DNA damage. Free Radic. Biol. Med. 2001, 31, 490–498. [Google Scholar] [CrossRef]
- Driessens, N.; Versteyhe, S.; Ghaddhab, C.; Burniat, A.; De Deken, X.; Van Sande, J.; Dumont, J.E.; Miot, F.; Corvilain, B. Hydrogen peroxide induces DNA single- and double-strand breaks in thyroid cells and is therefore a potential mutagen for this organ. Endocr. Relat. Cancer 2009, 16, 845–856. [Google Scholar] [CrossRef]
- Boya, P.; Andreau, K.; Poncet, D.; Zamzami, N.; Perfettini, J.L.; Metivier, D.; Ojcius, D.M.; Jaattela, M.; Kroemer, G. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J. Exp. Med. 2003, 197, 1323–1334. [Google Scholar] [CrossRef]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef]
- Brunk, U.T.; Zhang, H.; Dalen, H.; Ollinger, K. Exposure of cells to nonlethal concentrations of hydrogen peroxide induces degeneration-repair mechanisms involving lysosomal destabilization. Free Radic. Biol. Med. 1995, 19, 813–822. [Google Scholar] [CrossRef]
- Brunk, U.T.; Zhang, H.; Roberg, K.; Ollinger, K. Lethal hydrogen peroxide toxicity involves lysosomal iron-catalyzed reactions with membrane damage. Redox Rep. 1995, 1, 267–277. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Gao, X.; Fu, L.; Xiao, M.; Xu, C.; Sun, L.; Zhang, T.; Zheng, F.; Mei, C. The nephroprotective effect of tauroursodeoxycholic acid on ischaemia/reperfusion-induced acute kidney injury by inhibiting endoplasmic reticulum stress. Basic Clin. Pharmacol. Toxicol. 2012, 111, 14–23. [Google Scholar] [CrossRef]
- Hosohata, K. Role of Oxidative Stress in Drug-Induced Kidney Injury. Int. J. Mol. Sci. 2016, 17, 1826. [Google Scholar] [CrossRef]
- Li, S.Y.; Gilbert, S.A.; Li, Q.; Ren, J. Aldehyde dehydrogenase-2 (ALDH2) ameliorates chronic alcohol ingestion-induced myocardial insulin resistance and endoplasmic reticulum stress. J. Mol. Cell. Cardiol. 2009, 47, 247–255. [Google Scholar] [CrossRef]
- Pallepati, P.; Averill-Bates, D.A. Activation of ER stress and apoptosis by hydrogen peroxide in HeLa cells: Protective role of mild heat preconditioning at 40 degrees C. Biochim. Biophys. Acta 2011, 1813, 1987–1999. [Google Scholar] [CrossRef]
- Jang, H.R.; Rabb, H. The innate immune response in ischemic acute kidney injury. Clin. Immunol. 2009, 130, 41–50. [Google Scholar] [CrossRef]
- Pulskens, W.P.; Teske, G.J.; Butter, L.M.; Roelofs, J.J.; van der Poll, T.; Florquin, S.; Leemans, J.C. Toll-like receptor-4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS ONE 2008, 3, e3596. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahedi, K.; Barone, S.; Soleimani, M. Polyamine Catabolism in Acute Kidney Injury. Int. J. Mol. Sci. 2019, 20, 4790. https://doi.org/10.3390/ijms20194790
Zahedi K, Barone S, Soleimani M. Polyamine Catabolism in Acute Kidney Injury. International Journal of Molecular Sciences. 2019; 20(19):4790. https://doi.org/10.3390/ijms20194790
Chicago/Turabian StyleZahedi, Kamyar, Sharon Barone, and Manoocher Soleimani. 2019. "Polyamine Catabolism in Acute Kidney Injury" International Journal of Molecular Sciences 20, no. 19: 4790. https://doi.org/10.3390/ijms20194790
APA StyleZahedi, K., Barone, S., & Soleimani, M. (2019). Polyamine Catabolism in Acute Kidney Injury. International Journal of Molecular Sciences, 20(19), 4790. https://doi.org/10.3390/ijms20194790